

Abstract
We compared two DNA extraction methods (a semiautomated method using a Maxwell kit and a modified Boom method) and three amplification procedures (a single-step PCR, a nested PCR, and a real-time quantitative PCR) on 74 surgical tissue specimens from patients with clinically suspected Buruli ulcer. All of these procedures were compared before and after decontamination. We observed that, among the procedures tested, real-time PCR after the modified Boom extraction method or a single-run PCR assay after the Maxwell 16 extraction method, performed on nondecontaminated suspensions, are the best for the molecular diagnosis of Mycobacterium ulcerans disease.
INTRODUCTION
Mycobacterium ulcerans disease, commonly called Buruli ulcer (BU), is a skin disease mainly endemic to certain riverine areas of West and Central Africa (4, 12). The disease may present with a diverse range of clinical symptoms, and, due to a possible confusion with other tropical skin diseases, a diagnosis based strictly on clinical observation is not always accurate, leading to the necessity of confirmatory tests such as microscopy, culture, histopathology, and PCR (3, 16).
Microscopy is comparatively straightforward to perform, and the materials and skills required are available in regions of endemicity; but diagnosis based on microscopy lacks sensitivity (16). Therefore, even when samples are negative when tested by microscopy, another test should be carried out to confirm the diagnosis. Cultivation of M. ulcerans also lacks sensitivity and takes time to give results (6), rendering it less useful for the routine management of patients. Histopathology is sensitive, specific for BU, and helpful for differential diagnosis but is rarely available in countries where BU is endemic due to a lack of specialists trained and experienced in this technique (16).
PCR has been shown to be sensitive as well as specific and is increasingly used for BU diagnosis in countries of endemicity (10, 14). Various commercial and in-house DNA extraction and amplification procedures are used, mainly targeting the insertion sequence IS2404 of the M. ulcerans genome. However, only a few studies have evaluated these methods (5). Nevertheless, such an evaluation would be essential to identify the best method for the detection of M. ulcerans by PCR.
Since several laboratories which perform PCR also routinely perform culture, PCR is sometimes carried out on suspensions of specimens that have been subjected to a decontamination protocol in preparation for culture. To our knowledge, the effects of this decontamination step on PCR results have not, to date, been investigated.
In this study, we compared in two separate laboratories two extraction methods (a semiautomated method using the Maxwell 16 kit [Promega, Leiden, The Netherlands] and a modification of the method of Boom [11]) and three amplification procedures (a single-step PCR, a nested PCR, and a real-time quantitative PCR) routinely performed on surgical tissue specimens from patients with suspected BU. All of these procedures were applied on suspensions of specimens before and after a relevant culture decontamination protocol.
MATERIALS AND METHODS
Specimens.
Seventy-four human surgical tissue specimens from patients with suspected BU consecutively received from the Centre Sanitaire et Nutritionel Gbemoten (Zagnanado, Benin) were included. Each 1-g tissue specimen was stored in a semisolid transport medium (6) and split into two subsamples: one subsample was processed at the National Mycobacteria Reference Laboratory (Laboratoire de Référence des Mycobactéries [LRM]) in Cotonou, Benin, and the second subsample was sent to the Mycobacteriology Unit of the Institute of Tropical Medicine ([ITM] Antwerp, Belgium) within an average delay of 50 days. All 74 specimens were subjected to the same flow of procedures summarized in Fig. 1.
Fig 1.

Summary of procedures used.
Analyses at LRM.
At the LRM, each specimen was ground in a mortar, suspended in 2 ml of sterile distilled water, and split into two equal parts. One was directly subjected to the Maxwell 16 DNA extraction method, followed by a single-run gel-based PCR assay, while the second was subjected to decontamination using the reversed Petroff method (13); the pellet was resuspended in 1 ml of sterile distilled water. The decontamination method included a centrifugation step with a relative centrifugal force of 3,000 × g. After decontamination, the same DNA extraction method followed by the same PCR assay was performed on the new suspension.
Analyses at ITM.
At the ITM, the specimen was ground, suspended in 2 ml of sterile distilled water, and split into two equal parts. One directly underwent DNA extraction by a modified Boom method followed by either a nested gel-based PCR assay or a real-time PCR assay. The second part was subjected to decontamination with the reversed Petroff method (13) as at the LRM, and the pellet was suspended in 0.5 ml of sterile distilled water before being subjected to the same DNA extraction method and PCR assays performed on the suspensions before decontamination.
DNA extraction methods.
The Maxwell 16 DNA purification kit and the Maxwell 16 instrument (Promega, Leiden, The Netherlands) were used as previously described (5); 200 μl of suspended specimen was used, and the final volume of DNA suspension was 300 μl. The modified Boom DNA extraction method was carried out as previously described (11); 250 μl of suspended specimen was used, and the final volume of DNA suspension was 100 μl.
PCR assays.
The following three PCR assays were performed as previously described: a single-run gel-based PCR (13), a nested gel-based PCR (11), and a quantitative real-time PCR (7). All PCR assays targeted the insertion sequence IS2404 (8, 15).
For the remainder of this paper we will refer to the different methodologies used thus (Fig. 1): the Maxwell 16 extraction method followed by the single-run conventional PCR assay is referred to as procedure A; the modified Boom extraction method followed by the nested conventional PCR assay is referred to as procedure B; the modified Boom extraction method followed by the real-time PCR assay is referred to as procedure C.
Other combinations such as Maxwell 16 extraction followed by a nested or real-time PCR or modified Boom extraction followed by a single-run PCR, using identical volumes for different methods, were not tested since the goal of the study was to assess the relative interest of procedures routinely performed in the two laboratories for diagnosis of BU.
Direct smear examination.
One drop of the suspension before decontamination at ITM was smeared on a slide, stained with the Ziehl-Neelsen technique, read microscopically, and graded according to the American Thoracic Society scale (2).
Quality control.
At each step (extraction and amplification), several quality control samples (negative as well as positive) were routinely analyzed together with the test specimens. Both laboratories participate in the external quality control assessments for M. ulcerans PCR organized by the World Health Organization.
Statistical analysis.
PCR positivity rates were calculated as the ratio of the number of positive PCR results over the number of tested samples from patients with clinically suspected BU. Data were analyzed with SPSS (version 18.0) software (IBM, Armonk, NY). Nonparametric Cochran's Q tests for related samples with pairwise comparisons were used to test whether the distributions of the methods were equal.
Ethical provisions.
The provisional medical ethical committee of Benin (registered under IRB 00006860) gave its authorization to analyze and report the clinical data collected from the treated patients (ethical authorization number 011 of the 10 December 2010). Written informed consent was obtained from all patients or their parents or guardians (for patients younger than 18 years).
RESULTS
Comparison before versus after decontamination.
As shown in Table 1, performing PCR analysis before decontamination rather than after gave higher positivity rates for all procedures although this was only significant for procedures A (Q = −0.216; P < 0.0001) and B (Q = −0.108; P = 0.029). The decrease in positivity rate after decontamination was more important for procedure A than for procedure B and procedure C.
Table 1.
PCR positivity rates of the different procedures
| Procedurea | No. of positive specimens (%)b |
|
|---|---|---|
| Before decontamination (n = 74 ) | After decontamination (n = 74) | |
| A | 48 (64.9)† | 32 (41.9)#† |
| B | 41 (55.4)*‡ | 33 (44.6)°‡ |
| C | 50 (67.6)* | 44 (59.5)°# |
Procedure A, Maxwell 16 extraction followed by single-run gel-based PCR; procedure B, modified Boom extraction followed by nested PCR; procedure C, modified Boom extraction followed by real-time PCR.
Only significant P values of pairwise comparisons are displayed: *, Q = 0.122, P = 0.014; °, Q = 0.149, P = 0.003; #, Q = 0.162, P = 0.001; †, Q = −0.216, P < 0.0001; ‡, Q = −0.108; P = 0.029.
Before decontamination.
Procedure C resulted in the highest positivity rate. This was statistically significant compared to procedure B (Q = 0.122; P = 0.014) but not significantly higher than the rate for procedure A (Q = 0.027; P = 0.586). Five specimens were positive only with procedure C (Fig. 2); all specimens were positive only after ≥38 amplification cycles, indicating that very little template DNA was present. Moreover, only two of these specimens had a “scanty” positive smear; the remaining three specimens were smear negative. Procedure A also had five unique positive specimens (Fig. 2).
Fig 2.

Distribution of specimens positive with at least one procedure before decontamination (n = 55).
When the grading of the smears of these specimens after Ziehl-Neelsen staining was compared with the quantification by real-time PCR, quantitative PCR (qPCR) gave a similar estimation of bacillary load as direct smear examination (Fig. 3).
Fig 3.
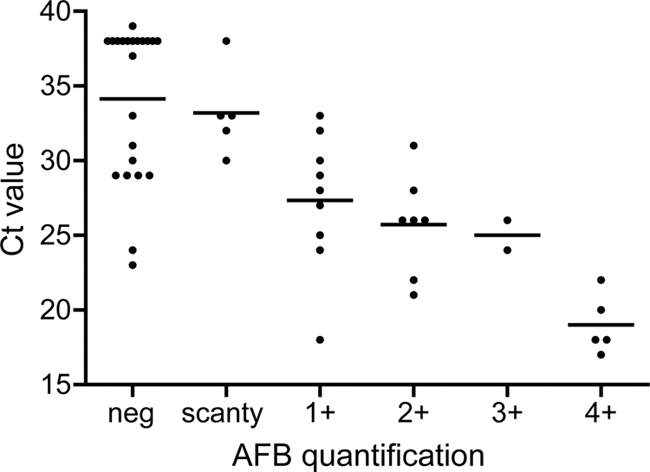
Quantification of bacillary load by direct-smear examination versus quantitative real-time PCR for the 50 specimens positive by real-time PCR before decontamination. The values of all specimens are plotted with the average cycle threshold (CT) value per acid-fast bacillus (AFB).
After decontamination.
Procedure C gave the highest positivity rate. This was statistically significant compared to results from both procedure A (Q = 0.162, P = 0.001) and procedure B (Q = 0.149; P = 0.003). Ten specimens were positive only with procedure C (all only after >36 amplification cycles with one scanty positive smear and one scored 1+) while procedure A had only two unique positive specimens. Procedure B had no unique positives (Fig. 4). The positivity rates of procedures A and B were not significantly different (Q = 0.014; P = 0.785).
Fig 4.

Distribution of specimens positive with at least one procedure after decontamination (n = 46).
DISCUSSION
The PCR positivity rates obtained in this study correspond to those obtained in previous studies, varying between 61% and 83%, depending on the type of lesion and specimen (3).
This study demonstrated that for all procedures some bacterial DNA is lost during the application of standard decontamination protocols. Some mycobacteria may be lysed, and the relative centrifugal force of 3,000 × g as recommended by the WHO may not be sufficient to spin down the released DNA (13).
Durnez et al. (5) concluded that, after decontamination, the modified Boom extraction method followed by a nested gel-based PCR performed better than Maxwell 16 extraction followed by the same PCR assay. In the present study, Maxwell 16 extraction followed by single-run PCR performed as well as the modified Boom extraction method followed by a nested PCR. Possibly the DNA may have been degraded en route to the ITM, resulting in a reduced positivity of procedures B and C, both of which use the modified Boom extraction method. The lower detection limit of real-time PCR may have compensated for this DNA degradation in procedure C.
Of the nondecontaminated suspensions, the single-run PCR had higher positivity rates than the nested PCR and rates very similar to those of the real-time quantitative PCR, known to be very sensitive (7). One would expect a nested PCR assay to be more sensitive than a single-step PCR assay. However, although both of these PCR assays target the insertion sequence IS2404, primers as well as PCR conditions are different. The single-run PCR conditions may be more optimal than those of the nested PCR. In order to test whether the different results were actually due to the PCR assays, Maxwell 16 DNA extracts could have been tested by the nested PCR in the ITM, and modified Boom DNA extracts could have been tested by the single-run PCR in the LRM.
Real-time PCR is relatively easy to perform but expensive and not available in most settings where BU is endemic. However, as shown by our results, performing a gel-based PCR with a high degree of quality control on nondecontaminated specimens gives similar results to quantitative real-time PCR and could be routinely used in these settings. Specimens positive using procedure C but negative using procedure A were only weakly positive with cycle threshold values of 38 or higher.
In all PCR and extraction runs, negative- and positive-control samples were included with results that were in every case valid. In addition, a contamination check (tube with water left open for hours in the pre-PCR and extraction rooms) was regularly performed. An international external quality control recently organized showed an agreement of 97% and 100%, indicating a minimal difference between their performances. It was difficult to calculate the sensitivity and specificity of the methods compared since no method can reasonably be used as a reference. For the assessment of specificity, it would be necessary to collect tissue specimens from patients who were confirmed to be BU negative, such as patients with diseases other than BU or healthy persons.
All smear-positive specimens were PCR positive, indicating that microscopy can be used as a screening test in peripheral centers of countries where BU is endemic (1, 17). Therefore, for cost-effectiveness, PCR can be reserved only for specimens judged negative by microscopy. Direct-smear examination can be used to quantify bacillary load in strong-positive specimens while real-time PCR can quantify bacillary load in weakly positive specimens. No conclusion can be drawn on bacterial viability by these tests. Nevertheless, the estimations can be used to predict disease outcome. Indeed, Lagarrigue et al. (9) found that bacterial load in cutaneous BU lesions quantified by AFB grading was associated with the development of bone dissemination.
The procedures investigated in this study used different volumes (either for specimen suspensions or DNA extraction solutions) but were routinely performed in the two laboratories. Other combinations such as Maxwell 16 extraction followed by a nested or real-time PCR or modified Boom extraction followed by a single-run PCR, using identical volumes for the different methods, also could have been tested. However, the goal of the study was to assess the relative interest of procedures routinely performed in the two laboratories for diagnosis of BU. Further studies could assess the value of other combinations in order to identify the optimal combination of DNA extraction procedure and PCR assay.
In conclusion, among the procedures tested, real-time PCR after the modified Boom extraction method and a single-run PCR assay after the Maxwell 16 extraction method, performed on nondecontaminated suspensions, are the best methods for the molecular diagnosis of M. ulcerans disease and are now implemented routinely for the testing of specimens from patients with suspected BU in the ITM and the LRM, respectively.
ACKNOWLEDGMENTS
This study was supported by the Directorate General for Development Cooperation (DGDC, Brussels, Belgium) through the Project 3.05: Buruli Ulcer Control (Benin, West Africa), by the Stop Buruli Initiative funded by the UBS Optimus Foundation (Zurich, Switzerland), by the European Commission (project HEALTH-F3-2010-241500-BuruliVac), and by the Fund for Scientific Research, Flanders (Belgium) (FWO grant number G.0301.01).
Footnotes
Published ahead of print 18 January 2012
REFERENCES
- 1. Affolabi D. 2009. Development of simple and inexpensive tools for the control of mycobacterial diseases in a low-resource country. Ph.D. dissertation University of Antwerp, Antwerp, Belgium [Google Scholar]
- 2. American Thoracic Society 1981. Diagnostic standards and classification of tuberculosis and other mycobacterial diseases. Am. Rev. Respir. Dis. 123:343–358 [DOI] [PubMed] [Google Scholar]
- 3. Beissner M, Herbinger KH, Bretzel G. 2010. Laboratory diagnosis of Buruli ulcer disease. Future Microbiol. 5:363–370 [DOI] [PubMed] [Google Scholar]
- 4. Debacker M, et al. 2004. Mycobacterium ulcerans disease (Buruli ulcer) in rural hospital, southern Benin, 1997–2001. Emerg. Infect. Dis. 10:1391–1398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Durnez L, et al. 2009. A comparison of DNA extraction procedures for the detection of Mycobacterium ulcerans, the causative agent of Buruli ulcer, in clinical and environmental specimens. J. Microbiol. Methods 76:152–158 [DOI] [PubMed] [Google Scholar]
- 6. Eddyani M, et al. 2008. Primary culture of Mycobacterium ulcerans from human tissue specimens after storage in semisolid transport medium. J. Clin. Microbiol. 46:69–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fyfe JAM, et al. 2007. Development and application of two multiplex real-time PCR assays for the detection of Mycobacterium ulcerans in clinical and environmental samples. Appl. Environ. Microbiol. 73:4733–4740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Guimaraes-Peres A, et al. 1999. Comparison of two PCRs for detection of Mycobacterium ulcerans. J. Clin. Microbiol. 37:206–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lagarrigue V, Portaels F, Meyers WM, Aguiar J. 2000. L'ulcère de Buruli: attention aux atteintes osseuses! A propos de 33 cas observés au Bénin. Med. Trop. (Mars.) 60:262–266 [PubMed] [Google Scholar]
- 10. Phillips R, et al. 2005. Sensitivity of PCR targeting the IS2404 insertion sequence of Mycobacterium ulcerans in an assay using punch biopsy specimens for diagnosis of Buruli ulcer. J. Clin. Microbiol. 43:3650–3656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Portaels F, et al. 2008. First cultivation and characterization of Mycobacterium ulcerans from the environment. PLoS Negl. Trop. Dis. 2:e178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Portaels F, Silva MT, Meyers WM. 2009. Buruli ulcer. Clin. Dermatol. 27:291–305 [DOI] [PubMed] [Google Scholar]
- 13. Portaels F, Johnson P, Meyers WM. (ed). 2001. Buruli ulcer. Diagnosis of Mycobacterium ulcerans disease. A manual for health care providers. WHO/CDS/GPE/GBUI/2001.4. World Health Organization, Geneva, Switzerland [Google Scholar]
- 14. Siegmund V, et al. 2005. Dry-reagent-based PCR as a novel tool for laboratory confirmation of clinically diagnosed Mycobacterium ulcerans-associated disease in areas in the tropics where M. ulcerans is endemic. J. Clin. Microbiol. 43:271–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stienstra Y, et al. 2003. Analysis of an IS2404-based nested PCR for diagnosis of Buruli ulcer disease in regions of Ghana where the disease is endemic. J. Clin. Microbiol. 41:794–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. van der Werf TS, et al. 2005. Mycobacterium ulcerans disease. Bull. World Health Organ. 83:785–791 [PMC free article] [PubMed] [Google Scholar]
- 17. Yeboah-Manu D, Asante-Poku A, Asan-Ampah K, Ampadu ED, Pluschke G. 2011. Combining PCR with microscopy to reduce costs of laboratory diagnosis of Buruli ulcer. Am. J. Trop. Med. Hyg. 85:900–904 [DOI] [PMC free article] [PubMed] [Google Scholar]