

Abstract
Our knowledge regarding immune-protective and immunopathogenic events in severe acute respiratory syndrome coronavirus (SARS-CoV) infection is limited, and little is known about the dynamics of the immune response at the primary site of disease. Here, an African green monkey (AGM) model was used to elucidate immune mechanisms that facilitate viral clearance but may also contribute to persistent lung inflammation following SARS-CoV infection. During primary infection, SARS-CoV replicated in the AGM lung for up to 10 days. Interestingly, lung inflammation was more prevalent following viral clearance, as leukocyte numbers peaked at 14 days postinfection (dpi) and remained elevated at 28 dpi compared to those of mock-infected controls. Lung macrophages but not dendritic cells were rapidly activated, and both cell types had high activation marker expression at late infection time points. Lung proinflammatory cytokines were induced at 1 to 14 dpi, but most returned to baseline by 28 dpi except interleukin 12 (IL-12) and gamma interferon. In SARS-CoV homologous rechallenge studies, 11 of the 12 animals were free of replicating virus at day 5 after rechallenge. However, incidence and severity of lung inflammation was not reduced despite the limited viral replication upon rechallenge. Evaluating the role of antibodies in immune protection or potentiation revealed a progressive increase in anti-SARS-CoV antibodies in lung and serum that did not correlate temporally or spatially with enhanced viral replication. This study represents one of the first comprehensive analyses of lung immunity, including changes in leukocyte populations, lung-specific cytokines, and antibody responses following SARS-CoV rechallenge in AGMs.
INTRODUCTION
Anovel coronavirus (CoV) emerged in 2002 as the etiologic agent of severe acute respiratory syndrome (SARS) and spread to more than 30 countries in a 6-month period (51). This zoonotic virus is thought to have passed from the Chinese horseshoe bat (23, 26) and, in contrast to the limited host range of other CoVs, has been shown to replicate in many different species, including humans, palm civets, raccoon dogs, monkeys, ferrets, and hamsters (10, 22, 27, 29, 40, 41, 47). Another unique feature of SARS-CoV is its high pathogenicity and ability to induce acute respiratory distress syndrome, which is in contrast to other identified human CoVs that are generally associated with only mild illness (35). Although the first SARS-CoV epidemic was successfully controlled largely through quarantine and sanitation measures, SARS-CoV remains a potential public health threat. There are currently no approved antiviral drugs that effectively target SARS-CoV, and no vaccines have been licensed for any of the human CoVs.
Damage to the lung in SARS-CoV infection is thought to occur via direct viral destruction of respiratory epithelium and by aberrant immune responses (4, 38). However, the relative contribution of these mechanisms to the disease remains controversial. Several immune-mediated mechanisms of SARS-CoV pathogenesis have been proposed, including antibody-dependent enhancement of infection, immune subversion (13, 15, 21, 30), immune evasion, as well as viral disruption of immune cell function (2, 38, 61). Still, our knowledge regarding the immune-protective versus immunopathogenic responses to SARS-CoV remains limited and warrants further study in established animal models.
Neutralizing antibodies to SARS-CoV spike (S) protein are thought to play a major role in host protection. Higher levels correlated with shorter disease duration in SARS-CoV-infected patients (46), and suboptimal neutralizing antibodies were detected in patients with more severe disease (32, 33, 52). Homologous rechallenge with SARS-CoV in ferrets reduced viral load and fever upon secondary infection, suggesting a protective memory response that correlated with increased neutralizing antibody titers (10). Furthermore, prophylactic administration of monoclonal anti-SARS-CoV antibodies to rodents was shown to reduce viral burden and associated lung pathology (17, 47). However, humoral responses to viral infections are complex, as antibodies have also been shown to increase viral replication and severity of disease in several models, including dengue virus, flavivirus, and feline infectious peritonitis virus (34, 45). Although similar mechanisms have not been observed in most SARS-CoV immunization studies (38, 40), severe hepatitis was reported in immunized ferrets and was thought to be mediated by antibody enhancement of SARS-CoV infection in the liver (50). In addition, recombinant viral vectors coated with SARS-CoV S protein showed antibody-dependent increased entry into 786-O cells, and therefore the possibility of immunopotentiation in SARS-CoV infection and vaccination must be fully investigated (57).
In addition to humoral immunity, the T lymphocyte-mediated response plays a key role in the defense against viral respiratory infections. However, the role of cell-mediated immunity in SARS-CoV infection is still not clear. The rapid development of lymphopenia during acute SARS-CoV infection in patients has been well documented and is associated with an adverse outcome of the disease (4). Despite the reduced numbers of total circulating T lymphocytes, effector and memory T cells specific for SARS-CoV structural proteins have been detected in convalescent SARS-CoV patients and have been shown to persist long after infection (3, 36, 37, 53, 54).
Monocytes/macrophages have also been implicated in SARS-CoV disease pathogenesis (8, 38). In SARS-CoV patients, infiltrating monocytes/macrophages have been shown to persist long after the virus has been eradicated from the lung, and the excessive accumulation of these cells is a prominent pathological feature in most SARS-CoV autopsy cases (9). Although SARS-CoV does not productively infect monocytes/macrophages (7), both in vitro and, more recently, in vivo studies have demonstrated that SARS-CoV can directly and indirectly impair several of their functions, including cytokine secretion, endocytic capacity, and ability to initiate adaptive immune responses (24, 48, 59, 60). Further demonstration of the central role for monocytes/macrophages in SARS-CoV immunopathogenesis is the prevention of lethal disease by depletion of lung macrophages prior to infecting mice with the SARS-CoV MA15 strain (61).
In the present study, several key questions regarding the host immune response to both primary and secondary homologous SARS-CoV infection are addressed. Herein, we show that although viral replication upon homologous rechallenge is severely limited compared to primary infection, the lung inflammation and histological changes are not reduced and may persist long after virus has been cleared. Lastly, the elucidation of the immune responses presented here will be crucial in the evaluation of future vaccines and immunomodulating therapies against SARS.
MATERIALS AND METHODS
Nonhuman primate (NHP) studies.
SARS-CoV human strain HKU-39849 derived from a clinical isolate was kindly provided by Leo Poon (Department of Microbiology, The University of Hong Kong, Hong Kong, China), and viral stocks for inoculating animals were generated in Vero E6 cells.
Animal studies were carried out in accordance with the “NIH Guide to Care and Use of Laboratory Animals.” All protocols were approved by the LRRI Animal Use Committee. Wild-caught, adult African green monkeys (Chlorocebus aethiops) (AGMs) were obtained from Barbados Primate Research Centre, Barbados, West Indies. Animals were quarantined at LRRI for 6 weeks. All were negative for tuberculosis and vaccinated against measles virus before exposure to SARS-CoV. AGMs were instilled with 1 ml of SARS-CoV at 107 PFU or virus-free cell culture medium via the intranasal route and sequentially sacrificed at 1, 3, 5, 10, 14, and 28 dpi (Fig. 1; 6 animals per time point except at 1 dpi [n = 5 animals]). In challenge-rechallenge studies, animals were inoculated with a second dose of SARS-CoV HKU-39849 via the intranasal route 28 days after primary infection, and animals were sacrificed at day 5 or 28 after primary or secondary infection (n = 6 animals per time point). All data for day 5 after SARS-CoV infection, except for histopathology scoring, were derived exclusively from the challenge-rechallenge studies.
Fig 1.

Experimental design for SARS-CoV challenge and rechallenge studies. African green monkeys were instilled with 1 × 107 PFU of SARS-CoV strain HKU-39849 intranasally (first inverted triangle) followed by sacrifice at 1, 3, 5, 10, and 14 days postinoculation. In rechallenge studies, animals were reinoculated intranasally with the same dose and strain of SARS-CoV 28 days after primary infection (second inverted triangle), and animals were sacrificed at days 5 or 28 after primary or secondary challenge. (For each study, n = 6 animals at the indicated time points, except at 1 dpi [n = 5 animals]).
Swabs of the nasal and pharyngeal areas were collected into fresh medium longitudinally and at the time of sacrifice. A complete necropsy was performed, and several tissue specimens were collected, including the nasal turbinates, lung, lymph nodes, and peripheral blood for virology, immunology, and pathology assessment.
Plaque and neutralization assays.
Virus titers were determined by applying serial dilutions of homogenized tissue suspensions onto Vero E6 cell monolayers in 24-well plates. Wells with dilutions that yielded 2 to 20 plaques were evaluated for number of plaques formed, and results were reported as PFU per gram of lung tissue. In neutralization assays, serum samples were serially diluted prior to incubating with 2,000 PFU/ml SARS-CoV overnight. The virus and serum mixture was inoculated onto 96-well plates of Vero E6 cells, and cultures were held at 37°C and 6% CO2 for 3 days. Cytopathic effect (CPE) was examined microscopically, and titers are expressed as the reciprocal of the highest dilution at which the CPE was completely inhibited. All in vitro SARS-CoV manipulations and instillation of the virus were performed inside a biosafety cabinet in biosafety containment level 3 facilities.
RNA isolation and quantitative RT-PCR analysis.
Swabs from mock- or SARS-CoV-infected NHPs were collected in buffer AVL (Qiagen, Valencia, CA) for viral nucleic acid purification, with GlycoBlue added as a coprecipitant (Applied Biosystems, Foster City, CA). RNAs were extracted with the phase separation reagent 1-bromo-3-chloropropane (Molecular Research Inc., Cincinnati, OH) according to the manufacturer's specifications. The full-length SARS-CoV N coding sequence was amplified through reverse transcription (RT)-PCR with primers NF (AGGATCCATGTCTGATAATGGACCCCAATCAAACC) and NR (AGAATTCTTATGCCTGAGTTGAATCAGCAGAAGCTCC), and the PCR products were cloned into pTarget (Promega, Madison, WI) for in vitro transcription with the MEGAshortscript kit (Applied Biosystems) to set up the standard curve. Primers and probes to detect SARS-CoV mRNA were specific for the nucleoprotein (IDT, Coralville, IA) (12), and reactions were carried out using the TaqMan 2× PCR universal Master Mix (Applied Biosystems). SARS-CoV N gene copies per milliliter were calculated based on inclusion of the SARS-CoV N gene standard curve.
Histopathology.
Sampling of tissues for histopathology studies was performed in a standardized manner such that a random assessment of pathology was enabled. Organs were fixed in 4% paraformaldehyde and embedded in paraffin for histological examination. Sections of the nose (n = 4), trachea (n = 2), left and right lung lobes (n = 6), tracheobronchial lymph node, spleen, mesentery lymph node, kidney, liver, colon, and rectum were obtained from each animal, paraffin embedded, and stained with hematoxylin and eosin (H&E). For comparison of lung injury, a grading was performed by a board-certified veterinary pathologist. Histopathology slides were read using a BX41 microscope with an attached digital DP20 camera or a BX51 microscope (Olympus America, PA). Histologic lesions were evaluated for severity and distribution using the following scale: 0, normal; 1, minimal; 2, mild; 3, moderate; 4, marked.
Flow cytometry.
Single-cell suspensions of the lung were prepared from standardized collected lung tissue from the proximal portion of the right caudal lobe in a manner similar to previously described methods (39). One to two grams of tissue was cut into small pieces followed by an enzymatic digestion with Liberase (Roche, Pleasanton, CA) and DNase solution (Sigma, St. Louis, MO) at 37°C for 90 min. Digested tissue was put through sterile wire mesh prior to lysing red blood cells and overlaying the remaining sample onto a 30% Percoll gradient (Sigma) followed by centrifugation for 20 min at 500 × g with no brake. Pelleted cells were washed, counted, and resuspended in 90% fetal bovine serum (FBS) and 10% dimethyl sulfoxide for cryopreservation. Tracheobronchial lymph nodes were collected and weighed, and single-cell suspensions were prepared at necropsy by mechanical disruption. Previously frozen lung leukocytes and lymph node cells were resuscitated and immunophenotyped by flow cytometry as previously described (11) in 4- or 6-color staining panels that included CD3 (clone SDP34-2), CD4 (clone MT477), CD8 (clone SK1), CD14 (clone M5E2), CD11c (clone SHCL-3), CD20 (clone L27), CD23 (clone ML233), CD86 (clone 2331Fun1), Granzyme B (clone GB11; Invitrogen, Carlsbad, CA), HLA-DR (clone L243), Ki67 (clone B56), and FcεR1alpha (clone AER-37; eBiosciences, San Diego, CA) antibodies, all conjugated to fluorochromes fluorescein isothiocyanate (FITC), phycoerythrin (PE), PerCP, allophycocyanin (APC), PECy7, or APCCy7 (BD Biosciences, San Jose, CA, unless specified). Following antibody staining, cells were fixed for 16 h in 1% paraformaldehyde and 3% FBS in phosphate-buffered saline (PBS). Sample data were acquired on a fluorescence-activated cell sorting (FACS)-Calibur or FACS-Canto flow cytometer instrument (BD Biosciences), and data files were analyzed utilizing FlowJo software (TreeStar, Medford, OR).
ELISAs and bead-based array assays.
Enzyme-linked immunosorbent assay (ELISA) plates were coated with purified recombinant SARS-CoV S protein in carbonate-coating buffer, 100 ng/well, overnight at 4°C (S protein NR-686 obtained through NIH Biodefense and Emerging Infections Research Resources Repository). Nonspecific binding was blocked with PowerBlock (Biogenex, San Ramon, CA). Serially diluted lung tissue homogenate suspensions, nasal swabs, or serum samples (clarified by centrifugation) were incubated on S-protein-coated plates overnight at 37°C. The starting dilution for lung homogenates was 1:25 with 3-fold serial dilutions, the starting dilution for nasal swabs was 1:10 with 2-fold serial dilutions, and the starting dilution for serum was 1:25 with 3-fold serial dilutions. Plates were washed with PBS-0.1% Tween 20 (Sigma) and incubated for 1 h with anti-monkey IgG or IgA horseradish peroxidase (HRP)-conjugated antibody (KPL Inc., Gaithersburg, MD) followed by substrate development with the ABTS Microwell peroxidase substrate system (KPL). Absorbance was read at 405 nm using a Thermo Electron Corporation plate reader (Thermo Electron Corporation, Houston, TX), and data were acquired with Ascent software (Ascent Software, London, United Kingdom). The ELISA antibody titer recorded for each sample was the reciprocal of the highest dilution in which the optical density (OD) reading for S protein-bound wells was at least 2-fold higher than that of the nonfat milk control. The OD of the highest titer chosen also had to fall within the linear range of the serial dilutions.
CXCL13, CCL3, CXCL11, interleukin 1β (IL-1β), gamma interferon (IFN-γ), IL-2, IL-8, CCL19, CXCL12, IL-18, IL-15, IL-6, CCL21, transforming growth factor β (TGF-β), IL-12, CCL2, CCL5, CXCL10, and tumor necrosis factor alpha (TNF-α) protein were measured in lung tissue homogenates using both human and NHP multiplex bead-based array kits (Millipore, Billerica, MA). The assays were performed according to the manufacturer's instructions with an overnight incubation of the samples in antibody-immobilized beads. The median fluorescent intensity was measured with the Bio-Plex system (Bio-Rad, Hercules, CA), and a weighted 5-parameter logistic curve-fitting method was used to calculate the concentration of individual analytes. All measurements were performed in duplicate. The average picogram/milliliter value for each chemokine was used to generate a heat map to show the fold change in protein levels compared to that of mock-infected controls using the gplots package in R/Bioconductor (49). Expression patterns were clustered using the hierarchical method, with a Euclidean distance metric and a complete linkage. The heat map color scheme was chosen, with red indicating 4-fold induction and green representing 4-fold reduction of protein levels compared to that of mock-infected controls. Data are expressed as the means ± standard errors in Table 5.
Table 5.
Cytokines and chemokines in the lungs of AGMs following SARS-CoV primary and secondary infection
| Cytokine or chemokine | Concn (pg/ml) cytokine ± SEM |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Mock infection | Primary infection |
Rechallenge |
|||||||
| Day 1 | Day 3 | Day 5 | Day 10 | Day 14 | Day 28 | Day 5 | Day 28 | ||
| CXCL13 | 460.7 ± 148.7 | 429.1 ± 132.8 | 897.6 ± 239.0 | 1,016 ± 555.2 | 2,057 ± 148.3* | 689.3 ± 148.3 | 79.3 ± 14.22 | 1,063 ± 377.2 | 829.5 ± 300.6 |
| CCL3 | 477.0 ± 62.92 | 856.7 ± 336.4 | 1,001 ± 338.1 | 442.9 ± 84.68 | 2,419 ± 1,793 | 430.8 ± 70.60 | 377.3 ± 43.74 | 101.6 ± 18.03 | 183.5 ± 62.98 |
| CXCL11 | 1,247 ± 225.8 | 3,035 ± 1,118 | 10,288 ± 3,943* | 4,422 ± 798.8* | 1,966 ± 567.3 | 1,140 ± 285.9 | 127.7 ± 43.99* | 1,203 ± 229.3 | 1,621 ± 567.2 |
| IL-1B | 0.9,550 ± 0.1,050 | 1.075 ± 0.9,250 | 8.353 ± 8.074 | 25.21 ± 10.63 | 4.216 ± 3.336 | 10.68 ± 1.183* | 0.2,800 ± 0.1,300 | 1.767 ± 0.7,219 | 1.050 ± 0.9,201 |
| IFN-γ | 31.20 ± 3.901 | 38.91 ± 14.74 | 57.00 ± 7.518* | 131.5 ± 56.85* | 94.52 ± 60.24 | 44.40 ± 4.596 | 56.60 ± 37.48 | 15.07 ± 7.533 | 62.95 ± 4.750 |
| IL-2 | 34.35 ± 12.23 | 82.72 ± 35.80 | 40.37 ± 15.07 | 78.71 ± 17.45 | 35.75 ± 7.572 | 64.35 ± 17.14 | ND | ND | 5.250 ± 4.922 |
| IL-8 | 7,908 ± 2,485 | 15,907 ± 8,439 | 4,214 ± 1,689 | 3,622 ± 1,063 | 19,994 ± 11,459 | 8,726 ± 3,588 | 3,837 ± 2,439 | 409.5 ± 151.1 | 303.4 ± 105.0 |
| CCL19 | 1,919 ± 575.7 | 2,015 ± 777.7 | 3,517 ± 1,678 | 3,478 ± 859.6 | 753.7 ± 116.0 | 2,331 ± 536.9 | 204.6 ± 54.96 | 2,449 ± 704.9 | 1,635 ± 638.9 |
| CXCL12 | 1,500 ± 362.0 | 1,841 ± 501.8 | 2,005 ± 606.2 | 1,753 ± 457.3 | 2,220 ± 380.6 | 1,521 ± 342.3 | 1,351 ± 432.0 | 1,196 ± 258.6 | 6,469 ± 1,680* |
| IL-18 | 1,492 ± 437.0 | 1,212 ± 104.9 | 986.5 ± 344.0 | 1,051 ± 188.3 | 1,469 ± 285.4 | 1,467 ± 194.3 | 902.2 ± 165.1 | 601.9 ± 70.32 | 1,168 ± 246.9 |
| IL-15 | 2,691 ± 447.2 | 2,395 ± 347.6 | 2,866 ± 265.2 | 2,502 ± 173.5 | 3,699 ± 375.2 | 3,043 ± 434.0 | 2,749 ± 460.9 | 1,299 ± 68.46 | 1,378 ± 214.2 |
| IL-6 | 212.2 ± 111.3 | 167.6 ± 83.52 | 207.4 ± 70.39 | 102.4 ± 33.09 | 170.9 ± 68.98 | 37.75 ± 9.911 | 22.3 ± 13.62 | 22.12 ± 10.01 | 24.97 ± 15.14 |
| CCL21 | 259.8 ± 56.97 | 184.7 ± 18.91 | 117.8 ± 40.62 | 259.5 ± 60.80 | 198.5 ± 40.04 | 215.3 ± 53.96 | 201 ± 24.32 | 315.0 ± 70.23 | 508.7 ± 107.9 |
| TGF-β | 208.9 ± 22.67 | 135.4 ± 18.90 | 578.4 ± 299.3 | 236.2 ± 42.90 | 164.5 ± 40.10 | 259.9 ± 22.78 | 227.2 ± 33.95 | 171.7 ± 29.26 | 392.1 ± 111.1 |
| IL-12 | 145.0 ± 19.27 | 63.8 ± 15.04 | 68.04 ± 17.05 | 2,277 ± 950.7 | 167.1 ± 36.38 | 8,596 ± 2,146* | 238.0 ± 180.2 | 42.22 ± 15.31 | 27.92 ± 5.554 |
| CCL2 | 33,290 ± 12,996 | 3,834 ± 106.0 | 1,147 ± 525.9 | 40,834 ± 11,665 | 2,898 ± 2,230 | 4,549 ± 559.4 | 40,902 ± 10,144 | 43,931 ± 11,338 | 68,376 ± 4,157 |
| CCL5 | 45,719 ± 9,230 | 10,473 ± 905.8 | 23,818 ± 3,948 | 16,954 ± 3,467 | 20,385 ± 1,961 | 16,189 ± 2,751 | 58,073 ± 5,412 | 49,934 ± 4,980 | 56,725 ± 4,738 |
Values represent picograms/milliliter cytokine ± SEM. Asterisks indicate values that are significantly different from those of mock-infected controls (P < 0.05). ND, not done.
Statistical analysis.
For statistical evaluation, unpaired, two-tailed nonparametric Mann-Whitney U tests were performed on sample data. Values from animals at various infection time points were compared to those from mock-infected controls for antibody measurements, flow cytometric analysis, and protein arrays. P values of <0.05 were considered statistically significant.
RESULTS
Presence of anti-SARS-CoV antibodies in the lung does not enhance infection.
The kinetics and magnitude of SARS-CoV replication and associated immune responses were evaluated in a natural history of primary infection as well as a homologous rechallenge study. The natural history study involved timed necropsies at 1, 3, 5, 10, and 14 days postinfection (dpi) (Fig. 1). In the rechallenge experiments, animals were reinoculated with SARS-CoV at day 28 after primary infection with sacrifice at days 5 and 28 after primary or secondary infection. The spatiotemporal analysis of SARS-CoV replication in AGMs indicated that virus was detectable in both swabs and respiratory tract tissues as early as 1 dpi (Fig. 2A to F). High levels of SARS-CoV were detected at days 1 and 3 in the trachea and proximal airways of the left caudal lung lobe (Fig. 2D and E). SARS-CoV inoculation resulted in productive infection of all AGMs, as virus was recovered from every animal at early infection time points (between days 1 to 5 pi). SARS-CoV titers peaked at 5 dpi in the swabs (Fig. 2A and B) and at 1 dpi in the lung (Fig. 2E and F). By 10 dpi, SARS-CoV replication in the lung had decreased, with a decline in overall titer and in the number of samples with detectable virus per animal. Interestingly, 4 of the 6 animals were SARS-CoV positive in the lung at 10 dpi (Fig. 2E and F); however, only 1 of these had detectable virus in its swab samples (Fig. 2A and B). No replicating virus was detected in any lung or swab sample collected at 14 dpi, at 28 dpi, or in 11 of the 12 rechallenged AGMs. Following secondary SARS-CoV infection, only one sample, the nasal swab of AGM B9292 at day 5 after rechallenge, tested positive for virus by plaque assay (Fig. 2A). Two additional animals (AGMs B5026 and B5078) had detectable SARS-CoV viral RNA in pharyngeal swab samples (∼400 to 800 N gene copies/ml) at day 5 after rechallenge as determined by RT-PCR (data not shown).
Fig 2.

Spatiotemporal analysis of SARS-CoV replication and antibody responses following primary and secondary challenge. (A to F) SARS-CoV replication was assessed in various respiratory tract tissues by plaque-forming assays. Shown in each graph are the virus levels of individual animals at the time of euthanasia for nasal swabs (A), pharyngeal swabs (B), as well as homogenized nasal turbinates (C), trachea (D), and portions of the right caudal lung lobe (proximal [E], distal [F]). Note that the data shown in panel A for B9292 (open gray triangle) at day 5 after rechallenge is not the necropsy time point for this animal but is included, as this was the only SARS-CoV plaque-positive sample recovered from any animal following rechallenge. (G) Anti-SARS-CoV S protein-specific IgG was measured in lung tissue by ELISA. The asterisk indicates that the mean titers are significantly different (P < 0.05). (H) SARS-CoV neutralizing antibodies were measured in the sera throughout infection, with the arrow marking the reinfection time point. Data are shown for animals that were followed until day 28 after rechallenge only (in gray), with AGM numbers indicated in the legend. The unique symbols representing each animal at the different time points are kept consistent in all of the graphs so as to enable tracking of virus and antibody in the distinct samples of each individual animal. Values are plotted on a log scale.
Antibodies to the spike (S) glycoprotein of SARS-CoV have been shown to block infection of target cells in vitro (1, 18, 42, 62); however, anti-S protein antibodies may also play a role in SARS-CoV immunopotentiation (38, 40). To determine if anti-SARS-CoV antibodies contribute to immune protection or pathogenesis in primary and secondary infection, SARS-CoV S protein-specific IgG levels were measured in lung tissue homogenates throughout the infection time course by ELISA (Fig. 2G). All animals had detectable anti-SARS-CoV antibodies in the lung by 14 dpi, and titers continued to rise up to 28 dpi despite the absence of productive infection after 10 dpi. A significant increase in mean antibody titers was observed at day 5 after rechallenge, followed by a decline in anti-SARS antibodies by day 28 after rechallenge to levels similar to those measured during primary infection. Anti-SARS-CoV IgA antibodies were monitored longitudinally in the mucosal secretions of rechallenge animals (Table 1). Not all had detectable anti-SARS-CoV IgA in nasal swabs; however, IgA titers were boosted in 3 of the 6 rechallenged animals. Systemic anti-SARS-CoV antibodies were also monitored in the sera following primary and secondary infection (Fig. 2H, Table 2). SARS-CoV neutralizing and anti-SARS-CoV S protein IgG antibody levels in sera varied between animals but showed a continuously increasing trend from days 5 to 28 after primary infection. Unlike lung anti-SARS-CoV IgG levels, no significant increase was detected in the SARS-CoV-specific antibody levels in the sera following reinfection.
Table 1.
Anti-SARS-CoV S protein-specific IgA antibodies measured in nasal swabs
| Sample | Dilutiona |
||||||
|---|---|---|---|---|---|---|---|
| Day 0 | Primary infection |
Rechallenge |
|||||
| Day 5 | Day 14 | Day 28 | Day 5 | Day 14 | Day 28 | ||
| B5026 | <10 | <10 | <10 | <10 | <10 | <10 | <10 |
| B5180 | <10 | <10 | <10 | <10 | <10 | <10 | <10 |
| B9392 | <10 | <10 | 160 | 640 | 2,560 | 40 | 40 |
| B5147 | <10 | <10 | 40 | <10 | <10 | 40 | 40 |
| B5149 | <10 | <10 | 2,560 | <10 | <10 | 2,560 | 2,560 |
| B9389 | <10 | <10 | 40 | <10 | <10 | <10 | <10 |
Values reported are the reciprocal of the highest dilution in which antibody binding was 2-fold higher than that of the nonspecific control and fell within the linear range of the serial dilutions.
Table 2.
Anti-SARS-CoV S protein-specific IgG measured in serum by ELISA
| Sample | Dilutiona |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Day 0 | Primary infection |
Rechallenge |
|||||||
| 5 | Day 5 | Day 14 | Day 28 | Day 5 | Day 14 | Day 28 | Day 5 | ||
| B5026 | <25 | <25 | 675 | 675 | 6,075 | 6,075 | 6,075 | 6,075 | 675 |
| B5180 | <25 | <25 | 225 | 675 | 6,075 | 6,075 | 54,675 | 18,225 | 54,675 |
| B9392 | <25 | <25 | 675 | 6,075 | 54,675 | 54,675 | 6,075 | <25 | 75 |
| B5147 | <25 | <25 | 75 | 675 | 75 | <25 | 75 | 75 | 75 |
| B5149 | <25 | <25 | 25 | 675 | 675 | 675 | <25 | 675 | 6,075 |
| B9389 | <25 | <25 | 675 | 675 | 675 | 6,075 | 675 | 6,075 | 6,075 |
Values reported are the reciprocal of the highest dilution in which antibody binding was 2-fold higher than that of the nonspecific control and fell within the linear range of the serial dilutions.
Inflammation in the lung observed long after viral clearance and following rechallenge.
To determine if viral replication levels correlated with severity of lung pathology, a comprehensive analysis of lung lesions in several regions of the respiratory tract at multiple time points postinfection was conducted (Fig. 3, Table 3). Histologically, inflammation in the lung parenchyma characterized by scattered foci of minimal lymphocyte and granulocyte infiltrations around small veins and minimal alveolitis was first evident at 3 dpi. At 5 dpi, interstitial pneumonia was present in 29% of the AGMs, in addition to the perivascular cuffing and alveolitis seen earlier. These changes were most severe and affected 100% of the monkeys at 10 dpi. At 14 dpi, septal fibrosis was present in 17% of the monkeys; other lung lesions of perivascular infiltrations and alveolitis were present but less severe. At 28 dpi, septal fibrosis was more prominent, affecting 33% of the animals, and other inflammatory lesions were present. At day 5 after rechallenge, perivascular cuffing, alveolitis, and interstitial pneumonia with fibrosis were present, with an incidence and severity similar to those of primary challenge but with a more chronic inflammatory response observed in the rechallenged group. Twenty-eight days after the rechallenge, interstitial pneumonia with fibrosis and alveolitis were slightly increased in severity but not incidence compared with those at day 5 after rechallenge. These AGMs examined at day 28 after rechallenge also exhibited increased septal fibrosis compared to that at day 5 after rechallenge. In all histopathology sections, regions of normal lung architecture were evident, indicating that lung pathology following infection was focal in nature.
Fig 3.

Representative histologic changes in the lung of AGM at specific times after SARS-CoV infection. H&E-stained sections from lung lesions at 1 day (A), 3 days (B), 5 days (C), 10 days (D), 14 days (E), and 28 days (F). Lung lesions from AGMs with SARS-CoV infection, 5 days after rechallenge (G) and 28 days after rechallenge (H). All photos were taken at 400× the original magnification.
Table 3.
Salient histologic changes after SARS-CoV intranasal instillation
| Group | No. of AGMs | Gradea | Value |
|||||
|---|---|---|---|---|---|---|---|---|
| Lung |
Trachea, tracheitis | TBLN, follicular hyperplasia | ||||||
| Perivascular cuffing | Alveolitis | Interstitial pneumonia | Septal fibrosis | |||||
| Mock | 6 | Severity | 0.17 | 0.17 | 0 | 0 | 0 | 0 |
| Incidence | 17% | 17% | 0 | 0 | 0 | 0 | ||
| 1 dpi | 3 | Severity | 0.33 | 0 | 0 | 0 | 0 | 0.25 |
| Incidence | 33% | 0 | 0 | 0 | 0 | 25% | ||
| 3 dpi | 6 | Severity | 1 | 0.5 | 0 | 0 | 1.5 | 0.5 |
| Incidence | 83% | 50% | 0 | 0 | 83% | 50% | ||
| 5 dpi | 17 | Severity | 1.3 | 1 | 0.47 | 0.06 | 0.63 | 0.47 |
| Incidence | 88% | 70% | 29% | 6% | 44% | 53% | ||
| 10 dpi | 6 | Severity | 1.7 | 1.3 | 1.5 | 0 | 0.67 | 1.3 |
| Incidence | 100% | 100% | 83% | 0 | 50% | 83% | ||
| 14 dpi | 6 | Severity | 1 | 0.33 | 0.5 | 0.5 | 0.5 | 0.6 |
| Incidence | 83% | 33% | 33% | 17% | 33% | 40% | ||
| 28 dpi | 6 | Severity | 1 | 1.2 | 0.5 | 0.33 | 0.2 | 0 |
| Incidence | 100% | 100% | 50% | 33% | 20% | 0 | ||
| 5 days after rechallenge | 6 | Severity | 1.5 | 0.67 | 0.67 | 0.17 | 1 | 1.7 |
| Incidence | 83% | 67% | 50% | 17% | 83% | 100% | ||
| 28 days after rechallenge | 6 | Severity | 1 | 0.83 | 1 | 0.5 | 0.3 | 0.67 |
| Incidence | 67% | 67% | 50% | 33% | 33% | 50% | ||
Severity is the average grade for the group (0 = normal, 1 = minimal, 2 = mild, 3 = moderate, 4 = marked); incidence is the percentage of the group affected.
Inflammatory lung infiltrates in SAR-CoV infection are predominantly composed of T lymphocytes and monocyte/macrophages.
To characterize and quantify the infiltrating immune cell populations that may contribute to SARS-CoV clearance and/or immunopathogenesis, lung leukocytes were isolated from mock- and SARS-CoV-infected animals and analyzed by flow cytometry. The absolute number of lung leukocytes per gram of tissue was significantly increased over that of mock-infected controls at 14 dpi (Fig. 4A), and this peak in inflammation was consistent with histopathology data (Fig. 3, Table 3). CD3+ T cells and CD14+CD11c+ alveolar macrophages (aMϕs) made up the greatest proportion of lung leukocytes at every time point postinfection, and their frequency in the lung was altered by SARS-CoV infection (Fig. 4B and C, Table 4). When total lung leukocyte numbers were at their highest (14 dpi), the T cell frequency is reduced, whereas the percentage of aMϕs was increased at this time point.
Fig 4.
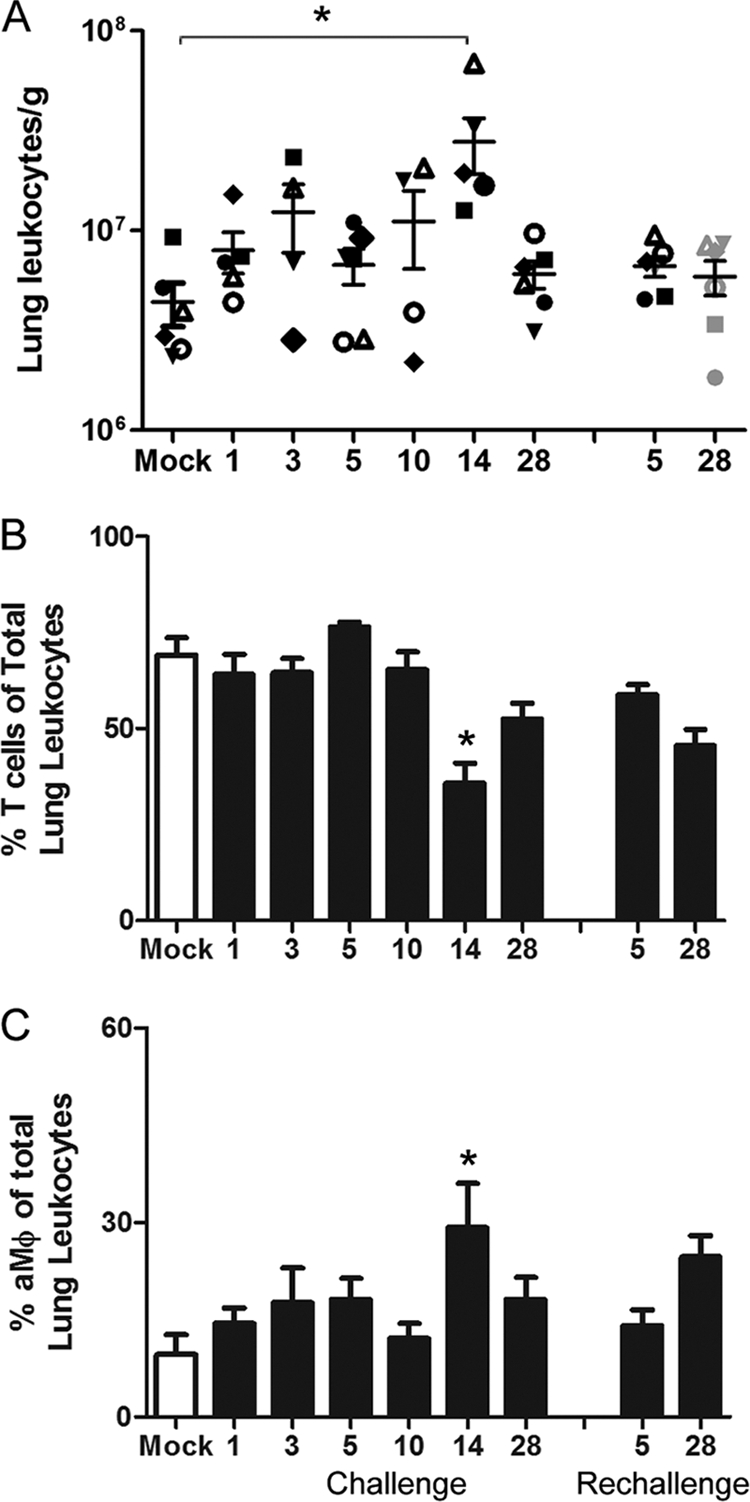
Flow cytometric characterization and leukocyte quantitation of SARS-CoV-induced inflammatory lung infiltrates. (A) The number of total lung leukocytes was determined per gram of tissue from standardized collected lung samples in mock-infected animals and at specific time points after primary and secondary SARS-CoV infection. The unique symbols at each time point represent the same animals in which virus and antibody levels were reported in Fig. 2. (B and C) Average percent frequencies of CD3+ T cells (B) and CD14+CD11c+ aMϕs (C) of total lung leukocytes were determined for mock (open bars) and SARS-CoV (solid bars)-infected animals by flow cytometry. Asterisks indicate that values are significantly different from those of mock-infected controls (P < 0.05).
Table 4.
Average cell numbers and relative frequency of leukocyte populations in the lung in SARS-CoV infection
| Cell type | No. of cellsa |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Mock infection | Primary infection |
Rechallenge |
|||||||
| Day 1 | Day 3 | Day 5 | Day 10 | Day 14 | Day 28 | Day 5 | Day 28 | ||
| Leukocytes | 4.4E6 ± 1E6 | 7.9E6 ± 1E6 | 1.2E7 ± 4E6 | 6.7E6 ± 1E6 | 1.1E7 ± 4E6 | 2.8E7 ± 8E6* | 6.0E6 ± 9E5 | 6.64E6 ± 7E5 | 5.88E6 ± 1E6 |
| T cells | 69.2 ± 4.4 | 64.1 ± 5.2 | 64.7 ± 3.6 | 76.5 ± 1.2 | 65.4 ± 4.6 | 35.7 ± 5.3* | 52.4 ± 4.2 | 58.9 ± 2.6 | 45.6 ± 4.2 |
| B cells | 1.5 ± 0.2 | 4.1 ± 0.3 | 2.4 ± 0.5 | 4.1 ± 0.6 | 3.4 ± 0.5 | 3.7 ± 0.9 | 2.2 ± 0.6 | 3.2 ± 1.2 | 4.2 ± 0.5* |
| aMϕ | 6.6 ± 3.1 | 14.4 ± 2.4 | 16.4 ± 4.1 | 17.2 ± 2.9 | 12.1 ± 2.2 | 25.1 ± 5.7* | 18.2 ± 3.3 | 5.3 ± 1.1 | 5.0 ± 1.3 |
| mDCs | 8.2 ± 1.4 | 7.1 ± 2.1 | 11.3 ± 1.9 | 14.0 ± 3.4 | 8.1 ± 1.1 | 16.1 ± 2.1 | 8.3 ± 2.9 | 8.6 ± 0.8 | 11.3 ± 1.7 |
| Eosinophils | 1.9 ± 1.0 | 3.7 ± 0.4 | 8.7 ± 3.4 | 2.1 ± 1.0 | 2.8 ± 0.1 | 4.0 ± 0.8 | 5.1 ± 1.0 | 5.8 ± 1.0 | 9.3 ± 0.9* |
Average absolute numbers of leukocytes per gram tissue ± SEM and the average frequency of T cells (CD3+), B cells (CD20+), aMϕ (CD14+ CD11c+), mDCs (CD14−CD11c+), and eosinophils (CD14−FcεR1+CD23+) of total lung leukocytes as determined by flow cytometry. Asterisks indicate values that are significantly different from those of mock-infected controls (P < 0.05).
Proinflammatory cytokines and chemokines elevated in the lung early in SARS-CoV infection.
As dysregulation of cytokines and chemokines is thought to play a critical role in the pathogenesis of SARS-CoV infection, we sought to define the expression profile of these proteins in the lung during primary and secondary infection. Bead-based protein arrays were used to measure levels of inflammatory as well as anti-inflammatory cytokines and chemokines in standardized collected lung tissue homogenates (Fig. 5 and Table 5). Several proinflammatory cytokines and chemokines were significantly upregulated early, between days 1 and 14 postinfection, including gamma interferon, IL-12, IL-1β, CXCL11, and CXCL13. Most cytokines returned to mock-infected control levels by 28 dpi, except IL-12 and gamma interferon. Interestingly, although productive infection was not detected in 11 of the 12 animals, several cytokines were elevated in the lung upon rechallenge, including the lymphocyte chemoattractants CXCL13 and CCL21 as well as monocyte chemoattractant, CCL2, gamma interferon, CXCL12, and the immunosuppressive cytokine transforming growth factor beta. However, only protein levels measured for CXCL12 in rechallenged animals were significantly increased over mock-infected controls. CXCL10 and TNF-α were included in the arrays; however, these cytokines were below the levels of detection in both mock- and SARS-CoV-infected animals.
Fig 5.

Cytokine and chemokine profile in the lung during SARS-CoV primary and secondary infection. Protein levels of cytokines and chemokines were measured in lung tissue homogenates across the primary infection time course (1, 3, 5, 10, 14, 28 dpi) and at days 5 and 28 after rechallenge with bead-based arrays. The fold-induction of average protein levels over those of mock-infected controls is represented in a heat map, with black representing no change, red indicating a 4-fold induction, and green indicating a 4-fold reduction in the average cytokine levels (n = 6 animals for each time point, except at 1 dpi [n = 5 animals]). Average cytokine and chemokine levels (pg/ml) ± standard errors of the means (SEM) at each time point are given in Table 5.
Early activation of lung macrophages but not dendritic cells in SARS-CoV infection.
Another mechanism thought to contribute to SARS-CoV pathogenesis is viral evasion of the host immune response. SARS-CoV has been shown to compromise macrophage and dendritic cell functions in vitro, and mice infected with the lethal MA15 strain show inefficient innate immune activation as well as poor T cell responses (61). Costimulatory and antigen-presenting molecule expression was evaluated on lung macrophages and myeloid dendritic cells (mDCs) by flow cytometry over the SARS-CoV infection time course (Fig. 6). The frequency of activation marker CD86+ lung macrophages increased with SARS-CoV infection, peaking at 14 dpi and remaining elevated even after rechallenge (although not reaching significance for the rechallenge time point) (Fig. 6B). In contrast, the proportion of CD86+ mDCs was not significantly altered in the lung following primary or secondary SARS-CoV infection. Expression of the antigen-presenting molecule HLA-DR (major histocompatibility complex class II [MHC-II]) was also low on lung mDCs during SARS-CoV infection, where the frequency of mDCs with high HLA-DR surface marker infection was lower than that of mock-infected controls until late in infection, day 28 pi (Fig. 6C). This HLA-DR suppression is consistent with results from in vitro SARS-CoV infection of DCs (44). SARS-CoV infection also failed to elevate the frequency of HLA-DR-high macrophages over the level of mock-infected controls. The expression of CD209/DC-SIGN, which is one of the cellular receptors for SARS-CoV (28, 56), was also evaluated on lung macrophages and mDCs during primary and secondary infection (Fig. 6D). CD209+ macrophages significantly increased at 5 dpi and had a higher trend following rechallenge, although not significant. Levels of CD209 were higher on macrophages than mDCs prior to and at most time points postinfection. Similar to HLA-DR expression, the frequency of CD209+ mDCs was not elevated until day 28 pi.
Fig 6.

Expression of activation markers on lung Mϕs and DCs in primary and secondary SARS-CoV challenge. Mϕs (CD14+) and DCs (CD14−CD11c+) isolated from the lungs of SARS-CoV-infected and mock-infected animals were evaluated by flow cytometry for activation marker CD86, antigen-presenting molecule HLA-DR, and SARS-CoV receptor CD209/DC-SIGN. (A) Representative FACS plots are shown for Mϕs and DCs from mock- and SARS-CoV-infected animals (14 dpi), depicting the gates used for evaluation of these cell surface antigens. The percent frequencies of receptor-positive cells of total Mϕs or DCs are indicated in each plot. (B to D) Flow cytometric results are summarized for activated Mϕs (solid bars) and DCs (open bars) during primary and secondary SARS-CoV infection. Percent frequencies of CD86 (B), HLA-DR high (C), and CD209/DC-SIGN (D) of total Mϕs or DCs are plotted. Asterisks indicate that values are significantly different from those of mock-infected controls for that particular cell type (P < 0.05).
To determine if reduced DC activation affected the generation of an adaptive immune response, the dynamics of lymphocyte expansion and activation were evaluated in the lung-draining lymph nodes. The absolute number of cells per gram of tracheobronchial lymph node tissue was determined, and the immune phenotype of lymph node cells was examined by flow cytometry (Fig. 7). The greatest expansion of lymphocytes during primary infection was observed at 5 dpi and a similarly high level of lymphocytes was also seen following rechallenge. The cellular composition remains consistent throughout primary infection, with T cells and mDCs making up the predominant portion of the lymph node cells (Fig. 7B and C). Evaluation of activation markers on lymph node DCs revealed reduced frequency of HLA-DR-high cells at days 5 and 14 after primary infection and at day 5 after rechallenge compared to that of mock-infected controls (Fig. 7D). An increase in HLA-DR-high DCs was not observed until 28 dpi, similar to results for lung DCs. Despite the reduction in costimulatory and antigen-presenting capacity of DCs in SARS-CoV infection, a significant increase in T cell proliferation was observed at 14 dpi, indicative of T cell activation and initiation of an adaptive immune response (Fig. 7E). Furthermore, a slight elevation of Granzyme B+ T cells was also observed at this time point (Fig. 7F).
Fig 7.

Impact of SARS-CoV on lymphocyte numbers and activation in the lung-draining lymph node. (A) The number of lymph node cells was determined per gram of tracheobronchial lymph node tissue. Lymph node cell numbers at necropsy time points after primary and secondary infection are graphed with the same symbols for individual animals as those used in Fig. 2 and 4. (B and C) The relative frequency of lymph node T cells (CD3+) and mDCs (CD14−CD11c+) for mock (open bars) and SARS-CoV (closed bars)-infected animals was determined by flow cytometry. (D to F) mDCs and lymph node T cells were further characterized for expression of activation markers, including HLA-DR (D), proliferation marker Ki67 (E), and Granzyme B (F). Asterisks indicate that values are significantly different from those of mock-infected controls (P < 0.05).
DISCUSSION
Our understanding of SARS-CoV infection is based largely on autopsy cases from the 2002 to 2003 outbreaks. Thus, our knowledge regarding the kinetics, magnitude, and duration of the immune events in primates, particularly at the site of SARS-CoV infection, is limited. Animal models that mimic the human disease are critical for elucidating the immune mechanisms that provide protection and add to our understanding of those responses that may contribute to disease pathogenesis in the lung. In this study, the host response to SARS-CoV infection in AGMs was examined with a specific focus on the local lung immunity.
AGMs were found to support productive viral replication for up to 10 days in the lung. However, unlike the ∼10% of human fatal cases, the animals were able to clear the virus and do not progress to fatal acute respiratory distress syndrome. AGMs were highly permissive for SARS-CoV infection, with replicating virus detected throughout the respiratory tract up to 10 dpi. Interestingly, evidence of SARS-CoV infection in the large airways was observed only very early after experimental infection, with little or no SARS-CoV detected at day 10 when virus continued to replicate in the distal lung. The transient nature of the infection in the airways is intriguing and consistent with reported clinical cases in which SARS-CoV can be isolated from upper airways during early but not later stages of disease (6, 14). Our findings suggest that although recoverable virus may be absent from nasopharyngeal aspirates or induced sputum, active SARS-CoV infection is likely occurring in the lower respiratory tract. Furthermore, the evidence that SARS-CoV may initially infect large airways before progressing to the distal alveolar epithelium has important implications for potential intervention strategies.
The pathogenesis of SARS through immune-mediated mechanisms has been proposed; however, clear evidence of immunopathogenesis in vivo with human CoVs is lacking (4, 38, 58). Findings from this study support a role for the immune response in contributing to SARS-CoV pathogenesis, as enhanced inflammation and lung leukocyte activation was observed long after viral clearance. Mild interstitial pneumonia and inflammation were observed in animals even at day 28 after rechallenge. Primary infection limited SARS-CoV replication upon rechallenge such that all but one animal was free of replicating SARS-CoV by day 5 after rechallenge, suggesting prevention of reinfection. However, analysis of viral replication at earlier time points after reinfection is needed to confirm this protection. If the majority of the AGMs were completely protected from reinfection, the pulmonary responses observed in these rechallenged animals may have been residual from primary infection (58 days prior). A large proportion of the inflammatory infiltrates were monocytes/macrophages, many of which continued to express activation markers at later time points postinfection and after rechallenge. Activated monocytes/macrophages are a major source of proinflammatory mediators that have the potential to activate immune cells and promote recruitment of additional leukocytes. Indeed, many proinflammatory cytokines and chemokines were upregulated in the lung between days 1 and 14 pi. Although this response does not appear to be indicative of a “cytokine storm” that has been reported in other coronavirus infections (38), there were several proinflammatory cytokines whose levels remained elevated over those of mock-infected controls after the virus had been cleared from the lung. This slow resolution of lung inflammation following SARS-CoV infection may contribute to the prolonged impairment of pulmonary function that was observed in SARS survivors months after discharge (20, 31).
Another immune-mediated mechanism of SARS-CoV pathogenesis that was explored in this study is the inefficient activation of DCs and defective T cell responses. Unlike the lethal models of mouse-adapted SARS-CoV (61), the experimentally infected AGMs in these studies were able to activate T lymphocytes in the lymph node and eradicate virus from the lungs. However, there was evidence for suppressed and/or delayed DCs compared to macrophage activation in the lung and lymph node during SARS-CoV infection. Costimulatory and antigen-presenting molecule expression on DCs from SARS-CoV-infected animals was similar to (or occasionally lower than) that of mock-infected controls at several time points postinfection. It is possible that lung and lymph node DC activation occurred at time points not sampled in this study. Another important factor to consider is that these animals were wild caught and, although tested for specific pathogens before inclusion, may have had previous respiratory infections (16) that could impact the timing and magnitude of the DC response to experimental SARS-CoV infection. Ultimately, it will be imperative to compare the kinetics of the SARS-CoV DC response to DC activation during other respiratory virus infections in this NHP model in future studies.
Lymphopenia in patients was a common hematological feature in acute SARS-CoV infection, with several studies documenting reduced numbers of both CD4 and CD8 T lymphocytes (25). Although the AGMs in our studies showed no significant changes in peripheral blood populations following experimental SARS-CoV infection, there was a significant reduction in the proportion of T lymphocytes of total lung leukocytes at 14 dpi. The cause of the lung T cell decline in our studies is unclear; however, the timing coincides with the observed peak in lung injury following infection. Activation of cytoplasmic caspase 3 has been associated with enhanced T cell death during SARS-CoV infection (5), and SARS-CoV E protein has been shown to induce apoptosis of a T cell line in vitro (55). However, no replicating virus could be recovered from the lung at day 14 postinfection, so the T cell decline may reflect the normal contraction of the effector T cell pool following pathogen clearance. Ultimately, additional studies are necessary to identify the mechanisms contributing to reduced T cell frequencies in the lungs of SARS-CoV-infected AGMs.
Anti-SARS-CoV antibodies progressively increased in the lung and sera between days 1 and 28 pi. Importantly, as lung anti-SARS-CoV IgG and serum antibody levels increased, viral replication declined, demonstrating that SARS-CoV antibodies in this model do not enhance viral infection in the lung. It was surprising to detect antibodies capable of neutralizing SARS-CoV in the serum as early as 1 dpi, as previous exposure in these AGMs is unlikely and preexposure antibodies showed no specificity for recombinant SARS-CoV proteins. However, the presence of preexisting SARS-CoV neutralizing antibodies has previously been reported in the serum of AGMs (29). It is also important to note that even in SARS-CoV-infected humans, seroconversion could be detected as early as 4 days after the onset of illness (19). Despite the existence of low levels of antibodies capable of neutralizing SARS-CoV in the sera of several AGMs at 1 dpi, the anti-SARS-CoV antibody titers were significantly higher (at least 4-fold) by 28 dpi. Another surprising aspect of the systemic immune response was the lack of antibody boost following rechallenge. Levels of circulating antibodies at the reinfection time point (28 dpi) were high, and perhaps if the intervening period between infections had been greater, a boost may have been observed. Another possibility is that an increase in circulating antibodies occurred at time points not sampled in our reinfection studies. Despite the lack of immune boost in the periphery following rechallenge, lung antibody levels were significantly elevated, and a dramatic expansion of lymphocytes was observed in the tracheobronchial lymph nodes with reinfection.
Taken together, the data indicate that in experimentally infected AGMs, local lung immune responses are capable of limiting SARS-CoV replication. Interestingly, previous exposure to SARS-CoV conferred protection by significantly limiting viral replication upon rechallenge; however, the incidence and severity of perivascular cuffing, alveolitis, and interstitial pneumonia was not reduced but remained similar to that observed during primary challenge. The prolonged activation of lung leukocytes and chronic inflammation suggests that mechanisms to resolve the immune response may be delayed or dysregulated during SARS-CoV. Our findings provide novel information regarding the protective and potentially pathogenic aspects of the immune response that is elicited in the lung upon SARS-CoV infection. This knowledge will be important to consider in the design of effective intervention strategies for SARS-CoV and potentially other severe respiratory infections.
ACKNOWLEDGMENTS
This work was supported by the NIAID contract number N01-AI-40095.
We thank the ABSL3 animal care and pathology staffs and Katie Overheim for assistance with these studies. We appreciate Linda Saif and her lab for their help with characterization of African green monkey serum antibodies. We thank Fred Cassels, NIAID, for helpful comments and suggestions on experimental design.
Footnotes
Published ahead of print 15 February 2012
REFERENCES
- 1. Berry JD, et al. 2004. Development and characterisation of neutralising monoclonal antibody to the SARS-coronavirus. J. Virol. Methods 120:87–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cameron MJ, et al. 2007. Interferon-mediated immunopathological events are associated with atypical innate and adaptive immune responses in patients with severe acute respiratory syndrome. J. Virol. 81:8692–8706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chen H, et al. 2005. Response of memory CD8+ T cells to severe acute respiratory syndrome (SARS) coronavirus in recovered SARS patients and healthy individuals. J. Immunol. 175:591–598 [DOI] [PubMed] [Google Scholar]
- 4. Chen J, Subbarao K. 2007. The immunobiology of SARS*. Annu. Rev. Immunol. 25:443–472 [DOI] [PubMed] [Google Scholar]
- 5. Chen RF, et al. 2006. Role of vascular cell adhesion molecules and leukocyte apoptosis in the lymphopenia and thrombocytopenia of patients with severe acute respiratory syndrome (SARS). Microbes Infect. 8:122–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cheng PK, et al. 2004. Viral shedding patterns of coronavirus in patients with probable severe acute respiratory syndrome. Lancet 363:1699–1700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cheung CY, et al. 2005. Cytokine responses in severe acute respiratory syndrome coronavirus-infected macrophages in vitro: possible relevance to pathogenesis. J. Virol. 79:7819–7826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chiang SF, Lin TY, Chow KC, Chiou SH. 2010. SARS spike protein induces phenotypic conversion of human B cells to macrophage-like cells. Mol. Immunol. 47:2575–2586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chow KC, Hsiao CH, Lin TY, Chen CL, Chiou SH. 2004. Detection of severe acute respiratory syndrome-associated coronavirus in pneumocytes of the lung. Am. J. Clin. Pathol. 121:574–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chu YK, et al. 2008. The SARS-CoV ferret model in an infection-challenge study. Virology 374:151–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Clay CC, et al. 2004. Chemokine networks and in vivo T-lymphocyte trafficking in nonhuman primates. J. Immunol. Methods 293:23–42 [DOI] [PubMed] [Google Scholar]
- 12. de Lang A, et al. 2007. Functional genomics highlights differential induction of antiviral pathways in the lungs of SARS-CoV-infected macaques. PLoS Pathog. 3:e112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Devaraj SG, et al. 2007. Regulation of IRF-3-dependent innate immunity by the papain-like protease domain of the severe acute respiratory syndrome coronavirus. J. Biol. Chem. 282:32208–32221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Drosten C, et al. 2004. Evaluation of advanced reverse transcription-PCR assays and an alternative PCR target region for detection of severe acute respiratory syndrome-associated coronavirus. J. Clin. Microbiol. 42:2043–2047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Frieman M, et al. 2007. Severe acute respiratory syndrome coronavirus ORF6 antagonizes STAT1 function by sequestering nuclear import factors on the rough endoplasmic reticulum/Golgi membrane. J. Virol. 81:9812–9824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Goulding J, et al. 2007. Respiratory infections: do we ever recover? Proc. Am. Thorac. Soc. 4:618–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Haagmans BL, Osterhaus AD. 2006. Coronaviruses and their therapy. Antiviral Res. 71:397–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. He Y, et al. 2004. Receptor-binding domain of SARS-CoV spike protein induces highly potent neutralizing antibodies: implication for developing subunit vaccine. Biochem. Biophys. Res. Commun. 324:773–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hsueh PR, Huang LM, Chen PJ, Kao CL, Yang PC. 2004. Chronological evolution of IgM, IgA, IgG and neutralisation antibodies after infection with SARS-associated coronavirus. Clin. Microbiol. Infect. 10:1062–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hui DS, et al. 2005. The 1-year impact of severe acute respiratory syndrome on pulmonary function, exercise capacity, and quality of life in a cohort of survivors. Chest 128:2247–2261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kopecky-Bromberg SA, Martinez-Sobrido L, Frieman M, Baric RA, Palese P. 2007. Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists. J. Virol. 81:548–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kuiken T, et al. 2003. Newly discovered coronavirus as the primary cause of severe acute respiratory syndrome. Lancet 362:263–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lau SK, et al. 2005. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc. Natl. Acad. Sci. U. S. A. 102:14040–14045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Law HK, et al. 2005. Chemokine up-regulation in SARS-coronavirus-infected, monocyte-derived human dendritic cells. Blood 106:2366–2374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li C, Xu X. 2010. Host immune response to SARS coronavirus in humans. In Lal SK. (ed), Molecular biology of the SARS-coronavirus, vol 1 Springer-Verlag, New York, NY [Google Scholar]
- 26. Li W, et al. 2005. Bats are natural reservoirs of SARS-like coronaviruses. Science 310:676–679 [DOI] [PubMed] [Google Scholar]
- 27. Martina BE, et al. 2003. Virology: SARS virus infection of cats and ferrets. Nature 425:915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Marzi A, et al. 2004. DC-SIGN and DC-SIGNR interact with the glycoprotein of Marburg virus and the S protein of severe acute respiratory syndrome coronavirus. J. Virol. 78:12090–12095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McAuliffe J, et al. 2004. Replication of SARS coronavirus administered into the respiratory tract of African green, rhesus and cynomolgus monkeys. Virology 330:8–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Narayanan K, et al. 2008. Severe acute respiratory syndrome coronavirus nsp1 suppresses host gene expression, including that of type I interferon, in infected cells. J. Virol. 82:4471–4479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ong KC, et al. 2004. Pulmonary function and exercise capacity in survivors of severe acute respiratory syndrome. Eur. Respir. J. 24:436–442 [DOI] [PubMed] [Google Scholar]
- 32. Peiris JS, et al. 2003. Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: a prospective study. Lancet 361:1767–1772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Peiris JS, et al. 2003. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 361:1319–1325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Peiris JS, Porterfield JS. 1979. Antibody-mediated enhancement of flavivirus replication in macrophage-like cell lines. Nature 282:509–511 [DOI] [PubMed] [Google Scholar]
- 35. Peiris JS, Yuen KY, Osterhaus AD, Stohr K. 2003. The severe acute respiratory syndrome. N. Engl. J. Med. 349:2431–2441 [DOI] [PubMed] [Google Scholar]
- 36. Peng H, et al. 2006. Human memory T cell responses to SARS-CoV E protein. Microbes Infect. 8:2424–2431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Peng H, et al. 2006. Long-lived memory T lymphocyte responses against SARS coronavirus nucleocapsid protein in SARS-recovered patients. Virology 351:466–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Perlman S, Dandekar AA. 2005. Immunopathogenesis of coronavirus infections: implications for SARS. Nat. Rev. Immunol. 5:917–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pollard AM, Lipscomb MF. 1990. Characterization of murine lung dendritic cells: similarities to Langerhans cells and thymic dendritic cells. J. Exp. Med. 172:159–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Roberts A, et al. 2007. Animal models and vaccines for SARS-CoV infection. Virus Res. 133:20–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Roberts A, et al. 2005. Severe acute respiratory syndrome coronavirus infection of golden Syrian hamsters. J. Virol. 79:503–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rockx B, et al. 2008. Structural basis for potent cross-neutralizing human monoclonal antibody protection against lethal human and zoonotic severe acute respiratory syndrome coronavirus challenge. J. Virol. 82:3220–3235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Reference deleted.
- 44. Spiegel M, Schneider K, Weber F, Weidmann M, Hufert FT. 2006. Interaction of severe acute respiratory syndrome-associated coronavirus with dendritic cells. J. Gen. Virol. 87:1953–1960 [DOI] [PubMed] [Google Scholar]
- 45. Sullivan NJ. 2001. Antibody-mediated enhancement of viral disease. Curr. Top. Microbiol. Immunol. 260:145–169 [DOI] [PubMed] [Google Scholar]
- 46. Temperton NJ, et al. 2005. Longitudinally profiling neutralizing antibody response to SARS coronavirus with pseudotypes. Emerg. Infect. Dis. 11:411–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. ter Meulen J, et al. 2004. Human monoclonal antibody as prophylaxis for SARS coronavirus infection in ferrets. Lancet 363:2139–2141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tseng CT, Perrone LA, Zhu H, Makino S, Peters CJ. 2005. Severe acute respiratory syndrome and the innate immune responses: modulation of effector cell function without productive infection. J. Immunol. 174:7977–7985 [DOI] [PubMed] [Google Scholar]
- 49. Warnes GR, et al. 2009. gplots: various R programming tools for plotting data. The Comprehensive R Archive Network. http://cran.r-project.org/package=gplots
- 50. Weingartl H, et al. 2004. Immunization with modified vaccinia virus Ankara-based recombinant vaccine against severe acute respiratory syndrome is associated with enhanced hepatitis in ferrets. J. Virol. 78:12672–12676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. WHO 2003. The world health report 2003—shaping the future. World Health Organization, Geneva, Switzerland [Google Scholar]
- 52. Wong RS, et al. 2003. Haematological manifestations in patients with severe acute respiratory syndrome: retrospective analysis. BMJ 326:1358–1362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yang L, et al. 2007. Persistent memory CD4+ and CD8+ T-cell responses in recovered severe acute respiratory syndrome (SARS) patients to SARS coronavirus M antigen. J. Gen. Virol. 88:2740–2748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yang LT, et al. 2006. Long-lived effector/central memory T-cell responses to severe acute respiratory syndrome coronavirus (SARS-CoV) S antigen in recovered SARS patients. Clin. Immunol. 120:171–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yang Y, et al. 2005. Bcl-xL inhibits T-cell apoptosis induced by expression of SARS coronavirus E protein in the absence of growth factors. Biochem. J. 392:135–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yang ZY, et al. 2004. pH-dependent entry of severe acute respiratory syndrome coronavirus is mediated by the spike glycoprotein and enhanced by dendritic cell transfer through DC-SIGN. J. Virol. 78:5642–5650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yang ZY, et al. 2005. Evasion of antibody neutralization in emerging severe acute respiratory syndrome coronaviruses. Proc. Natl. Acad. Sci. U. S. A. 102:797–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yasui F, et al. 2008. Prior immunization with severe acute respiratory syndrome (SARS)-associated coronavirus (SARS-CoV) nucleocapsid protein causes severe pneumonia in mice infected with SARS-CoV. J. Immunol. 181:6337–6348 [DOI] [PubMed] [Google Scholar]
- 59. Yilla M, et al. 2005. SARS-coronavirus replication in human peripheral monocytes/macrophages. Virus Res. 107:93–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yoshikawa T, Hill T, Li K, Peters CJ, Tseng CT. 2009. Severe acute respiratory syndrome (SARS) coronavirus-induced lung epithelial cytokines exacerbate SARS pathogenesis by modulating intrinsic functions of monocyte-derived macrophages and dendritic cells. J. Virol. 83:3039–3048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zhao J, Van Rooijen N, Perlman S. 2009. Evasion by stealth: inefficient immune activation underlies poor T cell response and severe disease in SARS-CoV-infected mice. PLoS Pathog. 5:e1000636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zhu Z, et al. 2007. Potent cross-reactive neutralization of SARS coronavirus isolates by human monoclonal antibodies. Proc. Natl. Acad. Sci. U. S. A. 104:12123–12128 [DOI] [PMC free article] [PubMed] [Google Scholar]