

Abstract
The serpin protease nexin-1 (PN-1) is expressed by vascular cells and secreted by platelets upon activation, and it is known to interact with several modulators of angiogenesis, such as proteases, matrix proteins, and glycosaminoglycans. We therefore investigated the impact of PN-1 on endothelial cell angiogenic responses in vitro and ex vivo and in vivo in PN-1-deficient mice. We found that PN-1 is antiangiogenic in vitro: it inhibited vascular endothelial growth factor (VEGF)-induced endothelial cell responses, including proliferation, migration, and capillary tube formation, and decreased cell spreading on vitronectin. These effects do not require the antiprotease activity of PN-1 but involve PN-1 binding to glycosaminoglycans. In addition, our results indicated that PN-1 does not act by blocking VEGF binding to its heparan sulfate proteoglycan coreceptors. The results obtained in vitro were supported ex vivo in PN-1-deficient mice, where the microvascular network sprouting from aortic rings was significantly enhanced. Moreover, in vivo, neovessel formation was promoted in the Matrigel plug assay in PN-1-deficient mice compared to wild-type mice, and these effects were reversed by the addition of recombinant PN-1. Taken together, our results demonstrate that PN-1 has direct antiangiogenic properties and is a yet-unrecognized player in the angiogenic balance.
INTRODUCTION
Serpins form a large family of structurally related proteins present in the plasma or in tissues and play a central role in the regulation of protease activity. Among them, serpin E2, or protease nexin-1 (PN-1), is a serine protease inhibitor that recently emerged as a potent regulator in the vascular system, as it is often found overexpressed at sites of tissue injury. PN-1 is barely detectable in plasma but is produced by most cell types (9). In the vascular system, PN-1 is expressed by smooth muscle cells (SMCs) (6, 30), endothelial cells (7), and fibroblasts (2). PN-1 was first identified as glia-derived nexin in the central nervous system, where it regulates the effects of thrombin on neuronal as well as glial cells (25). Indeed, PN-1 is the most efficient tissue inhibitor of thrombin, but it is also a powerful inhibitor of plasminogen activators and of plasmin, proteases largely involved in tissue remodeling. Furthermore, PN-1 avidly binds to glycosaminoglycans (GAGs), such as heparan sulfates (2), which potentiate its activity and target it to the pericellular space (30). Previous studies from our laboratory have demonstrated that PN-1 is expressed by vascular cells in a tightly regulated manner, well correlated with the susceptibility of cells to exogenous (thrombin) and endogenous (plasminogen activator) proteases (30, 33). Although PN-1 is not a plasma protein, a circulating pool of PN-1 is stored in platelets and secreted upon activation (4, 13). Importantly, we have demonstrated that blood platelets behave as a reservoir of PN-1 with major consequences on thrombosis and fibrinolysis (4, 5). Furthermore, platelets along with monocyte/macrophages are a major source of PN-1 accumulation in human atherosclerotic plaques (23). The regulating role of PN-1 on thrombosis may also involve an effect on endothelial cells. Indeed, PN-1 interacts with the endothelial membrane glycoprotein thrombomodulin and enhances its capacity to inhibit thrombin (7).
The development of new vessels in adults is a very complex process in which multiple factors are involved. Nesting of endothelial cells requires remodeling of the surrounding tissue by finely tuned proteolytic processes in which serine proteases, matrix metalloproteinases (MMPs), and their inhibitors are key players. A number of serpins have recently been identified as potential regulators of angiogenesis (3, 28). Despite its expression by endothelial cells and its delivery by activated platelets at sites of wounding, an effect of PN-1 on angiogenesis has so far not been reported. In this study, we addressed the possibility that PN-1 exhibits antiangiogenic properties, not only via its interaction with proangiogenic molecules, such as thrombin, plasmin, or vitronectin, but also directly, as has been reported for the latent and cleaved forms of antithrombin (AT) (28). For this purpose, we have assessed the effect of exogenous PN-1 on in vitro angiogenic responses of endothelial cells, examined its interaction with these cells, and determined the consequences of PN-1 deficiency in mouse angiogenic models. We found that PN-1 inhibits vascular endothelial growth factor (VEGF)-induced proliferation and migration of human umbilical vein endothelial cells (HUVECs) as well as capillary tube formation in Matrigel. In addition, ex vivo and in vivo angiogenic responses were enhanced in PN-1-deficient mice. Taken together, these results demonstrate that PN-1 has an antiangiogenic activity.
MATERIALS AND METHODS
Materials.
Heparin was obtained from Sanofi-Aventis (France). Bovine serum albumin (BSA), human serum albumin (HSA), heparan sulfate, dermatan sulfate, and chondroitin sulfate, protease inhibitor cocktail for mammalian tissues, vitronectin, gelatin, fibronectin, and p-nitrophenylphosphate were all obtained from Sigma-Aldrich (France). PET (polyethylene terephthalate) membrane inserts with an 8-μm pore size, rat tail collagen type I, and growth factor-reduced Matrigel were obtained from BD Biosciences. CellTiter 96 AQueous One cell proliferation assay reagent was obtained from Promega. RAP (receptor-associated protein) was obtained from Calbiochem (Germany). Alexa Fluor 488-phalloidin was obtained from Life Technologies, biotinylated isolectin B4 was obtained from Vector Laboratories (Paris, France), and Cy3-streptavidin was obtained from GE Healthcare (United Kingdom). Anti-PN-1 polyclonal antibody was kindly provided by D. Hantai (U975, Centre de Recherche en Neurosciences de la Pitié-Salpêtrière, Paris, France). VEGF 165 was obtained from Ozyme (France), and VEGF 121 came from Miltenyi Biotech (Germany). Polyclonal anti-phospho-extracellular signal-regulated kinase 1 and 2 antibodies (ERK1&2; Trp185/187) were obtained from Invitrogen, monoclonal anti-ERK2 came from Santa Cruz Biotechnology (Germany), and monoclonal anti-Akt 11E7 and polyclonal anti-phospho-Akt(Ser473) were obtained from Cell Signaling.
Expression and purification of recombinant PN-1 variants.
Expression of PN-1 was achieved essentially as previously described (8) with some modifications. The cDNA coding sequence for human PN-1 was amplified by PCR using the previously described pcDNA3/PN-1 vector as a matrix (7) and the following primers: upstream, 5′-CTTGAATTCTCCCACTTCAATCCTCTGTC-3′; downstream, 5′-CTTGAATTCTCAGGGTTTGTTTATCTGCC-3′. A single cDNA band was amplified and purified using the Nucleospin extract kit (Macherey Nagel, Germany). The cDNA was then treated with EcoRI under proper buffer conditions and inserted into the pGEX-6P-1 vector (GE Healthcare, United Kingdom) at the corresponding site, resulting in a fusion gene construct that encoded glutathione S-transferase (GST)–PN-1. The sequence of the amplified PN-1 portion of the fusion gene was verified by sequencing (Genome Express, France). PN-1 mutagenesis was achieved using the QuikChange site-directed mutagenesis kit (Stratagene). The K7Q variant of PN-1 was produced by changing the seven lysine residues of the heparin-binding site (K71, -74, -75, -78, -83, -84, and -86) to glutamine and the inactive R346A variant by changing the arginine (R346) residue of the reactive site to alanine. All the mutations were verified by sequence analysis (Genome Express, France). The GST–PN-1 constructs were expressed in the BL21(DE3) Escherichia coli strain (Life Technologies), grown to an optical density of 0.5 at 600 nm, and induced with 0.2 mM IPTG (isopropylthio-β-galactoside; GE Healthcare, Sweden) at 37°C for 2 h before harvesting. GST–PN-1 was purified from the cell lysate by glutathione-Sepharose affinity chromatography. On-column cleavage of the protein from GST was achieved using PreScission protease (GE Healthcare, Sweden), the site-specific protease whose recognition sequence is encoded between the GST domain and the multiple cloning site.
Recombinant PN-1 characterization.
PN-1 purity was assessed by SDS-PAGE and Coomassie blue staining. PN-1 activity toward thrombin was measured as previously described (31). Progress curve kinetics were used to estimate the accelerating effect of heparin on thrombin inhibition, under pseudo-first-order conditions. Wild-type (WT) and K7Q PN-1 (2 nM active concentration) were incubated with heparin (5 nM) in 20 mM phosphate, 100 mM NaCl, 0.1 mM EDTA, 0.1% polyethylene glycol 8000 (PEG 8000), pH 7.5, for 10 min at 37°C before the addition of the thrombin chromogenic substrate S-2238 (H-D-Phe-Pip-Arg-pNa at 0.3 mM; Chromogenix). The reactions were started by the addition of thrombin (0.1 nM). Thrombin activity was determined by measuring the rate of substrate hydrolysis at 405 nm using a microtiter plate reader and the Biolyse 2 application (Labsystem, France). Values for kon were calculated as described previously (7). The uncatalyzed second-order rate constant, i.e., in the absence of heparin, was determined using the above method but with 50 nM PN-1. WT and R346A PN-1 binding to heparin were investigated using (p-toluidinyl)naphthalene-2-sulfonic acid (TNS) fluorescence as described previously (31). Spectra were recorded using 15 to 30 μg/ml PN-1 (375 to 750 nM) and 12.5 μM TNS in the presence or absence of 5 μM heparin. Addition of polysaccharides alone to TNS had no effect on TNS intrinsic fluorescence.
Cell culture.
HUVECs (from pooled donors) were purchased from Cambrex and cultured according to the manufacturer's procedures in endothelial basal medium (EBM) supplemented with the endothelial cell growth supplement pack (Promocell, Germany) and antibiotic-antimycotic solution (PAA, France). All experiments were carried out between the second and the fifth passage.
Cell adhesion assay.
Adhesion of endothelial cells to immobilized fibronectin (50 μg/ml) or vitronectin (5 μg/ml) in the presence or absence of PN-1 (20 μg/ml) was quantified by measuring the phosphatase activity with p-nitrophenylphosphate as previously described (32). Alternatively, cells were fixed and spreading was quantified by staining with Alexa 488-conjugated phalloidin (Life Technologies), as described by the manufacturer, followed by densitometry analysis using Image J software. All experiments were performed in triplicate (n ≥ 3 per group).
Cell proliferation assay.
HUVEC proliferation induced by basic fibroblast growth factor (bFGF; 10 ng/ml), VEGF (10 ng/ml), or VEGF 121 (25 ng/ml) was analyzed after 48 h using the CellTiter 96 AQueous One cell proliferation assay reagent (Promega), as described previously (37). All experiments were performed at least in triplicate (n = 4 per group).
Western blotting.
To detect phospho-ERK1/2 and phospho-Akt levels, HUVECs were treated with 10 ng/ml VEGF for 10 min in the presence or absence of 20 μg/ml PN-1 and lysed with phospho-Tyr protecting lysis buffer (1% Triton X-100, 10% glycerol, 50 mM NaCl, 50 mM HEPES, 2 mM EDTA, 1 mM Na3VO4, 10 mM NaF, 10 mM NaPO4, 10 mM p-nitrophenylphosphate, 10 mM β-glycerol phosphate, and protease inhibitor cocktail). Samples were subjected to SDS-PAGE and immunoblotted with anti-phospho-ERK1/2 and anti-total ERK1/2 or anti-phospho-pAkt and anti-total pAkt antibodies.
Chemotaxis assay.
The chemotactic motility of HUVECs induced by 20 ng/ml VEGF was assessed using PET membrane inserts with an 8-μm pore size. HUVECs were allowed to migrate for 4 h at 37°C in the presence or absence of PN-1. The cells remaining on the upper surface of the insert membrane were removed with a swab. The cells that migrated to the lower surface of the insert membrane were fixed with methanol for 20 min, stained with 0.1% crystal violet for 20 min, photographed, and counted at 200× magnification. Six random fields in each well were counted. Experiments were performed in duplicate and repeated at least 3 times.
In vitro angiogenesis assay.
In vitro formation of capillary-like structures was examined using growth factor reduced BD Matrigel matrix as described previously (19). Cell suspensions containing 50 ng/ml VEGF in the presence or absence of 20 μg/ml PN-1 were plated on Matrigel-coated wells at a density of 25,000 cells per well in EBM containing 0.2% fetal calf serum (FCS). Cells were directly photographed after 18 h with a Leica camera (40× magnification), and the relative capillary-like structure density was quantified using Image J software. Experiments were repeated at least 3 times.
Cell-binding assay.
Binding experiments were performed at 4°C and at 37°C to, respectively, prevent and allow potential internalization processes during the binding assay time frame. PN-1 (1 μg/ml) was incubated for 2 h with serum-starved HUVECs with or without heparin (200 μg/ml), RAP (13 μg/ml), or different GAGs and, in some experiments, an additional 30-min incubation with heparin (200 μg/ml) was carried out. The PN-1 contents in cell lysates or cell fractions obtained by ultracentrifugation were analyzed by SDS-PAGE and immunoblotting with a polyclonal anti-PN-1 antibody, using the same amount of protein for each sample, as determined using the bicinchoninic acid protein assay kit (Pierce, Rockford, IL).
Mice.
Mice homozygous for null mutations in the PN-1 gene were generated as previously described (22). PN-1-deficient mice were back-crossed for 12 generations into the C57BL/6 line. Heterozygous matings generated PN-1-deficient mice and wild-type mice, all genotyped by PCR. Mice were bred and maintained in our own laboratory (Paris, France), and all experiments were performed in accordance with French ethical law.
Aortic ring assay.
Aortas of 8-week-old wild-type or PN-1-deficient mice were dissected, cut into short segments, and incubated overnight in EBM containing 0.2% FCS in the absence or presence of 50 ng/ml VEGF before being placed into collagen gels. Aortic explants were imaged 7 days after embedding using an inverted microscope (Leica, France), and the sprout area was quantified using Image J software. The explants were then fixed, and endothelial cells were stained with biotinylated-isolectin B4 followed by streptavidin-Cy3 as described previously (11).
Matrigel plug angiogenesis assay.
Angiogenesis was evaluated using the Matrigel plug assay, as previously described (29). Growth factor-reduced Matrigel, in liquid form at 4°C, was mixed with 300 ng/ml bFGF and injected (0.3 ml) subcutaneously into 8- to 9-week-old mice. Each mouse received two Matrigel implants. Animals were killed after 6 days, and Matrigel plugs were carefully dissected away from the surrounding adherent tissue, fixed, embedded in paraffin, and sectioned for histological studies.
Statistical analysis.
Data are presented as means ± standard errors of the means and were analyzed using an unpaired t test (P levels of <0.05, <0.01, and <0.005).
RESULTS
Expression and characterization of recombinant PN-1 variants.
Wild-type recombinant human PN-1, the inactive mutant form R346A, and the K7Q variant of PN-1 in which the heparin site is nonfunctional (34) were produced as described previously in a prokaryotic system as fusion proteins with GST (8). PN-1 was purified from bacterial lysates by affinity chromatography using glutathione-Sepharose 4B and cleaved from GST with a site-specific protease, followed by gel filtration chromatography on Sephadex G75 (GE Healthcare) (Fig. 1, inset).
Fig 1.

Characterization of PN-1 variants. (Inset) SDS-PAGE Coomassie staining of PN-1 after purification. (A and B) Thrombin inhibition by WT and K7Q PN-1 and catalytic effect of heparin. Thrombin and chromogenic substrate concentrations were 100 pM and 300 μM, respectively. Progress curves are shown. (A) Curves obtained with 50 nM WT (○) and K7Q (□) PN-1, used to determine uncatalyzed rate constants; (B) catalytic effect of 5 nM on thrombin inhibition by 2 nM PN-1. Dashed superimposed lines correspond to WT PN-1, K7Q PN-1, and K7Q PN-1 plus heparin. ●, WT PN1 plus heparin. (C) Representative fluorescence spectra of three of the TNS-bound PN-1 variants in the absence (solid lines) or presence (dashed lines) of heparin, determined with 500 nM WT or K7Q PN-1 and 750 nM R346A. Spectra are difference spectra between TNS plus PN-1 with or without heparin and TNS alone.
The stoichiometries for WT and K7Q PN-1 binding to thrombin were determined by measuring the loss of thrombin activity on the chromogenic substrate S-2238 and gave values from 3.3 to 4.1. As expected, R346A PN-1 had no inhibitory effect on thrombin activity toward S-2238 (data not shown). The uncatalyzed rate constant for thrombin inhibition by PN-1 was of 3.0 ± 0.3 105 M−1 s−1 for both WT and K7Q PN-1 (Fig. 1A, 50 nM, ○ and □, respectively), in agreement with previous studies (31). The catalytic activity of heparin on thrombin inhibition was analyzed with 2 nM WT and K7Q PN-1 (Fig. 1B). As expected, the curves obtained with K7Q PN-1 in the absence and presence of heparin were superimposed with the curve obtained with WT PN-1 in the absence of heparin (dashed lines), whereas heparin increased the rate of thrombin inhibition by WT PN-1 (●) (34). The abilities of the PN-1 variants to bind heparin were determined using the fluorescent probe TNS. As shown previously (31), binding of the probe to WT and mutant PN-1 was accompanied by a great increase in TNS fluorescence. Heparin caused a change in the environment of WT and R346A PN-1-bound TNS, resulting in a 50 to 60% decrease in TNS fluorescence intensity (Fig. 1C) and reflecting the binding of heparin to both PN-1 variants. In contrast, heparin had no effect on K7Q PN-1-bound TNS fluorescence, confirming the heparin-binding defect of this variant.
PN-1 inhibits HUVEC spreading and FGF- and VEGF-induced cell proliferation and migration.
To investigate the role of PN-1 in angiogenesis, we examined the effect of the PN-1 variants on VEGF- and FGF-induced HUVEC responses in vitro. As shown in Fig. 2A, WT PN-1 significantly reduced both FGF- and VEGF-induced proliferation in a concentration-dependent manner. Increasing the concentration of FGF or VEGF reduced the inhibitory effect of a fixed concentration of WT PN-1 (20 μg/ml), which was used at a concentration that significantly inhibited both VEGF- and FGF-induced cell proliferation (Fig. 2B). PN-1 had no cytotoxic effect, since it neither significantly reduced endothelial proliferation by itself (Fig. 2A) nor had a proapoptotic effect alone or in the presence of VEGF, as determined by terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling analysis (more than 95% cell viability in all experiments [data not shown]).
Fig 2.

Effects of PN-1 on angiogenic responses of HUVECs. (A) Concentration-dependent effect of PN-1 on 10 ng/ml FGF- or VEGF-induced HUVEC proliferation (n = 4 to 10). (B) Effect of PN-1 (20 μg/ml) on cell proliferation induced by increasing growth factor (GF) concentrations (n = 4). Results are expressed as the percentage of inhibition of proliferation, calculated from the ratio of the proliferation induced by each growth factor concentration measured in the presence of recombinant PN-1 to that measured in the absence of PN-1. (C and D) HUVEC adhesion (C) and HUVEC spreading on vitronectin and fibronectin (D), in the absence or presence of 20 μg/ml PN-1 (n = 3 to 4). (E) HUVEC migration in the absence or presence of 20 ng/ml VEGF. *, P < 0.05; **, P < 0.01; ***, P < 0.0001 versus VEGF or FGF alone in panels A and E, versus control (CTL) in panels C and D, and versus 10 ng/ml VEGF or FGF in panel B.
HUVEC adhesion to the matrix proteins vitronectin and fibronectin was not modified by the addition of PN-1 (Fig. 2C). However, PN-1 slightly but significantly reduced the spreading of HUVECs on vitronectin (76.9 ± 2.0% of control; P < 0.0001) and to a lesser extent on fibronectin (89.0 ± 2.2% of control; P < 0.005) (Fig. 2D).
VEGF-directed cell migration was significantly inhibited by PN-1 in a concentration-dependent manner (Fig. 2E). In the presence of PN-1 (50 μg/ml), growth factor-induced migration was inhibited by 80.5 ± 4.4% compared to the control without PN-1. The low level of cell migration observed in the absence of VEGF was not modified in the presence of PN-1.
PN-1 inhibits vascular tube formation on Matrigel.
Our data suggested that the PN-1 interaction with endothelial cells could impair angiogenesis; we thus examined the morphological differentiation of HUVECs in Matrigel. Under control conditions, after 18 h HUVECs formed a small number of capillary-like structures, arranged in an incomplete network (Fig. 3A) that was not modified by the addition of PN-1. VEGF (50 ng/ml) increased the capillary density by 214% ± 8% of control without VEGF and formed elongated capillary-like structures, arranged in complete networks. In contrast, VEGF-induced capillary formation was blocked in the presence of recombinant PN-1, with a capillary density close to the value obtained in the absence of VEGF (131% ± 7% of control without VEGF).
Fig 3.

Effects of PN-1 on HUVEC responses to VEGF. (A) Capillary tube formation in Matrigel in the presence or absence of 50 ng/ml VEGF and 20 μg/ml PN-1. Results are expressed as the mean tube density relative to the control without VEGF, measured on 3 fields per well from 3 to 4 experiments. ***, P < 0.0001 versus VEGF alone. (B) Akt and Erk phosphorylation. Whole-cell lysates of HUVECs incubated for 10 min in the presence or absence of 10 ng/ml VEGF and/or PN-1 at 20 μg/ml were immunoblotted with specific anti P-ERK and P-Akt antibodies and with antibodies to the whole protein. The results are representative of at least 3 independent experiments.
The potential impact of PN-1 on VEGF downstream signaling in HUVECs based on the phosphorylation of ERK and Akt was analyzed in cells incubated or not with VEGF in the presence or absence of PN-1. ERK and Akt were mostly in an inactive dephosphorylated state in resting HUVECs, and both were significantly activated by tyrosine phosphorylation after treatment with VEGF (Fig. 3B). Neither the Erk nor Akt phosphorylation state was modified by PN-1, in the presence or in the absence of VEGF.
Binding of PN-1 to HUVECs.
Binding of PN-1 to the cell surface was measured at 4°C. Cell-bound PN-1 was detected by immunoblotting on whole-cell lysates using a polyclonal antibody to PN-1 (Fig. 4). A 42-kDa band was clearly detected in lysates of cells incubated with recombinant PN-1 (Fig. 4A, WT) compared to control cell lysates (Fig. 4A, CTL), indicating that added PN-1 firmly bound to the cells. In addition, we examined whether the fully glycosylated PN-1 that is released by human platelets upon activation (4) could also bind to endothelial cells. A 50-kDa band was clearly observed in lysates of cells incubated with the fraction released by activated platelets, indicating that the platelet PN-1 bound to HUVECs (Fig. 4B).
Fig 4.

PN-1 binding to HUVECs at 4°C. (A to C) Cells were incubated at 4°C with recombinant PN-1 variants (1 μg/ml) in the presence or not of heparin (Hep), chondroitin sulfate (CS), or dermatan sulfate (DS) (A and C) or with supernatant of resting or activated platelets (B). Whole-cell lysates were submitted to immunoblotting with a polyclonal anti-PN-1 antibody and an anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody as a protein loading control. Results are shown with representative immunoblots from at least 3 independent experiments (A, B, and C). (D) Densitometric quantification, expressed as the mean intensity (PN-1/GAPDH ratio) relative to control (WT binding), determined for each immunoblot. NS, not significant; **, P < 0.01; ***, P < 0.0001 versus WT or the variants without heparin.
The role played by membrane glycosaminoglycans in the binding of PN-1 to HUVECs was examined. Binding experiments were conducted in the presence of exogenous GAGs (heparin, dermatan sulfate, or chondroitin sulfate) as competitors of membrane GAGs. As shown in Fig. 4A and the densitometric analysis results in Fig. 4D, heparin and dermatan sulfate greatly inhibited the binding of PN-1 to HUVECs (by 95% ± 2% and 73% ± 4%, respectively), whereas chondroitin sulfate, at the same concentration, had a lower inhibitory effect (17% ± 6% of inhibition). In addition, protamine sulfate, a basic protein which binds to heparan sulfate proteoglycans (HSPGs) (15), also prevented PN-1 binding to HUVECs (data not shown), ruling out the possibility that inhibition by heparin was due to a conformational change of PN-1 (31).
Functional domains of PN-1 required for binding to HUVECs and antiangiogenic effects.
Mutant forms of PN-1 were used to identify the PN-1 functional domains involved in its effects on HUVECs. R346A PN-1, which lacks antiprotease activity, and K7Q PN-1, in which the heparin site is nonfunctional (34), were examined for their binding activity with HUVECs and effects on the angiogenic responses. At 4°C, R346A PN-1 bound to HUVECs as WT PN-1 did (Fig. 4C). Cell binding of the inactive variant was also fully inhibited by heparin. K7Q PN-1 also bound to HUVECs but to a lesser extent than WT and R346A PN-1, as indicated by the lower intensity of the corresponding band, and this binding was heparin resistant. Densitometric analysis of the blots obtained from at least 3 independent experiments gave 92.9 ± 2.4% and 41.2 ± 4.3% of WT binding for R346A and K7Q PN-1, respectively (Fig. 4D). These data indicate that GAGs constitute the major binding site for WT and inactive PN-1 at the surface of HUVECs. In addition, they also suggest that an accessory, distinct site allows the binding of K7Q PN-1.
To determine whether PN-1 could be internalized, binding experiments were performed at 37°C, and whole-cell lysates and cytosolic and membrane fractions were analyzed for the presence of PN-1 (Fig. 5). The data obtained for whole-cell lysates were similar to those obtained at 4°C for WT, R346A, and K7Q PN-1 (Fig. 5A and C). When added simultaneously to PN-1 at 37°C, heparin also inhibited the binding of WT PN-1 to HUVECs (Fig. 5B, WT Hep). The same effect was observed when heparin was added 2 h after PN-1 (Fig. 5B and C, WT then Hep), indicating that even after 2 h at 37°C, PN-1 was still largely associated with the cell surface and could be removed by heparin. In addition, PN-1 was observed in the membrane fraction but was not detectable in the cytosolic fraction (Fig. 5A, Mb and Cyto), suggesting that PN-1 was not internalized. However, as PN-1 has been shown to be internalized, free or in complex with proteases, by a low-density lipoprotein receptor-related protein 1 (LRP1) mechanism in fibroblasts (17, 20, 21), we used the high-affinity ligand of LRP, RAP as an antagonist for binding to LRP. However, RAP had no effect on PN-1 binding to HUVECs (Fig. 5B). Together, these data thus indicate that binding of PN-1 to surface GAGs on HUVECs did not lead to the internalization of the serpin.
Fig 5.

PN-1 binding to HUVECs at 37°C. (A) Whole-cell lysates and cell fractions were obtained after a 2-h incubation of PN-1 variants with HUVECs. Mb, membrane; Cyto, cytosolic. Samples were analyzed as described for Fig. 4, and no signal corresponding to PN-1 was detected in the cytosolic fractions. (B) Whole-cell lysates prepared from HUVECs incubated in the presence or not of heparin (Hep) added at the same time (WT Hep) or 2 h after (WT then Hep), WT PN-1, or of RAP were analyzed as described above. Results are shown in representative blots from at least 3 independent experiments (A and B) or expressed as the mean intensity (PN-1/GAPDH ratio) relative to control WT binding, determined for each immunoblot (C). *, P < 0.05; ***, P < 0.0001, versus WT.
R346A PN-1 retained the capacity to inhibit FGF- and VEGF-induced HUVEC proliferation (Fig. 6A), VEGF-induced HUVEC migration (Fig. 6B), and tube formation (Fig. 6C). In contrast, K7Q PN-1 had only a limited effect on capillary tube formation and did not significantly reduce either cell proliferation or migration (Fig. 6A to C). These results indicate that the binding of PN-1 to the cell surface GAGs is essential for its antiangiogenic activity in vitro, whereas the antiprotease activity of PN-1 is dispensable. The possibility that PN-1 could act by blocking the binding of VEGF to its surface HSPG coreceptors was ruled out by the observation that PN-1 also efficiently inhibited the mitogenic activity of VEGF (121 amino acids) (Fig. 6D), which does not bind heparin (14).
Fig 6.

Role of PN-1 functional sites on PN-1 HUVEC angiogenic responses. Effects of PN-1 variants (20 μg/ml [A and C] or 50 μg/ml [B]) on FGF- and VEGF-induced HUVEC proliferation (n = 4 to 10) (A), VEGF-induced HUVEC migration (n = 3) (B), and VEGF-induced HUVEC organization in Matrigel (C). The tube density was measured on 3 fields per well from 3 to 4 experiments and averaged relative to the control without PN-1 and VEGF. (D) Effects of WT PN-1 on VEGF 121-induced HUVEC proliferation. **, P < 0.01; ***, P < 0.001, versus VEGF or FGF alone.
PN-1 deficiency enhances ex vivo and in vivo angiogenesis.
Sprouting of vascular tubes from WT and PN-1-deficient aortic rings into a collagen gel was analyzed. The tubulogenic responses of WT and PN-1-deficient aortic explants in the absence of VEGF were weak. Nevertheless, sprouting from PN-1-deficient rings was significantly higher than from WT rings (Fig. 7, isolectin B4 labeling of endothelial cells is shown in red). VEGF enhanced the tubulogenic response of aortic explants, and quantitative analysis revealed that the mean density of the neovessels was significantly higher for the PN-1-deficient aorta (1.7-fold) than for WT aorta, with more and thicker sprouts. In addition, treatment of PN-1-deficient aortic rings with recombinant PN-1 reversed the effect of VEGF, with a tubulogenic response close to that of WT explants. These data indicate that the enhanced angiogenic capacity of PN-1-deficient endothelial cells was rescued by the addition of exogenous PN-1.
Fig 7.

Impact of PN-1 deficiency on ex vivo angiogenesis. Thoracic aortic rings from PN-1-deficient (-/-) and WT (+/+) mice were cultured in a collagen gel in the presence or absence of 50 ng/ml VEGF and 20 μg/ml recombinant WT PN-1. The microvascular networks sprouting from the rings were observed by phase-contrast microscopy and after isolectin B4 labeling (in red), and results were quantified by densitometric analysis. Representative micrographs and results are shown for 6 to 20 rings from 3 to 7 mice. ***, P < 0.001 WT versus PN-1-deficient mice in the presence or absence of recombinant PN-1 (rPN-1).
The murine model of the Matrigel plug was used to further evaluate the role of PN-1 in angiogenesis in vivo. Six days after Matrigel implantation, the plugs of PN-1-deficient and WT mice were removed, photographed, sectioned, and stained (Fig. 8). Histological examination of the Matrigel implants showed the formation of vessels connected with the murine vasculature, as demonstrated by the presence of blood-filled spaces and vessels. This was confirmed by fluorescence of the vessels within the Matrigel plugs from mice that received an intravenous injection of fluorescein isothiocyanate (FITC)-dextran 20 min before sacrifice (Fig. 8A). Plugs implanted in PN-1-deficient mice appeared globally bloodier (Fig. 8B). Matrigel section analysis after hematoxylin and eosin staining (Fig. 8C) revealed a significantly increased cell infiltration in plugs from PN-1-deficient compared to WT mice (Fig. 8D), indicating an enhanced vascularization of these plugs. Addition of recombinant PN-1 to the Matrigel plugs reversed the angiogenic response observed in PN-1-deficient mice to levels close to those observed in WT mice.
Fig 8.
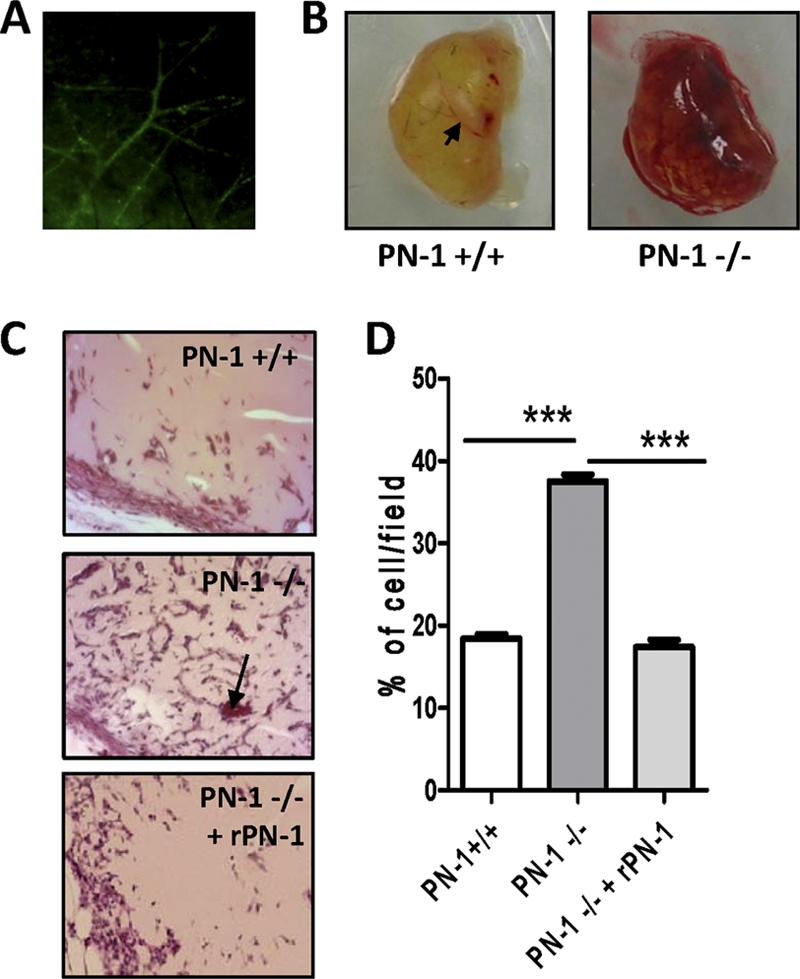
Impact of PN-1 deficiency on in vivo angiogenesis: vessel formation in Matrigel plugs. Control plugs were implanted in 9 to 10 wild-type mice (PN-1 +/+) or PN-1-deficient mice (PN-1 -/-), and plugs supplemented with 20 μg/ml recombinant PN-1 (rPN-1) were implanted in 3 PN-1-deficient mice (PN-1−/− + rPN-1). (A) Representative fluorescence of the vessels following retroorbital injection of FITC-dextran. (B) Representative plugs from PN-1+/+ and PN-1−/− mice. (C) Representative microphotographs of sections of Matrigel plugs stained with hematoxylin-eosin. Magnification, ×200. Arrows indicate erythrocyte-containing neovessels. (D) Quantification of cell infiltration (4 fields/plug). ***, P < 0.001 for WT versus PN-1-deficient mice and for PN-1-deficient mice versus PN-1-deficient mice administered rPN-1.
DISCUSSION
The role of the regulatory protein PN-1 in vascular biology has not been investigated until recently. It is now assumed that PN-1, expressed by vascular and blood cells, is not essential under physiological conditions but is required for appropriate responses to pathological conditions. As PN-1 can interact with numerous partners, including molecules involved in angiogenesis, it appears to be a good candidate for the regulation of angiogenic responses. In the present study, we analyzed the relation between the interaction of PN-1 with endothelial cells and its effects on their angiogenic responses in vitro as well as in ex vivo and in vivo mouse models. Our data provide the first evidence that PN-1 may play a role in modulating angiogenesis. Endothelial cells have already been reported to express PN-1 (7). In the present study, we found that exogenous recombinant PN-1, or PN-1 secreted by platelets, has the ability to interact with endothelial cells, indicating that all potential PN-1-binding sites on endothelial cells are not saturated under basal conditions. The reactive site of PN-1 was not required for binding to HUVECs, excluding a surface-exposed serine protease that could be the acceptor under nonstimulated conditions. In contrast, we obtained clear evidence that the major binding site for recombinant PN-1 on HUVECs is constituted by surface-exposed GAGs: (i) heparin and other HSPGs prevented PN-1 binding to HUVECs; (ii) protamine sulfate, which blocks cell surface GAGs, also prevented PN-1 binding; and (iii) cell-bound PN-1 was eluted by heparin. As previously suggested by studies from our group (31) and others (36), our results indicated that PN-1 binding to the cell surface does not require a specific GAG sequence. In addition, the findings agree with previous studies showing that endogenous PN-1 expressed by endothelial cells localizes at the cell surface, at least partly bound to the chondroitin chain of thrombomodulin (7). Surprisingly, however, we observed that the K7Q PN-1 variant, which is unable to bind heparin, still bound to HUVECs, although to a much lesser extent than WT PN-1. The normal antithrombin activity of this K7Q PN-1 variant (Fig. 1) excludes the possibility that it was wrongly folded. These data thus suggest that PN-1 can interact with an accessory binding site distinct from GAGs. However, this interaction only occurs when the PN-1 heparin-binding site is nonfunctional, and it has no consequence on angiogenic cell responses, indicating that it probably plays a minor role in the regulation of angiogenesis by PN-1.
PN-1 binding to cell surface GAGs was not followed by PN-1 internalization in cultured endothelial cells, as evidenced by studies performed at 37°C and on cell fractions. These findings contrast with the previously reported internalization of PN-1, either free or complexed with target proteases, by fibroblasts (16, 26) and SMCs (26) via an LRP mechanism. An alternative mechanism of PN-1 internalization, involving syndecan-1 as a receptor, has also been reported for LRP-deficient fibroblasts (20). However, LRP has previously been reported to be poorly expressed by endothelial cells (24), in agreement with the fact that we did not observe any effect of its antagonist RAP on the PN-1 interaction with HUVECs.
Binding of exogenous PN-1 was accompanied by the inhibition of angiogenic responses in vitro. PN-1 did not modify HUVEC adhesion but decreased cell spreading on different matrix proteins and inhibited cell proliferation, migration, and tube formation. When PN-1 binding to cell surface GAGs was prevented by the K7Q mutation, it no longer had any effect on their angiogenic responses. In contrast, R346A PN-1 consistently inhibited VEGF responses of HUVECs, indicating that the modulation of cellular responses by PN-1 is mostly independent of its protease-inhibitory activity. PN-1 has been reported to inhibit developmental steps in early Xenopus laevis embryos (27), also independently of its serpin activity. Our observations are also in agreement with the antiangiogenic effects reported for other serpins, in particular antithrombin (28), whose antiangiogenic activity has also been reported to be independent of its antiprotease activity.
A number of pro- and antiangiogenic factors have been shown to bind to HSPGs or heparin, including the key angiogenic growth factors FGF and VEGF (35). Binding of FGF and VEGF to HSPGs at the cell surface is known to promote their binding to their respective receptors (12). The observation that increasing the concentrations of FGF or VEGF decreased the inhibitory effects of PN-1 on cell proliferation suggested that PN-1 binding to GAGs could prevent binding of these angiogenic factors to their endothelial receptors, as suggested for antithrombin (38). However, PN-1 still efficiently inhibited the mitogenic activity of VEGF 121, an alternatively spliced variant of VEGF that lacks the heparin-binding domain but retains the capacity to induce HUVEC proliferation. In contrast to many other antiangiogenic agents, we observed that PN-1 inhibition of HUVEC responses was not related to modification of ERK or Akt phosphorylation, either under basal conditions or after stimulation by VEGF. Together, these results indicated that PN-1 does not directly modulate VEGF- or FGF-induced responses by blocking their interactions with cell surface binding sites, but rather by interacting with another cell receptor through its heparin-binding site, which would in turn interfere downstream of the VEGF- or FGF-induced signals. Current investigations are in progress to identify the mechanism that couples PN-1 binding to HUVECs and PN-1-induced inhibition of angiogenic responses.
The concentrations of recombinant PN-1 used in the in vitro experiments were comparable to those used for other antiangiogenic serpins (1, 18). Moreover, PN-1 is known to be carried by platelets and released locally in large amounts upon activation. Local concentration of GAG-bound PN-1 is therefore likely to reach a high level at endothelial or SMC surfaces, and the serpin is likely to have a potent physiological impact on angiogenesis in vivo. Regardless of the mechanism of action of PN-1, the animal studies in fact clearly indicated that PN-1 inhibits endothelial angiogenic responses in vivo. PN-1 deficiency resulted in an increased endothelial sprouting from aortic rings and invasion of Matrigel plugs, effects reversed by the addition of recombinant PN-1, indicating that PN-1 directly regulates the angiogenic capacity of the endothelium in mice. Our in vivo studies thus demonstrate the physiological relevance of PN-1 in angiogenesis.
The origin of PN-1 involved in the modulation of angiogenic responses in vivo could come from different cell types. Endothelial cells themselves express PN-1, even though this appears to be at a low level. PN-1 is also stored in platelet alpha granules (4), and we have observed that PN-1 released during platelet plug formation binds to endothelial cells. Platelets are known to be key mediators in angiogenesis, and the secretion of pro- and antiangiogenic factors from platelet granules is crucial in this process (10). Furthermore, PN-1 is also expressed by SMCs (30) and is overexpressed by these cells under pathological conditions such as hypertension (6) or aneurysms. Endothelial cells are thus likely to be in contact with significant amounts of PN-1 of various origins in vivo. Local delivery of PN-1 by activated platelets or overexpression of PN-1 in pathological vessels could control excessive angiogenesis, especially under pathological conditions in which it would be beneficial to downregulate it, such as tumor growth, rheumatoid arthritis, or diabetic retinopathy. However, we cannot exclude that overexpression of PN-1 could also be deleterious in processes requiring correct angiogenesis, such as tissue regeneration. Regulation of PN-1 expression and/or function could thus lead to the modulation of angiogenic processes implicated in many diseases, and further studies are needed to determine whether PN-1 could constitute a therapeutic target.
In summary, PN-1 binds to endothelial cell surface GAGs, independently of its protease-inhibitory activity. This interaction leads to the negative regulation of endothelial cell responses and to a decreased angiogenesis process. PN-1 is thus a yet-unrecognized player in angiogenesis, a dynamic and multistep process controlled directly and indirectly by angiogenic stimulators and inhibitors. PN-1 thus appears to be an important player in the angiogenic balance.
ACKNOWLEDGMENTS
We thank Mary Osborne-Pellegrin for editing the manuscript.
This work was supported by INSERM, University Paris Diderot Sorbonne Paris Cité, the Leducq Foundation, the Bettencourt Schueller Foundation, and grant 2009002497 from the “Fondation de France.” Y. Boulaftali was supported by the “Fondation pour la Recherche Médicale,” and Sonia Selbonne was supported by the Région Ile de France (DIM Cardiovasculaire).
Footnotes
Published ahead of print 13 February 2012
REFERENCES
- 1. Asanuma K, et al. 2007. Protein C inhibitor inhibits breast cancer cell growth, metastasis and angiogenesis independently of its protease inhibitory activity. Int. J. Cancer 121: 955–965 [DOI] [PubMed] [Google Scholar]
- 2. Baker JB, Low DA, Simmer RL, Cunningham DD. 1980. Protease-nexin: a cellular component that links thrombin and plasminogen activator and mediates their binding to cells. Cell 21: 37–45 [DOI] [PubMed] [Google Scholar]
- 3. Balsara RD, Ploplis VA. 2008. Plasminogen activator inhibitor-1: the double-edged sword in apoptosis. Thromb. Haemost. 100: 1029–1036 [PMC free article] [PubMed] [Google Scholar]
- 4. Boulaftali Y, et al. 2010. Anticoagulant and antithrombotic properties of platelet protease nexin-1. Blood 115: 97–106 [DOI] [PubMed] [Google Scholar]
- 5. Boulaftali Y, et al. 2011. Platelet protease nexin-1, a serpin that strongly influences fibrinolysis and thrombolysis. Circulation 123: 1326–1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bouton MC, et al. 2003. The serpin protease-nexin 1 is present in rat aortic smooth muscle cells and is upregulated in L-NAME hypertensive rats. Arterioscler. Thromb. Vasc. Biol. 23: 142–147 [DOI] [PubMed] [Google Scholar]
- 7. Bouton MC, et al. 2007. Protease nexin-1 interacts with thrombomodulin and modulates its anticoagulant effect. Circ. Res. 100: 1174–1181 [DOI] [PubMed] [Google Scholar]
- 8. Chen LM, Zhang X, Chai KX. 2004. Regulation of prostasin expression and function in the prostate. Prostate 59: 1–12 [DOI] [PubMed] [Google Scholar]
- 9. Eaton DL, Baker JB. 1983. Evidence that a variety of cultured cells secrete protease nexin and produce a distinct cytoplasmic serine protease-binding factor. J. Cell Physiol. 117: 175–182 [DOI] [PubMed] [Google Scholar]
- 10. Feng W, et al. 2011. A novel role for platelet secretion in angiogenesis: mediating bone marrow-derived cell mobilization and homing. Blood 117: 3893–3902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gerhardt H, et al. 2003. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J. Cell Biol. 161: 1163–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gitay-Goren H, Soker S, Vlodavsky I, Neufeld G. 1992. The binding of vascular endothelial growth factor to its receptors is dependent on cell surface-associated heparin-like molecules. J. Biol. Chem. 267: 6093–6098 [PubMed] [Google Scholar]
- 13. Gronke RS, Knauer DJ, Veeraraghavan S, Baker JB. 1989. A form of protease nexin I is expressed on the platelet surface during platelet activation. Blood 73: 472–478 [PubMed] [Google Scholar]
- 14. Houck KA, Leung DW, Rowland AM, Winer J, Ferrara N. 1992. Dual regulation of vascular endothelial growth factor bioavailability by genetic and proteolytic mechanisms. J. Biol. Chem. 267: 26031–26037 [PubMed] [Google Scholar]
- 15. Jang JH, Wang F, Kan M. 1997. Heparan sulfate is required for interaction and activation of the epithelial cell fibroblast growth factor receptor-2IIIb with stromal-derived fibroblast growth factor-7. In Vitro Cell Dev. Biol. Anim. 33: 819–824 [DOI] [PubMed] [Google Scholar]
- 16. Knauer DJ, Majumdar D, Fong PC, Knauer MF. 2000. SERPIN regulation of factor XIa. The novel observation that protease nexin 1 in the presence of heparin is a more potent inhibitor of factor XIa than C1 inhibitor. J. Biol. Chem. 275: 37340–37346 [DOI] [PubMed] [Google Scholar]
- 17. Knauer MF, Kridel SJ, Hawley SB, Knauer DJ. 1997. The efficient catabolism of thrombin-protease nexin 1 complexes is a synergistic mechanism that requires both the LDL receptor-related protein and cell surface heparins. J. Biol. Chem. 272: 29039–29045 [DOI] [PubMed] [Google Scholar]
- 18. Larsson H, et al. 2000. Antiangiogenic effects of latent antithrombin through perturbed cell-matrix interactions and apoptosis of endothelial cells. Cancer Res. 60: 6723–6729 [PubMed] [Google Scholar]
- 19. Lee SY, et al. 2008. F2L, a peptide derived from heme-binding protein, inhibits LL-37-induced cell proliferation and tube formation in human umbilical vein endothelial cells. FEBS Lett. 582: 273–278 [DOI] [PubMed] [Google Scholar]
- 20. Li X, Herz J, Monard D. 2006. Activation of ERK signaling upon alternative protease nexin-1 internalization mediated by syndecan-1. J. Cell Biochem. 99: 936–951 [DOI] [PubMed] [Google Scholar]
- 21. Low DA, Baker JB, Koonce WC, Cunningham DD. 1981. Released protease-nexin regulates cellular binding, internalization, and degradation of serine proteases. Proc. Natl. Acad. Sci. U. S. A. 78: 2340–2344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Luthi A, et al. 1997. Endogenous serine protease inhibitor modulates epileptic activity and hippocampal long-term potentiation. J. Neurosci. 17: 4688–4699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mansilla S, et al. 2008. Macrophages and platelets are the major source of protease nexin-1 in human atherosclerotic plaque. Arterioscler. Thromb. Vasc. Biol. 28: 1844–1850 [DOI] [PubMed] [Google Scholar]
- 24. Moestrup SK, Gliemann J, Pallesen G. 1992. Distribution of the alpha 2-macroglobulin receptor/low density lipoprotein receptor-related protein in human tissues. Cell Tissue Res. 269: 375–382 [DOI] [PubMed] [Google Scholar]
- 25. Monard D, Suidan HS, Nitsch C. 1992. Relevance of the balance between glia-derived nexin and thrombin following lesion in the nervous system. Ann. N. Y. Acad. Sci. 674: 237–242 [DOI] [PubMed] [Google Scholar]
- 26. Muhl L, et al. 2007. Inhibition of PDGF-BB by factor VII-activating protease (FSAP) is neutralized by protease nexin-1, and the FSAP-inhibitor complexes are internalized via LRP. Biochem. J. 404: 191–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Onuma Y, Asashima M, Whitman M. 2006. A serpin family gene, protease nexin-1 has an activity distinct from protease inhibition in early Xenopus embryos. Mech. Dev. 123: 463–471 [DOI] [PubMed] [Google Scholar]
- 28. O'Reilly MS, Pirie-Shepherd S, Lane WS, Folkman J. 1999. Antiangiogenic activity of the cleaved conformation of the serpin antithrombin. Science 285: 1926–1928 [DOI] [PubMed] [Google Scholar]
- 29. Passaniti A, et al. 1992. A simple, quantitative method for assessing angiogenesis and antiangiogenic agents using reconstituted basement membrane, heparin, and fibroblast growth factor. Lab. Invest. 67: 519–528 [PubMed] [Google Scholar]
- 30. Richard B, et al. 2004. Protease nexin-1: a cellular serpin down-regulated by thrombin in rat aortic smooth muscle cells. J. Cell Physiol. 201: 138–145 [DOI] [PubMed] [Google Scholar]
- 31. Richard B, et al. 2006. Modulation of protease nexin-1 activity by polysaccharides. Thromb. Haemost. 95: 229–235 [DOI] [PubMed] [Google Scholar]
- 32. Richard B, et al. 2006. The serpin protease nexin-1 regulates vascular smooth muscle cell adhesion, spreading, migration and response to thrombin. J. Thromb. Haemost. 4: 322–328 [DOI] [PubMed] [Google Scholar]
- 33. Rossignol P, et al. 2004. Protease nexin-1 inhibits plasminogen activation-induced apoptosis of adherent cells. J. Biol. Chem. 279: 10346–10356 [DOI] [PubMed] [Google Scholar]
- 34. Stone SR, et al. 1994. Localization of the heparin-binding site of glia-derived nexin/protease nexin-1 by site-directed mutagenesis. Biochemistry 33: 7731–7735 [DOI] [PubMed] [Google Scholar]
- 35. Stringer SE. 2006. The role of heparan sulphate proteoglycans in angiogenesis. Biochem. Soc. Trans. 34: 451–453 [DOI] [PubMed] [Google Scholar]
- 36. Wallace A, Rovelli G, Hofsteenge J, Stone SR. 1989. Effect of heparin on the glia-derived nexin-thrombin interaction. Biochem. J. 257: 191–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang W, et al. 2005. The heparin-binding site of antithrombin is crucial for antiangiogenic activity. Blood 106: 1621–1628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang W, Swanson R, Xiong Y, Richard B, Olson ST. 2006. Antiangiogenic antithrombin blocks the heparan sulfate-dependent binding of proangiogenic growth factors to their endothelial cell receptors: evidence for differential binding of antiangiogenic and anticoagulant forms of antithrombin to proangiogenic heparan sulfate domains. J. Biol. Chem. 281: 37302–37310 [DOI] [PubMed] [Google Scholar]