

Abstract
Current hepatitis E virus (HEV) negative-sense RNA detection assays have the drawback of false positivity. cDNA synthesis using tag-based primer and Superscript RT-III followed by exonuclease I treatment increased the specificity. Assays could detect as few as 10 copies of negative-sense RNA and could be used in detecting low levels of HEV replication in cells. Virus particles in stool samples of hepatitis E patients showed encapsidation of negative-sense RNA along with HEV genomic RNA.
TEXT
Hepatitis E virus (HEV) is the major etiological agent of waterborne epidemics in India and of a significant proportion of sporadic hepatitis cases in adults. The genome of HEV is a single-stranded, positive-sense RNA molecule of ∼7,200 bases. Due to the lack of a cell culture system, HEV replicons are being used in studies of virus replication (4, 6, 7, 8, 10). HEV replication proceeds via a negative-sense intermediate (12, 13, 14) that acts as a template for synthesis of new positive-sense genomic RNA and subgenomic RNA molecules (8, 10). Detection of negative-sense RNA has been taken as evidence of viral replication. Several studies in HEV have used strand-specific assays (3, 11, 13, 14, 16, 17, 19, 20); however, except one (11), none have addressed the issue of the specificity and accuracy of correct strand detection. This report deals with various factors which might be responsible for false-positive results in detecting negative-sense RNA.
Generation of synthetic transcripts.
Partial HEV open reading frame 1 (ORF1) regions (nucleotides [nt] 2987 to 5698; 2,712 bp) cloned in pGEMT-EASY vector (Promega) and an HEV full-length cDNA clone (pGEMT-EASY-HEVT1FG) were linearized and used as templates for in vitro transcription (Ribomax in vitro transcription kit; Promega). Template DNA was removed by two rounds of RNase-free DNase treatment. Concentrations of purified RNA were determined using a NanoDrop ND-1000 spectrophotometer at 260 nm.
RT-PCR.
Primers used in the present study are listed in Table 1 and Table 2. RNA was denatured at 65°C for 5 min in the presence of 10 pmol of primer, and reverse transcription (RT) reactions were performed in a 20-μl volume containing 625 μM deoxynucleoside triphosphates (dNTPs), 40 U of RNase inhibitor, and either AMV-RT (Promega) or Superscript RT-III (Invitrogen) under the respective buffer conditions. Where mentioned, after RT, cDNA was incubated with 50 U of exonuclease I (USB) at 37°C for 30 min and purified (QIAquick PCR purification kit; Qiagen). First-round and second-round PCRs were carried out for 40 and 30 cycles, respectively.
Table 1.
Primers used for reverse transcription
| Procedure | Primer |
|
|---|---|---|
| Negative-strand assay | Positive-strand assay | |
| cDNA synthesis | HEVTagF1 | HEVTagR1 |
| First PCR | Tag as forward primer | F1 as forward primer |
| R1 as reverse primer | Tag as reverse primer | |
| Second PCR | F2 as forward primer | F2 as forward primer |
| R2 as reverse primer | R2 as reverse primer | |
Table 2.
Sequences and locations of primers
| Primer | Sequencea | Position (nt) |
|---|---|---|
| HEVF1 | 5′TGAGAATGATTTCTCTGAGTTTG3′ | 4398–4420 |
| HEVF2 | 5′ATACCGTCTGGAACATGGC3′ | 4604–4622 |
| HEVR1 | 5′ATGTTATTCATTCCACCCG3′ | 5116–5098 |
| HEVR2 | 5′AGCATCCCAATCAGGTTATG3′ | 5018–4999 |
| HEVTagF1 | 5′CGGTCATGGTGGCGAATAATGAGAATGATTTCTCTGAGTTTG3′ | 4398–4420 |
| HEVTagR1 | 5′CGGTCATGGTGGCGAATAAATGTTATTCATTCCACCCG3′ | 5116–5098 |
| Tag | 5′CGGTCATGGTGGCGAATAA3′ |
Underlined sequences in HEVTagF1 and HEVTag R1 represent non-HEV sequences.
Lack of strand specificity in detecting negative-sense RNA due to false priming during cDNA synthesis.
HEV diagnostic RT-PCR (2) was initially tested for strand specificity using synthetic HEV full-genome RNA (gRNA) as a template for cDNA synthesis, in two sets of reaction mixtures containing (i) forward primer (HEVF1; negative-sense RNA detection) and (ii) reverse primer (HEVR1; positive-sense RNA detection). For PCR, nested HEV-specific primer pair HEVF1 and HEVR1 (PCR I; 718 bp) and primer pair HEVF2 and HEVR2 (PCR II; 415 bp) were used. Amplifications were seen in all reactions with RT primer HEVF1 (Fig. 1, lanes 1, 4, 10, and 13) and with no primer (Fig. 1, lanes 3, 6, 12, and 15), indicating self-priming of RNA with both 106 and 103 gRNA copies. Lack of amplification in the RT reactions carried out without reverse transcriptase confirmed (i) false priming during the reverse transcription step and (ii) the absence of carryover contamination of the DNA template used for in vitro transcription.
Fig 1.

Lack of strand specificity in detecting negative-sense RNA due to false priming during cDNA synthesis: 106 (lanes 1 to 9) and 103 (lanes 10 to 18) HEVT1FG RNA copies were reverse transcribed in the presence of AMV-RT (lanes 1, 2, 3, 10, 11, and 12), Superscript RT-III (lanes 4, 5, 6, 13, 14, and 15), or no enzyme (lanes 7, 8, 9, 16, 17, and 18). Lanes 1, 4, 7, 10, 13, and 16 represent RT reactions with primer HEVF1, lanes 2, 5, 8, 11, 14, and 17 represent RT reactions with primer HEVR1, and lanes 3, 6, 9, 12, 15, and 18 represent RT reactions with no primer. A 10-μl volume of cDNA was used for the first-round PCR, and 10 μl of the first PCR product was used for the nested PCR.
Tag-based PCR to remove false priming.
The generation of falsely primed cDNA during the RT step originates mainly from those of the opposite strand (positive sense), as they are always in excess compared to negative-sense RNA intermediates in the cells. The false-priming events can be attributed to secondary structures of RNA (18) or to small RNA molecules present in cellular RNA (9). HEVF1 and HEVR1 primers were modified at their 5′ ends by adding 19 nt of non-HEV sequence (15) to generate HEVTagF1 and HEVTagR1. The assumption is that the modified primer can be used for RT to generate cDNA with a tag at the 5′ end which can be specifically amplified with tag primer and HEV-specific reverse primer by PCR. The prerequisite was to remove RT primers by exonuclease I treatment and column purification. Synthetic gRNAs (106, 105, and 103 copies) were reverse transcribed using HEVTagF1 or HEVTagR1 or no primer (Fig. 2). Nonspecific amplification was absent with 103 copies (Fig. 2C). Reactions with AMV-RT containing 105 and 106 copies and HEVTagF1 showed nonspecific amplification (lanes 1 of Fig. 2A and B). Reactions done with AMV-RT (42°C) containing 106 RNA copies showed amplification due to self-priming (lane 3, Fig. 2A). However, with Superscript RT-III (55°C), nonspecific amplifications were not observed, indicating better specificity of Superscript RT-III compared to AMV-RT at all tested template RNA concentrations (lanes 4 to 6 of Fig. 2A, B, and C).
Fig 2.
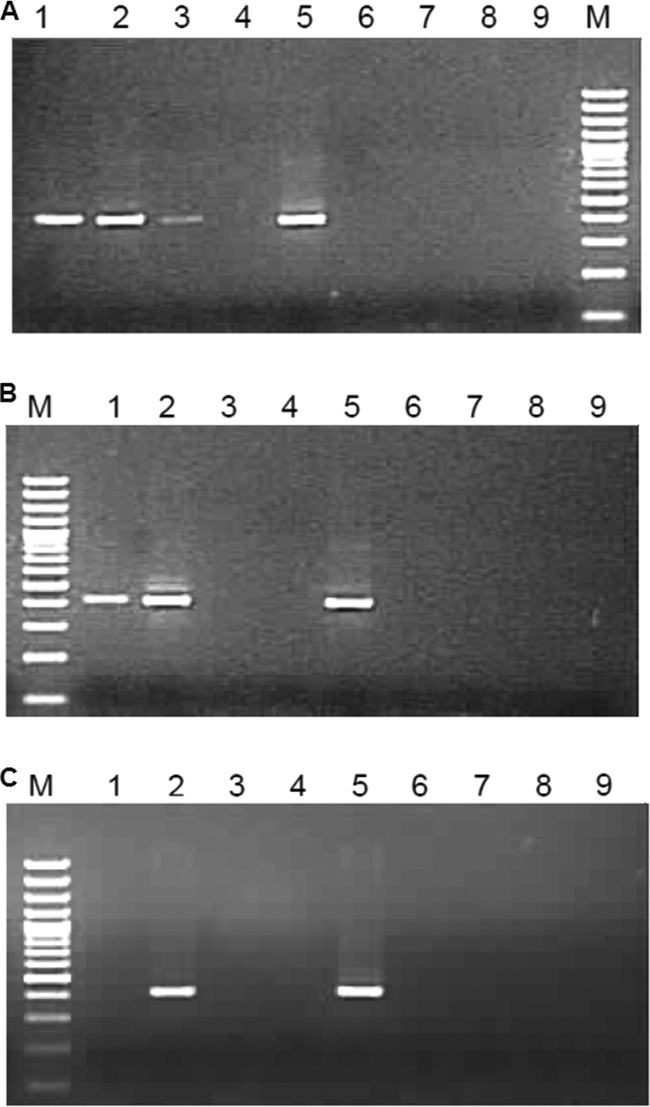
Tag-based PCR to remove false priming: strand-specific assays were performed on 106 (A), 105 (B), and 103 (C) full-genome positive-sense RNA copies by the use of two reverse transcriptase enzymes. For each gel, lanes 1 to 3 represent reactions with AMV-RT (lane 1, RT with HEVTagF1 primer; lane 2, RT with HEVTagR1 primer; lane 3, no primer), lanes 4 to 6 represent reactions with Superscript RT-III (lane 4, RT with HEVTagF1 primer; lane 5, RT with HEVTagR1 primer; lane 6, no primer), and lanes 7 to 9 represent reactions without any RT enzyme (lane 7, HEVTagF1 primer; lane 8, HEVTagR1 primer; lane 9, no primer). After reverse transcription and exonuclease I treatment, cDNAs were purified by the use of a silica-based column. Samples without reverse transcriptase enzyme were processed similarly to samples with RT enzymes.
Specificity and sensitivity of the strand-specific assays.
Ten-fold-diluted synthetic positive- and negative-sense partial ORF1 RNA templates (1010 to 10 copies) were subjected to strand-specific assays separately in the presence of 1 μg of cellular RNA (extracted from Huh-7 human hepatocarcinoma cells with a Ribopure kit [Ambion]).
With positive-sense RNA, the positive-strand-specific assay was positive until 10 RNA copies were reached (Fig. 3A). Similarly, the negative-strand-specific assay performed using negative-sense RNA was positive until 10 copies were reached (Fig. 3C). To see the specificity of each strand-specific assay, reactions were performed using only opposite-sense RNAs as templates. Negative-strand assay reactions showed false priming in the presence of 1010 to 107 copies of positive-sense RNA (lanes 1 to 4, Fig. 3D). However, reactions were completely negative from 106 to 10 copies. When same set of reactions were carried out using only column-purified cDNA without exonuclease I treatment, there was a 100-fold increase in the nonspecific positivity (Fig. 3E). With the positive-strand-specific assay performed with negative-sense RNA, false positivity was seen until 105 copies were reached (Fig. 3B), and reactions were negative below that.
Fig 3.

Specificity and sensitivity of the strand-specific assays. Synthetic positive-sense and negative-sense RNAs were diluted 10-fold, and RNA copies ranging from 1010 to 10 were subjected to strand-specific PCR separately. The cDNAs were synthesized in the presence of 1 μg of total cellular RNA with Superscript RT-III and treated with exonuclease I (where mentioned) for 30 min followed by silica-based column purification. A 10-μl volume of cDNA was used for the first round of PCR, and 5 μl of the first PCR product was further subjected to nested PCR. Lanes in all four gels represent the following: 1, 1010 RNA copies; 2, 109 RNA copies; 3, 108 RNA copies; 4, 107 RNA copies; 5, 106 RNA copies; 6, 100-bp ladder; 7, 105 RNA copies; 8, 104 RNA copies; 9, 103 RNA copies; 10, 102 RNA copies; 11, 10 RNA copies; 12, no RNA. (A) Positive-strand assay on synthetic positive strand with exonuclease. (B) Positive-strand assay on synthetic negative strand with exonuclease. (C) Negative-strand assay on synthetic negative strand with exonuclease. (D) Negative-strand assay on synthetic positive strand with exonuclease. (E) Negative-strand assay on synthetic positive strand without exonuclease.
Specificity of the assay with gRNA.
Taking into account that secondary structures of 5′ and 3′ nontranslated regions in the RNA genome may lead to self-priming, 107, 106, 105, 104, and 103 copies of synthetic gRNA were subjected to negative-strand detection in the presence of 1 μg of total cellular RNA. Amplification was seen at 107 copies, whereas the remaining reactions were negative (Fig. 4). These observations were in agreement with the results obtained with partial ORF1 RNA and also showed that nontranslated regions and the presence of cellular RNA were not altering the specificity.
Fig 4.
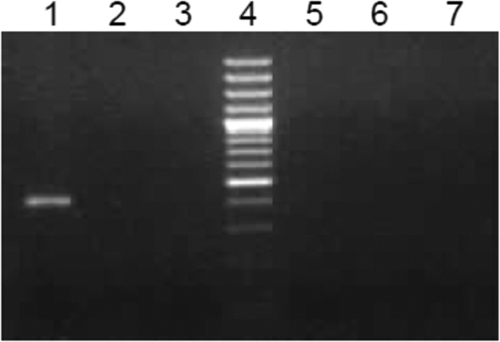
Specificity of the assay with full-length positive-strand RNA. A negative-strand assay was performed on 107, 106, 105, 104, and 103 (lanes 1, 2, 3, 5, and 6, respectively) copies of HEVT1FG positive-sense synthetic RNA in the presence of 1 μg of total cellular RNA. Nuclease-free water served as the negative control (lane 7).
Negative-strand assay in the presence of positive-sense RNA.
To check the sensitivity of negative-strand detection in the presence of the opposite strand, assays were performed on 105, 104, 103, 102, and 10 copies of negative-sense RNA in the presence of 105 copies of positive-sense RNA and 1 μg of cellular RNA. All samples were positive, indicating that the standardized assays are sufficiently sensitive to detect even 10 copies of negative-sense RNA (Fig. 5).
Fig 5.
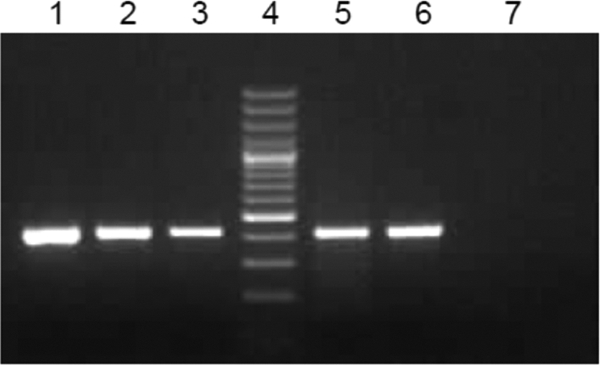
Negative-strand assay in the presence of positive-sense RNA: a negative-strand assay was performed on 105, 104, 103, 102, and 10 (lanes 1, 2, 3, 5, and 6, respectively) copies of negative-sense synthetic RNA in the presence of 105 copies of positive-sense synthetic RNA and 1 μg of total cellular RNA. Nuclease-free water served as the negative control (lane 7).
Screening of HEV RNA-positive stool samples.
Stool samples from 20 acute-phase hepatitis E cases (anti-HEV IgM positive) (2) were used to prepare 10% suspensions and processed for viral RNA isolation (viral RNA minikit; Qiagen) and HEV RNA copy number determination using a TaqMan real-time PCR assay (1). RNA-positive samples (6/20) were processed further for negative-strand-specific detection. Samples showing >105 copies/11.5 μl were diluted appropriately so that the input copy number did not exceed 105 copies/reaction. Three of the six samples (Fig. 6, lanes 2, 4, and 8) were positive, indicating packaging of replicative RNA intermediates in the excreted mature virus particles. To rule out the presence of detached alimentary canal cells in the stool samples, three independent HEV RNA-positive stool samples were processed for virus purification using a sucrose gradient (5) and tested. All (3/3) samples showed the presence of negative-sense RNA (Fig. 6, lanes 10, 11, and 12). With these results, it was clear that encapsidation of negative-sense RNA is a common phenomenon during HEV replication. All samples were negative when no primer was added during cDNA synthesis (data not shown).
Fig 6.

Screening of HEV RNA-positive stool samples from acute hepatitis E cases. Negative-strand detection was carried out on RNA isolated as follows: lane 1, stool sample 1 (539 copies of HEV positive-sense genomic RNA/reaction); lane 2, stool sample 2 (1 × 105 copies); lane 3, negative control; lane 4, stool sample 3 (1 × 105 copies); lane 5, stool sample 4 (619 copies); lane 6, negative control; lane 7, stool sample 5 (9,591 copies); lane 8, stool sample 6 (1 × 105 copies); lane 9, 100-bp ladder; lane 10, purified virus stock 1 (1.4 × 104 copies); lane 11, virus stock 2 (2,653 copies); lane 12, virus stock 3 (1,782 copies).
In conclusion, use of a tag primer for cDNA synthesis, followed by exonuclease I treatment and column purification of the cDNA products, was found to reduce false-positive signals significantly. Assay could detect as few as 10 copies of negative-sense RNA specifically even in the presence of 105 copies of positive-sense RNA and cellular RNA.
Footnotes
Published ahead of print 28 December 2011
REFERENCES
- 1. Arankalle VA, Lole KS, Deshmukh TM, Srivastava S, Shaligram US. 2009. Challenge studies in rhesus monkeys immunized with candidate hepatitis E vaccines: DNA, DNA-prime-protein boost and DNA-protein encapsulated in liposomes. Vaccine 27:1032–1039 [DOI] [PubMed] [Google Scholar]
- 2. Arankalle VA, Lole KS, Deshmukh TM, Chobe LP, Gandhe SS. 2007. Evaluation of human (genotype 1) and swine (genotype 4)-ORF-2-based ELISAs for anti-HEV IgM and IgG detection in an endemic country and search for type 4 human HEV infections. J. Viral. Hepat. 14:435–445 [DOI] [PubMed] [Google Scholar]
- 3. Billam P, et al. 2008. Development and validation of a negative-strand-specific reverse transcription-PCR assay for detection of a chicken strain of hepatitis E virus: identification of nonliver replication sites. J. Clin. Microbiol. 46:2630–2634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cao D, Huang YW, Meng XJ. 2010. The nucleotides on the stem-loop RNA structure in the junction region of the hepatitis E virus genome are critical for virus replication. J. Virol. 84:13040–13044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Emerson SU, Nguyen H, Torian U, Purcell RH. 2006. ORF3 protein of hepatitis E virus is not required for replication, virion assembly, or infection of hepatoma cells in vitro. J. Virol. 80:10457–10464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Emerson SU, et al. 2001. Recombinant hepatitis E virus genomes infectious for primates: importance of capping and discovery of a cis-reactive element. Proc. Natl. Acad. Sci. U. S. A. 98:15270–15275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Graff J, et al. 2008. Mutations within potential glycosylation sites in the capsid protein of hepatitis E virus prevent the formation of infectious virus particles. J. Virol. 82:1185–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Graff J, Torian U, Nguyen H, Emerson SU. 2006. A bicistronic subgenomic mRNA encodes both the ORF2 and ORF3 proteins of hepatitis E virus. J. Virol. 80:5919–5926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gunji T, et al. 1994. Specific detection of positive and negative stranded hepatitis C viral RNA using chemical RNA modification. Arch. Virol. 134:293–302 [DOI] [PubMed] [Google Scholar]
- 10. Huang YW, Opriessnig T, Halbur PG, Meng XJ. 2007. Initiation at the third in-frame AUG codon of open reading frame 3 of the hepatitis E virus is essential for viral infectivity in vivo. J. Virol. 81:3018–3026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ippagunta SK, Naik S, Jameel S, Ramana KN, Aggarwal R. 2011. Viral RNA but no evidence of replication can be detected in the peripheral blood mononuclear cells of hepatitis E virus-infected patients. J. Viral. Hepat. 18:668–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Meng XJ, et al. 1998. Experimental infection of pigs with the newly identified swine hepatitis E virus (swine HEV), but not with human strains of HEV. Arch. Virol. 143:1405–1415 [DOI] [PubMed] [Google Scholar]
- 13. Nanda SK, Panda SK, Durgapal H, Jameel S. 1994. Detection of the negative strand of hepatitis E virus RNA in the livers of experimentally infected rhesus monkeys: evidence for viral replication. J. Med. Virol. 42:237–240 [DOI] [PubMed] [Google Scholar]
- 14. Panda SK, Ansari IH, Durgapal H, Agrawal S, Jameel S. 2000. The in vitro-synthesized RNA from a cDNA clone of hepatitis E virus is infectious. J. Virol. 74:2430–2437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Peyrefitte CN, Pastorino B, Bessaud M, Tolou HJ, Couissinier-Paris P. 2003. Evidence for in vitro falsely-primed cDNAs that prevent specific detection of virus negative strand RNAs in dengue-infected cells: improvement by tagged RT-PCR. J. Virol. Methods 113:19–28 [DOI] [PubMed] [Google Scholar]
- 16. Tam AW, et al. 1996. In vitro propagation and production of hepatitis E virus from in vivo-infected primary macaque hepatocytes. Virology 215:1–9 [DOI] [PubMed] [Google Scholar]
- 17. Thakral D, Nayak B, Rehman S, Durgapal H, Panda SK. 2005. Replication of a recombinant hepatitis E virus genome tagged with reporter genes and generation of a short-term cell line producing viral RNA and proteins. J. Gen. Virol. 86:1189–1200 [DOI] [PubMed] [Google Scholar]
- 18. Timofeeva AV, Skrypina NA. 2001. Background activity of reverse transcriptases. Biotechniques 30:22–28 [DOI] [PubMed] [Google Scholar]
- 19. Varma SP, et al. 2011. Hepatitis E virus replication involves alternating negative- and positive-sense RNA synthesis. J. Gen. Virol. 92:572–581 [DOI] [PubMed] [Google Scholar]
- 20. Zhang HY, et al. 2011. Both swine and human cells are capable to support the replication of swine hepatitis E virus type 4 in vitro. Virus Res. 158:289–293 [DOI] [PubMed] [Google Scholar]