

Abstract
Several arenaviruses, chiefly Lassa virus (LASV), cause hemorrhagic fever (HF) disease in humans and pose a significant public health concern in regions where they are endemic. On the other hand, evidence indicates that the globally distributed prototypic arenavirus lymphocytic choriomeningitis virus (LCMV) is a neglected human pathogen. The phosphatidylinositol 3-kinase (PI3K)/Akt pathway participates in many cellular processes, including cell survival and differentiation, and also has been shown to play important roles in different steps of the life cycles of a variety of viruses. Here we report that the inhibition of the PI3K/Akt pathway inhibited budding and to a lesser extent RNA synthesis, but not cell entry, of LCMV. Accordingly, BEZ-235, a PI3K inhibitor currently in cancer clinical trials, inhibited LCMV multiplication in cultured cells. These findings, together with those previously reported for Junin virus (JUNV), indicate that targeting the PI3K/Akt pathway could represent a novel antiviral strategy to combat human-pathogenic arenaviruses.
INTRODUCTION
Several arenaviruses cause hemorrhagic fever (HF) disease in humans. Lassa virus (LASV) and Junin virus (JUNV) are the causative agents of Lassa fever (LF) and Argentine HF disease, respectively, which represent significant public health problems within their endemic geographic regions of West Africa (LASV) and Argentina (JUNV). In addition, evidence indicates that the globally distributed prototypic arenavirus lymphocytic choriomeningitis virus (LCMV) is a neglected human pathogen of clinical significance in congenital viral infections (3, 15, 24). Moreover, LCMV infection of immunosuppressed individuals can result in severe disease and death (13, 30). Public health concerns about arenavirus infections are aggravated by the lack of FDA-licensed vaccines and limited existing therapeutic options. The only arenavirus vaccine tested in humans is Candid 1, a live attenuated strain of JUNV that is licensed only in Argentina and is ineffective against LASV or LCMV. On the other hand, current arenavirus antiviral drug therapy is restricted to the use of the nucleoside analogue ribavirin, which is only partially effective and associated with significant side effects (9, 25, 26). Therefore, it is important to develop novel antiviral strategies to combat human-pathogenic arenaviruses, a task that would be facilitated by a detailed understanding of the molecular and cell biology of arenaviruses.
Arenaviruses are enveloped viruses with a bi-segmented, negative-strand (NS) RNA genome and a life cycle restricted to the cell cytoplasm (6). Each RNA genome segment uses an ambisense coding strategy to direct the expression of two gene products in opposite orientations and separated by a noncoding intergenic region (IGR). The large segment (L) (7.2 kb) encodes the L protein, an RNA-dependent RNA polymerase (RdRp), and the small RING finger protein Z that is the counterpart of the matrix (M) protein found in many enveloped NS RNA viruses. The small segment (S) (3.5 kb) encodes the viral nucleoprotein (NP) and the glycoprotein precursor (GPC) that is posttranslationally processed to yield the peripheral virion attachment protein GP1 and the fusion-active transmembrane protein GP2. Trimers of GP1/GP2 form the spikes that decorate the virus surface and mediate cell entry via receptor-mediated endocytosis (6).
Many viruses interfere with signaling pathways in their infected host cells to favor an environment conducive to a productive infection, which can also impact the host physiology and contribute to virus-associated pathogenesis and disease. Therefore, the identification and targeting of host cell factors and pathways involved in different steps of a virus life cycle may uncover novel antiviral strategies. In this regard, the phosphatidylinositol 3-kinase (PI3K)/Akt pathway, known to regulate a variety of cellular processes, including cell growth, proliferation, survival, and metabolism (14), has also been involved in the regulation of cell entry (34), as well as RNA replication and gene expression (38), for a variety of viruses. Thus, infection with the New World (NW) arenavirus JUNV was shown to activate the PI3K/Akt pathway (20), and inhibition of the PI3K/Akt pathway resulted in decreased production of infectious progeny due to a blockage of the recycling of the transferrin receptor involved in JUNV cell entry (20). Because significant biological differences have been observed among different arenaviruses (17), we examined whether the PI3K/Akt pathway also played a role in the life cycle of LCMV, the prototypic Old World (OW) arenavirus. OW arenaviruses are a group that includes LASV, the HF arenavirus with the highest impact in public health. To answer this question, we tested a variety of commercially available PI3K/Akt inhibitors. The PI3K/Akt signaling pathway is initiated by receptor-mediated recruitment of catalytically active PI3K to the membrane. Active PI3K converts phosphatidylinositol 4,5-biphosphate to phosphatidylinositol 3,4,5-triphosphate (PIP3). PIP3 facilitates colocalization of Akt with its activating kinase PDK1, which mediates phosphorylation of Akt at residue Thr 308 (T308); this results in the initial activation of Akt, which is subsequently fully activated by a second phosphorylation event at Ser 473 (S473). Consistent with previous findings, treatment with the PI3K inhibitor LY294002 (LY) resulted in strong inhibition of Akt phosphorylation (S473) that was associated with a robust inhibitory effect on LCMV multiplication in the absence of cell toxicity. Mechanism-of-action studies indicated that LY did not affect virus cell entry but rather viral budding and, to a lesser extent, viral RNA synthesis. To our knowledge, this is the first report showing a contribution of the PI3K/Akt pathway to virus budding. The PI3K/Akt pathway is often upregulated in tumors and therefore is being pursued as a target for antitumor therapy, and several inhibitors of the PI3K/Akt pathway are currently undergoing clinical trials as potential drugs for treating several different tumors (21). Our finding that BEZ-235, a dual PI3K/mammalian target of rapamycin (Rpm) (mTOR) inhibitor currently in cancer clinical trials, inhibited multiplication of LCMV in cultured cells provided further impetus to explore targeting of the PI3K/Akt pathway as a novel antiviral strategy to combat human-pathogenic arenaviruses.
MATERIALS AND METHODS
Plasmids.
LCMV- and LASV-Z-expressing plasmids have been described (40). These Z constructs were FLAG tagged at their C termini. p-T7, pMG-CAT, pCAGGS-NP, and pCAGGS-L have been described (18, 19).
Chemical inhibitors.
Akt-IV and Akt-VIII were purchased from Calbiochem (product numbers 124015 and 124018, respectively). LY294002 was purchased from Cell Signaling (product number 9901). BEZ-235 was purchased from Selleck Chemicals (product number s1009). Rapamycin was purchased from Sigma-Aldrich (product number R0395).
Cells and viruses.
293T cells were grown in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen) containing 10% fetal bovine serum, 2 mM l-glutamine, 100 mg/ml streptomycin, and 100 U/ml penicillin. Recombinant LCMV (rLCMV), Armstrong strain, and r3LCMV/green fluorescent protein (GFP) as well as rVSVΔG-GFP/VSV-G and rVSVΔG-GFP/LCMV-GP have been described (12, 41). The GP expressed by rVSVΔG-GFP/LCMV-GP corresponded to that of the Armstrong strain of LCMV.
Virus titration.
LCMV titers were determined using an immunofocus assay (4). Briefly, 10-fold serial virus dilutions were used to infect Vero cell monolayers in a 96-well plate, and at 20 h postinfection (p.i.), cells were fixed with 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS). After cell permeabilization by treatment with 0.3% Triton X-100 in PBS containing 3% bovine serum albumin (BSA), cells were stained by using an anti-NP mouse monoclonal antibody and an Alexa Fluor 568-labeled anti-mouse second antibody (Molecular Probes).
Detection of Akt phosphorylation.
Cells were treated with the indicated compounds and concentrations. After 4 or 24 h of treatment, cells were washed with PBS and cell lysates prepared using a lysis buffer (1% NP-40, 50 mM Tris-HCl [pH 8.0], 62.5 mM EDTA, 0.4% sodium deoxycholate) supplemented with phosphatase inhibitors Cocktail I (product number P2850; Sigma) and II (product number P5726; Sigma) just before use. Cell lysates were clarified by centrifugation (13,000 × g, 5 min at 4°C). Samples were analyzed by Western blotting (WB) to detect either total or phosphorylated (S473) Akt.
Cytotoxicity assay.
The effect of compounds tested in 293T cell viability was assessed using the CellTiter-Glo luminescent cell viability assay (Promega). This method determines the number of viable cells based on levels of ATP (8). Briefly, 5 × 104 cells were plated per 96-well plate and cultured overnight. Cells were treated with the indicated concentrations of each compound for 24 h before the CellTiter-Glo reagent was added. Thereafter the assay was performed according to the manufacturer's recommendations and readings were obtained using a luminometer (Centro LB 960; Berthold Technologies). Viability of compound-treated cells was calculated as the percentage of values obtained with dimethyl sulfoxide (DMSO)-treated cells (set at 100%).
Assessment of LCMV GP-mediated cell entry.
Recombinant vesicular stomatitis viruses (rVSV) (rVSVΔG-GFP/VSV-G and rVSVΔG-GFP/LCMV-GP) were used to assess the contribution of the PI3K/Akt pathway to cell entry mediated by the GP of the Armstrong strain of LCMV. 293T cells were pretreated with the indicated compounds and concentrations for 1 h prior to infection (multiplicity of infection [MOI], 1) with the indicated rVSV in the presence of drug. At 12 h p.i., cells were fixed (4% PFA/PBS) and numbers of GFP-positive cells determined by direct epifluorescence. Total cells and GFP-positive cells were counted in four different fields, and the average and standard deviation (SD) of the percentage of GFP-positive cells were determined. Results represent the average ± SD of the results from the two independent infections for each virus.
LCMV minigenome assay.
293T cells were seeded (4.5 × 105 per well) on M-12 well plates and the following day transfected with p-T7, pMG-CAT, pCAGGS-NP, and pCAGGS-L under conditions previously described (18, 19, 29, 31, 32). At 5 h posttransfection, the transfection medium was replaced with fresh medium containing the indicated compounds and concentrations. At 24 h posttransfection, cell lysates were prepared to determine levels of chloramphenicol acetyltransferase (CAT) protein by enzyme-linked immunosorbent assay (ELISA) using a CAT ELISA kit (product number 11363727001; Roche). Briefly, cells were lysed with 1 ml of lysis buffer and equal amounts (4 μl) of each sample were used for ELISA. Dilutions of a known amount of CAT were used to generate a calibration curve. CAT ELISA plates were incubated for 1 h at 37°C, followed by two washes with wash buffer, followed by reaction with an antibody to CAT for 1 h at 37°C. After reaction with the primary antibody, samples were washed twice and then reacted with the secondary antibody conjugated to peroxidase for 1 h at 37°C, followed by two washes prior to adding the substrate. After 20 min at room temperature, samples were analyzed using an ELISA reader (SPECTRA max plus 384; Molecular Devices) to determine the absorbance (405 nm for samples, 490 nm for the reference).
RNA analysis by Northern blot hybridization.
Total cellular RNA was isolated by using TRI Reagent (Molecular Research Center, Inc.) according to the manufacturer's instructions and was analyzed by Northern blot hybridization. Briefly, RNA samples were fractionated by 2.2 M formaldehyde-agarose (1.2%) gel electrophoresis followed by transfer (4 h) in 20× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate) of the RNA to a Magnagraph (0.22-μm-pore-size) membrane using the rapid downward transfer system (TurboBlotter). Membrane-bound RNA was cross-linked by exposure to UV, and the membrane was hybridized to a 32P-labeled strand-specific probe to the minigenome (MG)-derived CAT mRNA.
Polymerase II (Pol-II)-based transcription assay.
293T cells were seeded (8 × 104/well) on a 96-well plate and transfected the next day with pC-Fluc (27.5 ng/well) using Lipofectamine 2000 (LF) (Invitrogen); 12 h later, the transfection medium was replaced with fresh medium containing the indicated compounds and concentrations, and 24 h later, cell lysates were prepared for firefly luciferase (Fluc) assay using the Steady Glo lysis buffer (Promega) and conditions according to the manufacturer's recommendations. Values of Fluc activity were obtained using a luminometer (Centro LB 960; Berthold Technologies). Levels of Fluc activity obtained with lysates from compound-treated cells were determined as percentages of values obtained with lysates of DMSO-treated control cells (set at 100%).
Budding assay.
293T cells (2.5 × 105) were transfected with 0.25 μg of either pC-LCMV-Z or pC-LASV-Z using Lipofectamine 2000 (LF) (2.5 μl LF/μg DNA). At 5 h after transfection, the medium was replaced with fresh medium containing the indicated compounds and concentrations. At 24 h after treatment, virus-like particle (VLP)-containing tissue culture supernatants and cells were collected. After clarification from cell debris (1,500 × g, 5 min), VLPs were collected by ultracentrifugation (100,000 × g, 30 min at 4°C) through a 20% sucrose cushion. Cells and VLPs were resuspended in lysis buffer (1% NP-40, 50 mM Tris-HCl [pH 8.0], 62.5 mM EDTA, 0.4% sodium deoxycholate) and analyzed by Western blotting.
Immunoblotting.
Cell lysates or VLP samples were resolved by SDS-PAGE followed by Western blotting (WB) using the indicated antibodies. To detect FLAG-tagged Z proteins, we used a rabbit polyclonal serum to FLAG (product number 162150; Cayman) and an anti-rabbit IgG conjugated to horseradish peroxidase (HRP) as a secondary antibody. To detect either total Akt or phosphorylated Akt (S473), mouse monoclonal antibodies to Akt (product number 9272; Cell Signaling) and to phosphorylated (S473) Akt (product number 9271; Cell Signaling), respectively, were used combined with an anti-mouse IgG conjugated to HRP as a secondary antibody.
RESULTS
Effect of PI3K inhibitors on LCMV propagation.
We first tested the effect of several commercially available PI3K and Akt inhibitors on LCMV multiplication in cultured cells following infection at both low (0.001) and high (1.0) MOI. For this, we infected 293T cells with the Armstrong strain of LCMV in the absence of inhibitors during the adsorption time (90 min), followed by the addition of each inhibitor tested at the indicated concentrations (Fig. 1A). At the indicated h p.i., we determined titers of infectious virus in tissue culture supernatants (TCS). The PI3K inhibitor LY inhibited LCMV propagation following infection at both low and high MOI, but the drug's inhibitory effect was significantly stronger in cells infected at low MOI (Fig. 1A).
Fig 1.

Effect of PI3K/Akt inhibitors on multiplication of LCMV and Akt phosphorylation. (A) LY inhibits multiplication of LCMV in cultured cells. 293T cells were infected with LCMV (MOI, 0.001 or 1.0). After 90 min of adsorption time, the inoculum was removed, cell monolayers were washed, and fresh medium containing Akt-IV (0.5 μM or 2 μM), Akt-VIII (2 μM), or LY (20 or 50 μM) was added. At the indicated times p.i., virus titers were determined in TCS using an immune focus-forming (IFF) assay (see Materials and Methods). (B) Effect of LY on Akt phosphorylation. (Bi) 293T cells were treated with Akt-IV (0.5 or 2 μM), Akt-VIII (2 μM), LY (20 or 50 μM), or DMSO as a control. At 4 or 24 h posttreatment, cell lysates were prepared and total Akt and phosphorylated Akt (S473) were detected by Western blotting (WB). (Bii) Cell toxicity associated with the indicated drug treatments was assessed by determining cell viability after 24 h of treatment using the CellTiter-Glo luminescent cell viability assay (Promega). DMSO treatment was adjusted to 1.0. Data are averages and standard deviations from three independent experiments normalized with respect to DMSO treatment.
In contrast to the results observed with LY, the Akt inhibitors Akt-IV and Akt-VIII did not affect significantly LCMV propagation following infection at either low or high MOI. This finding led us to examine whether under our experimental conditions there was a correlation between the antiviral activity of the compound tested and its ability to inhibit Akt phosphorylation. For this, we treated 293T cells with the different inhibitors at the indicated concentrations and 4 or 24 h later prepared cell lysates to determine levels of Akt phosphorylation by Western blotting (Fig. 1Bi). Contrary to the commonly assumed properties of Akt-IV (16, 38), but consistent with a recent report (11), we observed that in 293T cells Akt-IV did not inhibit Akt phosphorylation at residue S473. Unexpectedly, under our experimental conditions Akt-VIII treatment also failed to inhibit Akt phosphorylation. In contrast, LY treatment inhibited efficiently the phosphorylation of Akt. At the concentrations tested, LY and Akt-VIII did not cause significant cell toxicity, whereas treatment with Akt-IV even at the lowest (0.5 μM) concentration tested reduced cell survival by about 60% as determined by the CellTiter-Glo assay (8) (Fig. 1Bii).
Our finding that LY inhibited very efficiently LCMV multiplication led us to explore the possible repurposing of PI3K inhibitors as antiarenaviral drugs. For this we used BEZ-235, which is currently in clinical trial for treatment of advanced solid tumors (21). BEZ-235 is a synthetic low-molecular-mass compound belonging to the class of imidazoquinolines that potently and reversibly inhibits class 1 PI3K catalytic activity by competing at its ATP-binding site. BEZ-235 also inhibits mTOR catalytic activity but does not target other protein kinases (22, 36). Under our experimental conditions BEZ-235 inhibited Akt phosphorylation in 293T cells in a dose- and time-dependent manner (Fig. 2A). Treatment with 1 nM BEZ-235 for 24 h resulted in significantly reduced levels of phosphorylated Akt at S473, whereas at 50 nM the inhibitory effect was already observed after 4 h of treatment. Within the dose range tested, BEZ-235 exhibited minimal cell toxicity (Fig. 2B). To examine the potential antiviral activity of BEZ-235 against LCMV, we infected 293T cells (MOI, 0.01) and treated them with BEZ-235 at the indicated concentrations, and at the indicated h p.i. we determined titers of infectious virus in TCS (Fig. 3). Within the 0.5 to 5 μM range, BEZ-235 caused about a 2- to 2.5-log reduction in the production of infectious progeny in the absence of noticeable cell toxicity.
Fig 2.
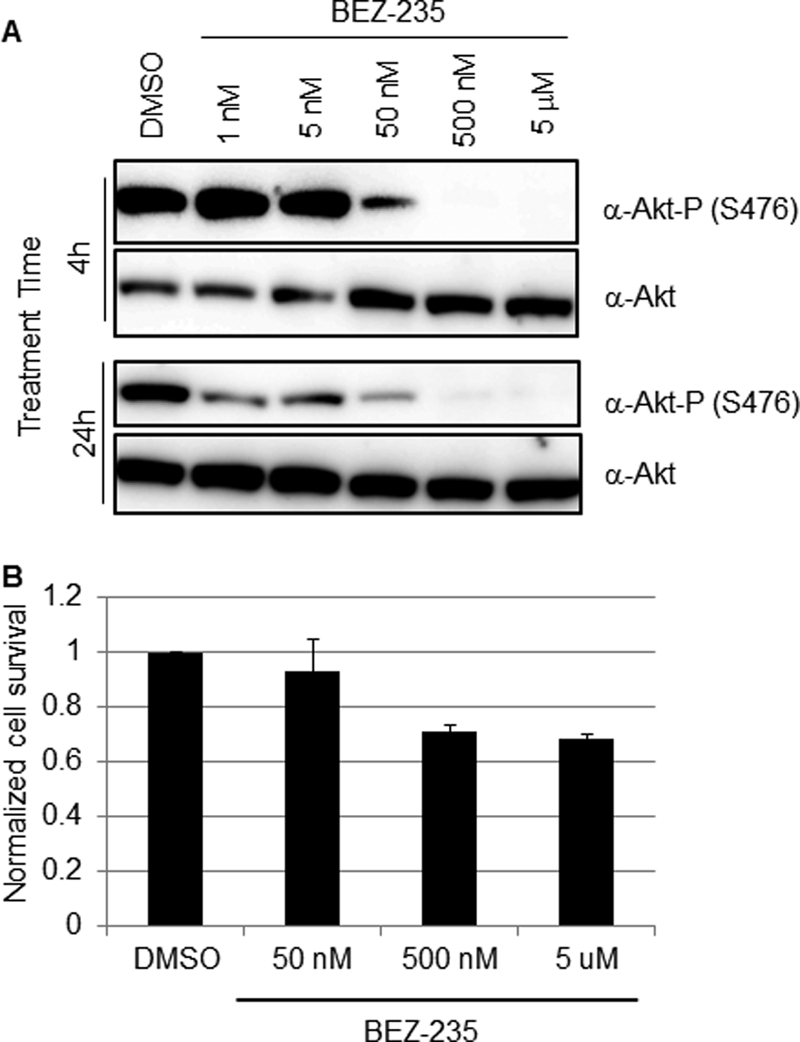
Effect of BEZ-235 on Akt phosphorylation in 293T cells. (A) 293T cells were treated with BEZ-235 (1, 5, 50, or 500 nM or 5 μM) or DMSO as a control. At 4 or 24 h posttreatment, cell lysates were prepared and total Akt and phosphorylated Akt (S473) were detected by Western blotting (WB). (B) Cell toxicity associated with BEZ-235 treatment. 293T cells were treated with the indicated BEZ-235 concentrations or with DMSO for 24 h, and then cell viability was determined using the CellTiter-Glo luminescent cell viability assay (Promega). Survival in DMSO-treated cells was adjusted to 1.0. Data are averages and standard deviations from three independent experiments.
Fig 3.

BEZ-235 inhibits LCMV multiplication in cultured cells. 293T cells were infected with LCMV (MOI, 0.01). After 90 min of adsorption time, the inoculum was removed, cell monolayers were washed, and fresh medium containing the indicated BEZ-235 concentration was added. At the indicated times p.i., virus titers were determined in TCS using an IFF assay (Materials and Methods).
Effect of PI3K inhibitors on LCMV entry.
To examine the effect of LY and BEZ-235 on LCMV GP-mediated cell entry we used a recombinant VSV (rVSVΔG-GFP/LCMV-GP) whose cell entry is mediated by the GP of the Armstrong strain of LCMV (41). The rationale for using this rVSV was that multiplication of VSV was shown not to be affected by LY (10). Therefore, an effect of LY or BEZ-235 treatment on rVSVΔG-GFP/LCMV-GP multiplication would reflect the drug's effect on LCMV GP-mediated cell entry. We treated 293T cells with LY (50 μM) or BEZ-235 (5 μM) for 1 h prior to infection (MOI, 1) with rVSVΔG-GFP/LCMV-GP or with the control rVSVΔG-GFP/VSV-G, an rVSV expressing its own glycoprotein (G). At 12 h p.i., cells were fixed and numbers of infected cells determined based on the presence of GFP-positive cells. Neither LY (Fig. 4A) nor BEZ-235 (Fig. 4B) treatment affected significantly the numbers of GFP+ cells following infection with either rVSVΔG-GFP/VSV-G or rVSVΔG-GFP/LCMV-GP. These results indicated that the PI3K/Akt pathway does not play a significant role in cell entry mediated by the GP of the Armstrong strain of LCMV. We obtained similar results when compounds (LY or BEZ-325) were added 4 h prior to starting virus adsorption or when adsorption was done at 4°C in the absence of drug, followed by changing to prewarmed (37°C) medium containing LY or BEZ-235 and transfer of cells to 37°C. Together these findings would suggest that an early step of cell entry was unlikely to be affected by LY or BEZ-235.
Fig 4.
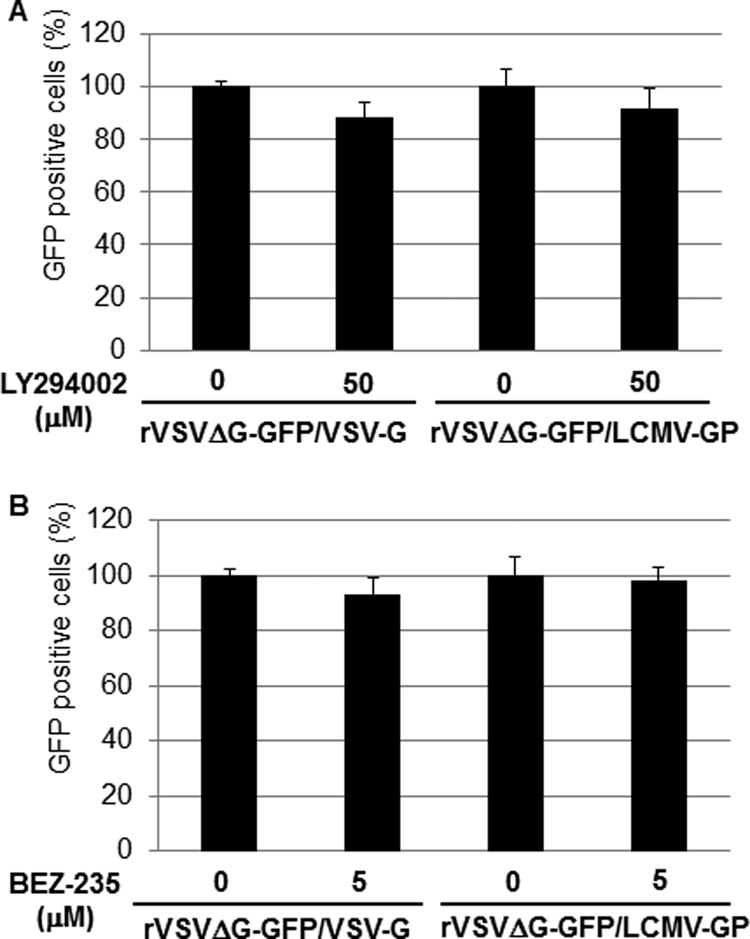
LY and BEZ-235 do not inhibit LCMV GP-mediated cell entry. 293T cells were pretreated with LY (50 μM) (A) or BEZ-235 (5 μM) (B) for 1 h prior to infection with either VSVΔG-GFP/VSV-G or rVSVΔG-GFP/LCMV-GP (MOI, 1.0). Infections were done in the presence of drugs. At 12 h p.i., cells were fixed and for each sample numbers of GFP-positive cells in four different fields determined by epifluorescence. Averages and standard deviations were obtained. Numbers of GFP-positive cells were normalized with respect to values obtained in nontreated cells, which were adjusted to 100%.
Effect of PI3K and Akt inhibitors on LCMV RNA replication and gene transcription.
To examine whether the PI3K/Akt pathway played a role in LCMV RNA replication and gene transcription we used an LCMV minigenome (MG) assay (18, 19, 29, 31, 32) that allowed us to separate the steps involved in virus RNA replication and transcription from those involved in cell entry, as well as virus particle assembly and cell egress. In this MG-based assay, expression of the CAT reporter gene was used as a surrogate to measure levels of RNA synthesis by the intracellularly reconstituted LCMV polymerase complex. LY exhibited a dose-dependent inhibitory effect on LCMV MG-derived reporter gene expression (30% and 70% reduction in CAT expression at 20 μM and 50 μM, respectively), whereas treatment with Akt-VIII had a very modest effect on MG-derived CAT expression (Fig. 5A). Intriguingly, although Akt-IV did not exhibit an inhibitory effect on LCMV propagation, treatment with Akt-IV resulted in a significant (40%) reduction of CAT expression (Fig. 5A). A plausible explanation for these apparently contradictory findings would be that Akt-IV treatment could have negatively impacted Pol-II-based transcription and thereby affected levels of plasmid-supplied LCMV NP and L proteins in the MG-based assay. To evaluate this possibility, we determined the effect of LY, Akt-VIII, and Akt-IV on firefly luciferase (Fluc) expression mediated by a Pol-II-based expression plasmid (Fig. 5B). LY (20 and 50 μM) and Akt-VIII (2.0 μM) did not affect significantly levels of Fluc expression, whereas treatment with Akt-IV caused a very significant reduction in levels of Fluc expression (50% at 0.5 μM). As with LY, we observed a dose-dependent inhibitory effect of BEZ-235 on MG-derived CAT expression (30%, 50%, and 65% reduction, at 50 nM, 500 nM, and 5 μM, respectively) (Fig. 5A). BEZ-235 had a minimal effect on Pol-II-mediated expression of Fluc (Fig. 5B), suggesting that the effect of BEZ-235 on LCMV MG expression was not the result of an overall drug's effect on Pol-II-mediated transcription.
Fig 5.

Effects of commercially available PI3K/Akt inhibitors and BEZ-235 on LCMV MG-derived reporter gene expression. (A) Drug effects on MG-derived reporter gene expression. 293T cells were transfected with pC-T7, pMG-CAT, pC-NP, and pC-L as described previously (18, 19, 29, 31, 32). After 5 h of transfection, medium was replaced with fresh medium containing Akt-IV (0.5 μM), Akt-VIII (2.0 μM), LY (20 or 50 μM), or BEZ-235 (0.05, 0.5, or 5 μM). At 24 h posttransfection, cell lysates were prepared for CAT ELISA. CAT expression levels from vehicle (DMSO)-treated cells were set to 1.0 to normalize CAT expression levels from the other samples. (B) Drug effects on Pol-II-based transcription. 293T cells (8 × 104/96-well plate) were transfected with pC-Fluc using Lipofectamine 2000 (Invitrogen), and 12 h later, medium was replaced with fresh medium containing the indicated compounds at the indicated concentrations. At 24 h post-compound treatment, levels of Fluc were determined using the Steady Glo assay (Promega) and a luminometer (Centro LB 960; Berthold Technologies). The viability of DMSO-treated control cells was set at 1.0. Data are averages and standard deviations from three independent experiments.
To further examine the effects of LY and BEZ-235 in RNA synthesis mediated by the LCMV polymerase, we used Northern blotting to determine levels of both RNA replication and transcription in the LCMV MG assay (Fig. 6). We observed a good correlation between the effect on CAT protein expression caused by either LY or BEZ-235 and the corresponding drug effect on levels of both MG replication and MG-derived CAT mRNA.
Fig 6.
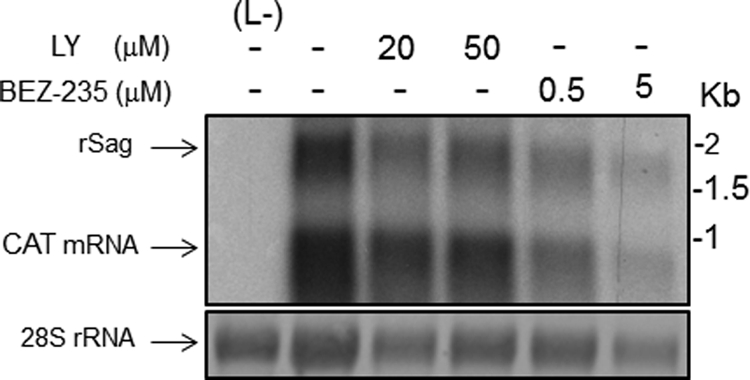
Effects of LY and BEZ-235 on LCMV-MG-derived RNA synthesis. 293T cells were transfected with pC-T7, pMG-CAT, pC-NP, and pC-L as described for Fig. 5. After 5 h, the transfection medium was replaced with fresh medium containing the indicated drugs and concentrations, and 24 h later, total cell RNA was isolated and analyzed by Northern blotting using a 32P-RNA probe that specifically hybridized to the CAT mRNA and the recombinant S antigenome (rSag) RNA species.
Effect of LY and BEZ-235 on LCMV and LASV Z-mediated budding.
Arenavirus Z protein has been shown to be the main driving force of virus budding (28, 37, 39). Since the magnitude of the inhibitory effect exerted by LY or BEZ-235 on LCMV multiplication was much greater than the corresponding effects of LY or BEZ-235 on viral RNA synthesis, we examined whether LY could inhibit Z-mediated budding, a key step of the virus life cycle required for production and propagation of infectious progeny. For this, we transfected 293T cells with either pC-LCMV-Z-FLAG or pC-LASV-Z-FLAG and, 12 h later, cells were washed and fresh medium containing the indicated concentration of LY or BEZ-235 was added. At 24 h posttreatment, we collected TCS and prepared cell lysates to detect levels of FLAG-tagged Z protein in both cell lysates and VLP recovered from TCS of transfected cells by WB using an anti-FLAG antibody. Both LY (Fig. 7A) and BEZ-235 (Fig. 7B) exerted a robust dose-dependent inhibitory effect on Z-mediated budding without significantly affecting levels of Z expression in transfected cells. The reasons for the apparent higher inhibitory effect of LY on budding mediated by LASV-Z, compared to that on LCMV-Z, remain to be determined.
Fig 7.

Effect of LY and BEZ-235 on Z-mediated budding. 293T cells were transfected with 0.25 μg of either pC-LCMV-Z-FLAG or pC-LASV-Z-FLAG, and 12 h posttransfection, the medium was replaced with fresh medium containing the indicated concentration of LY (A) or BEZ-235 (B); 24 h later, TCS were collected and total cell lysates prepared. VLPs were isolated from TCS as described previously (40). Levels of Z protein in total cell lysates and VLPs were detected by WB using an antibody to FLAG (product number 162150; Cayman).
Effect of rapamycin on LCMV multiplication.
BEZ-235 was shown to inhibit class 1 PI3K catalytic activity of both class 1 PI3K and mTOR (22, 36). To assess the possible contribution of mTOR inhibition to the antiviral activity associated with BEZ-235, we examined the effect of rapamycin (Rpm), a well-characterized mTOR inhibitor, on LCMV multiplication. Production of infectious virus in Rpm-treated cells was not significantly affected (Fig. 8A). Consistent with this observation, virus cell propagation (Fig. 8B) and virus RNA replication and gene transcription (Fig. 8C) were not significantly affected by Rpm treatment.
Fig 8.

Effect of Rpm on LCMV multiplication. BHK-21 cells were infected (MOI, 0.1) with r3LCMV/GFP (12) and treated with Rpm at the indicated concentrations. At the indicated h p.i., TCS were collected and cell monolayers fixed in 4% PFA/PBS. In addition, total cellular RNA was isolated from duplicate infections treated with the indicated concentration of Rpm. (A) Infectious progeny in TCS was determined using an IFF assay. (B) Numbers of virus-infected cells in each case were determined based on GFP expression. (C) Levels of viral RNA synthesis, both replication and transcription, were assessed by Northern blot hybridization using an NP-specific double-strand DNA probe that hybridized to the rS (replication) and NP mRNA (transcription) RNA species.
DISCUSSION
PI3K has been shown to have key regulatory roles in many cellular processes, including cell survival, proliferation, and differentiation. PI3K transduces signals from various growth factors and cytokines into intracellular messages by generating phospholipids, which activate the serine-threonine protein kinase Akt (also known as protein kinase B [PKB]) and other downstream effector pathways (14). Since many components of the PI3K and the Akt (PI3K/Akt) pathway are frequently targeted by germ line mutations or somatic mutations in a broad range of human cancers, PI3K has become an attractive target for therapeutic intervention in cancer (21). On the other hand, more recent findings have uncovered a relationship between the PI3K/Akt pathway and different steps of the life cycle for a variety of RNA viruses. Thus, recognition of cell surface receptors by a variety of virus surface glycoproteins can result in activation of the PI3K/Akt pathway to promote virus cell entry (34). In addition, some viral proteins like VSV M have been shown to downregulate PI3K/Akt signaling at an early stage of virus replication (10). Likewise, there is evidence that the PI3K/Akt pathway plays an important role in RNA replication of a variety of negative-stranded (NS) RNA viruses (38).
Infection with JUNV has been shown to activate Akt phosphorylation via a mechanism that did not require active virus replication or gene expression (20). Moreover, LY-mediated inhibition of the PI3K/Akt pathway resulted in decreased production of JUNV infectious progeny, which was proposed to be caused by an LY-induced blockage of the recycling of the transferring receptor proposed to mediate JUNV cell entry (20). Because significant differences have been reported regarding the biological features displayed by NW and OW arenaviruses, including the use of different receptors for cell entry (7, 17, 33), we investigated whether the PI3K/Akt pathway had also an effect on multiplication of the prototypic OW arenavirus LCMV.
The PI3K inhibitor LY, but not the Akt inhibitors Akt-IV and Akt-VIII, inhibited multiplication of LCMV following infection at low MOI (Fig. 1A). Akt-VIII has 50% inhibitory concentrations (IC50s) of 58 nM, 210 nM, and 2.12 μM for the Akt isoforms 1, 2, and 3, respectively. Therefore, in cells treated with 2 μM Akt-VIII, Akt3 would be predicted to retain significant activity that could have accounted for the lack of inhibition of LCMV multiplication in cells treated with 2 μM Akt-VIII. Nevertheless, we observed that levels of p27 phosphorylation, known to be mainly an Akt3 target (5), were not affected in 293T cells treated with 2 μM Akt-VIII as determined by Western blotting using a phospho-p27 (T157)-specific antibody (not shown). In addition, whereas LY treatment inhibited phosphorylation of Akt at S473 (Fig. 1B), under our experimental conditions Akt phosphorylation at S473 was not significantly affected by treatment with Akt-IV or Akt-VIII (Fig. 1B). The inability of Akt-IV to inhibit Akt phosphorylation was unexpected based on its published properties (38) but was consistent with a recent report showing that Akt-IV blocked VSV multiplication without affecting Akt phosphorylation (11). Akt-VIII has been shown to be a direct inhibitor of the kinase activity of Akt in several cell systems (2) and to have antiviral activity (1). The reason why under our experimental conditions we did not observe an Akt-VIII-mediated inhibition of Akt phosphorylation is presently unknown. One possibility is that the inhibitory activity of Akt-VIII may be cell dependent and at 2 μM it could robustly inhibit phosphorylation of Akt in BHK-21 (11) but not in the 293T cells used in our studies.
The lesser inhibitory effect of LY on LCMV multiplication following infection at high compared to low MOI suggested a more strict requirement of the integrity of the PI3K/Akt pathway in the late steps of the virus life cycle. Accordingly, LY and BEZ-235 treatment did not affect cell entry mediated by the GP of the Armstrong strain of LCMV (Fig. 4). However, a recent report by Pasqual and colleagues has documented that LASV and LCMV enter host cells via the multivesicular body (MVB) pathway (27), and biogenesis and functionality of the MVB require the lipid PI3P and hence also PI3K activity. Accordingly, treatment with the PI3K inhibitor wortmannin significantly affected cell entry by LASV or LCMV GP (27). A possible explanation for these apparent conflicting findings is that wortmannin is able to rapidly and specifically inhibit some of the nonclassical PI3K isoforms that might compensate for LY-mediated inhibition of major PI3K forms. Likewise, differences in cell entry mediated by GPs of the Armstrong (our study) and Cl-13 (study by Pasqual and colleagues) strains of LCMV may have also contributed to the differences between our results and those reported by Pasqual and colleagues (27). In this regard, it should be noted that VSV cell entry was shown to be dependent also on the MVB and endosomal sorting complex required for transport (ESCRT) pathways but was not affected by wortmannin treatment, suggesting that VSV-G-mediated fusion with membranes of the intraluminal vesicles (ILV) within the MVB takes place under mildly acidic pH and the virus ribonucleoprotein core is delivered to late endosomes in the lumen of the ILVs (23). Similarly, it is plausible that Armstrong GP2 may promote fusion between viral and cellular membranes at higher pH than GP2 from Cl-13 or LASV.
Both LY and BEZ-235 had a significant and specific inhibitory effect on MG-derived CAT expression levels (Fig. 5A). In contrast, the observed inhibitory effect of Akt-IV on MG-derived reporter gene activity was likely a reflection of a general effect of Akt-IV on cell viability and Pol-II-mediated transcription (Fig. 1Bii and 5B). These results suggested a role of the PI3K/Akt pathway in LCMV RNA replication and gene transcription, which was experimentally supported by Northern blot analysis of MG-derived RNA species in the absence or presence of PI3K inhibitors (Fig. 6). The mechanisms by which either LY or BEZ-235 affect the activity of the arenavirus polymerase remain to be elucidated. Previous studies have shown that Akt directly phosphorylates parainfluenza virus 5 (PIV5) and respiratory syncytial virus (RSV) polymerase cofactor P protein, and this phosphorylation of P has a critical role for virus polymerase activity (38). However, arenaviruses do not have a counterpart of the P protein found in many other NS RNA viruses.
Consistent with a more pronounced involvement of the PI3K/Akt pathway in late steps of the LCMV life cycle, both LY (Fig. 7A) and BEZ-235 (Fig. 7B) exhibited a strong dose-dependent inhibitory effect on Z-mediated budding, which documents for the first time a role of the PI3K/Akt pathway in virus budding. The degree of inhibition of Z-mediated budding appeared to be higher in BEZ-235-treated than in LY-treated cells. However, it seems unlikely that a PI3K-independent mTOR activation may have also contributed to Z-mediated budding, as the mTOR inhibitor Rpm did not affect significantly LCMV multiplication in cultured cells (Fig. 8). The PI3K/Akt pathway participates in the regulation of many cellular processes, including vesicular trafficking (35). PI3P, the product of the PI3K activity, is needed for efficient assembly of the ESCRT complex at the limiting membrane of the early endosome, and some ESCRT proteins contain modules that recognize PI3P. Budding of a variety of enveloped viruses, including arenaviruses, involves interactions between viral budding proteins and ESCRT. It is plausible that these interactions may be facilitated by virus-induced PI3P-rich “microdomains” at the site of budding and thereby inhibition of PI3K activity will disrupt normal budding. The PI3K/Akt pathway might be directly involved in phosphorylation of LCMV and LASV Z protein, which may be necessary for efficient Z-mediated budding. The use of NetPhos 2.0 software (http://www.cbs.dtu.dk/services/NetPhos/) identified six S residues in both LCMV and LASV-Z, as well as three (LCMV-Z) and one (LASV-Z) Y residues as highly likely substrates for phosphorylation (not shown). Whether Z is a target of PI3K/Akt-mediated phosphorylation and the possible role of phosphorylation in the regulation of the budding activity of Z are issues that remain to be resolved. Likewise, a similar analysis identified 21 S, four T, and four Y residues within NP as highly likely substrates for phosphorylation. Whether PI3K/Akt-mediated phosphorylation of NP could contribute to regulation of RNA synthesis by the arenavirus polymerase remains to be determined.
Our findings, together with those previously documented for JUNV, suggest that targeting the PI3K/Akt pathway may offer the possibility of inhibiting multiplication of both OW and NW HF arenaviruses. It should be also noted that strong evidence indicates that morbidity and mortality of HF arenaviruses correlate with high viral load due to the failure of both the host's innate and its adaptive immune responses to restrict virus multiplication. Targeting the PI3K/Akt pathway could reduce the virus load and rate of propagation and thereby provide the host with a window of opportunity to mount an efficient antiviral immune response. Targeting a cellular factor or pathway required for optimal viral growth would offer the advantage of overcoming the problem related to the emergence of drug-resistant viral variants commonly observed with antiviral drugs against RNA viruses characterized by their highly error-prone replication machineries. The detailed mechanisms by which inhibition of the PI3K/Akt pathway affects LCMV RNA synthesis and budding remain to be determined. The identification of specific cellular effectors contributing to impaired LCMV RNA synthesis and budding upon inhibition of the PI3K/Akt pathway, together with current efforts to develop cancer therapies based on targeting of the PI3K/Akt pathway, should facilitate the identification of anticancer drugs with potential repurposing value as antiviral drugs to combat human-pathogenic arenaviruses. This possibility is illustrated by BEZ-235, a synthetic small molecule that is a dual PI3K and mTOR inhibitor and is currently being tested in clinical trials for solid tumors (21, 22, 36), which also exhibited a potent dose-dependent antiviral activity against LCMV within a concentration range (0.5 to 5 μM) that had only very modest effects on cell viability.
ACKNOWLEDGMENTS
This work was supported by NIH grants R01 AI047140, R01 AI077719, and R01 AI079665 to J.C.T. S.U. was supported by NIH grant T32-AI0735419.
Footnotes
Published ahead of print 15 February 2012
This is The Scripps Research Institute publication 21443 from the Department of Immunology and Microbial Science.
REFERENCES
- 1. Arita M, Wakita T, Shimizu H. 2009. Cellular kinase inhibitors that suppress enterovirus replication have a conserved target in viral protein 3A similar to that of enviroxime. J. Gen. Virol. 90:1869–1879 [DOI] [PubMed] [Google Scholar]
- 2. Barnett SF, et al. 2005. Identification and characterization of pleckstrin-homology-domain-dependent and isoenzyme-specific Akt inhibitors. Biochem. J. 385:399–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Barton LL, Mets MB, Beauchamp CL. 2002. Lymphocytic choriomeningitis virus: emerging fetal teratogen. Am. J. Obstet. Gynecol. 187:1715–1716 [DOI] [PubMed] [Google Scholar]
- 4. Battegay M. 1993. Quantification of lymphocytic choriomeningitis virus with an immunological focus assay in 24 well plates. ALTEX 10:6–14 [PubMed] [Google Scholar]
- 5. Brognard J, Sierecki E, Gao T, Newton AC. 2007. PHLPP and a second isoform, PHLPP2, differentially attenuate the amplitude of Akt signaling by regulating distinct Akt isoforms. Mol. Cell 25:917–931 [DOI] [PubMed] [Google Scholar]
- 6. Buchmeier MJ, Peters CJ, de la Torre JC. 2007. Arenaviridae: the viruses and their replication, p 1791–1851 In Knipe DM, et al. (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 7. Cao W, et al. 1998. Identification of alpha-dystroglycan as a receptor for lymphocytic choriomeningitis virus and Lassa fever virus. Science 282:2079–2081 [DOI] [PubMed] [Google Scholar]
- 8. Crouch SP, Kozlowski R, Slater KJ, Fletcher J. 1993. The use of ATP bioluminescence as a measure of cell proliferation and cytotoxicity. J. Immunol. Methods 160:81–88 [DOI] [PubMed] [Google Scholar]
- 9. Damonte EB, Coto CE. 2002. Treatment of arenavirus infections: from basic studies to the challenge of antiviral therapy. Adv. Virus Res. 58:125–155 [DOI] [PubMed] [Google Scholar]
- 10. Dunn EF, Connor JH. 2011. Dominant inhibition of Akt/protein kinase B signaling by the matrix protein of a negative-strand RNA virus. J. Virol. 85:422–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dunn EF, Fearns R, Connor JH. 2009. Akt inhibitor Akt-IV blocks virus replication through an Akt-independent mechanism. J. Virol. 83:11665–11672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Emonet SF, Garidou L, McGavern DB, de la Torre JC. 2009. Generation of recombinant lymphocytic choriomeningitis viruses with trisegmented genomes stably expressing two additional genes of interest. Proc. Natl. Acad. Sci. U. S. A. 106:3473–3478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fischer SA, et al. 2006. Transmission of lymphocytic choriomeningitis virus by organ transplantation. N. Engl. J. Med. 354:2235–2249 [DOI] [PubMed] [Google Scholar]
- 14. Franke TF. 2008. Intracellular signaling by Akt: bound to be specific. Sci. Signal. 1:pe29. [DOI] [PubMed] [Google Scholar]
- 15. Jahrling PB, Peters CJ. 1992. Lymphocytic choriomeningitis virus. A neglected pathogen of man. Arch. Pathol. Lab. Med. 116:486–488 [PubMed] [Google Scholar]
- 16. Kau TR, et al. 2003. A chemical genetic screen identifies inhibitors of regulated nuclear export of a Forkhead transcription factor in PTEN-deficient tumor cells. Cancer Cell 4:463–476 [DOI] [PubMed] [Google Scholar]
- 17. Kunz S. 2009. Receptor binding and cell entry of Old World arenaviruses reveal novel aspects of virus-host interaction. Virology 387:245–249 [DOI] [PubMed] [Google Scholar]
- 18. Lee KJ, Novella IS, Teng MN, Oldstone MB, de La Torre JC. 2000. NP and L proteins of lymphocytic choriomeningitis virus (LCMV) are sufficient for efficient transcription and replication of LCMV genomic RNA analogs. J. Virol. 74:3470–3477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee KJ, Perez M, Pinschewer DD, de la Torre JC. 2002. Identification of the lymphocytic choriomeningitis virus (LCMV) proteins required to rescue LCMV RNA analogs into LCMV-like particles. J. Virol. 76:6393–6397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Linero FN, Scolaro LA. 2009. Participation of the phosphatidylinositol 3-kinase/Akt pathway in Junin virus replication in vitro. Virus Res. 145:166–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu P, Cheng H, Roberts TM, Zhao JJ. 2009. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov. 8:627–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu TJ, et al. 2009. NVP-BEZ235, a novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor, elicits multifaceted antitumor activities in human gliomas. Mol. Cancer Ther. 8:2204–2210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Luyet PP, Falguieres T, Pons V, Pattnaik AK, Gruenberg J. 2008. The ESCRT-I subunit TSG101 controls endosome-to-cytosol release of viral RNA. Traffic 9:2279–2290 [DOI] [PubMed] [Google Scholar]
- 24. Mets MB, Barton LL, Khan AS, Ksiazek TG. 2000. Lymphocytic choriomeningitis virus: an underdiagnosed cause of congenital chorioretinitis. Am. J. Ophthalmol. 130:209–215 [DOI] [PubMed] [Google Scholar]
- 25. Moreno H, et al. 2011. Ribavirin can be mutagenic for arenaviruses. J. Virol. 85:7246–7255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Parker WB. 2005. Metabolism and antiviral activity of ribavirin. Virus Res. 107:165–171 [DOI] [PubMed] [Google Scholar]
- 27. Pasqual G, Rojek JM, Masin M, Chatton JY, Kunz S. 2011. Old world arenaviruses enter the host cell via the multivesicular body and depend on the endosomal sorting complex required for transport. PLoS Pathog. 7:e1002232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Perez M, Craven RC, de la Torre JC. 2003. The small RING finger protein Z drives arenavirus budding: implications for antiviral strategies. Proc. Natl. Acad. Sci. U. S. A. 100:12978–12983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Perez M, de la Torre JC. 2003. Characterization of the genomic promoter of the prototypic arenavirus lymphocytic choriomeningitis virus. J. Virol. 77:1184–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Peters CJ. 2006. Lymphocytic choriomeningitis virus—an old enemy up to new tricks. N. Engl. J. Med. 354:2208–2211 [DOI] [PubMed] [Google Scholar]
- 31. Pinschewer DD, Perez M, de la Torre JC. 2005. Dual role of the lymphocytic choriomeningitis virus intergenic region in transcription termination and virus propagation. J. Virol. 79:4519–4526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pinschewer DD, Perez M, de la Torre JC. 2003. Role of the virus nucleoprotein in the regulation of lymphocytic choriomeningitis virus transcription and RNA replication. J. Virol. 77:3882–3887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Radoshitzky SR, et al. 2007. Transferrin receptor 1 is a cellular receptor for New World haemorrhagic fever arenaviruses. Nature 446:92–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Saeed MF, Kolokoltsov AA, Freiberg AN, Holbrook MR, Davey RA. 2008. Phosphoinositide-3 kinase-Akt pathway controls cellular entry of Ebola virus. PLoS Pathog. 4:e1000141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schu PV, et al. 1993. Phosphatidylinositol 3-kinase encoded by yeast VPS34 gene essential for protein sorting. Science 260:88–91 [DOI] [PubMed] [Google Scholar]
- 36. Serra V, et al. 2008. NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res. 68:8022–8030 [DOI] [PubMed] [Google Scholar]
- 37. Strecker T, et al. 2003. Lassa virus Z protein is a matrix protein and sufficient for the release of virus-like particles. J. Virol. 77:10700–10705 (Erratum, 77:12927.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sun M, et al. 2008. Akt plays a critical role in replication of nonsegmented negative-stranded RNA viruses. J. Virol. 82:105–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Urata S, Noda T, Kawaoka Y, Yokosawa H, Yasuda J. 2006. Cellular factors required for Lassa virus budding. J. Virol. 80:4191–4195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Urata S, Yasuda J, de la Torre JC. 2009. The Z protein of the New World arenavirus Tacaribe virus has bona fide budding activity that does not depend on known late domain motifs. J. Virol. 83:12651–12655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Urata S, et al. 2011. Antiviral activity of a small-molecule inhibitor of arenavirus glycoprotein processing by the cellular site 1 protease. J. Virol. 85:795–803 [DOI] [PMC free article] [PubMed] [Google Scholar]