

Abstract
Kaposi's sarcoma-associated herpesvirus (KSHV) is linked to several neoplastic diseases: Kaposi's sarcoma, primary effusion lymphoma (PEL), and multicentric Castleman's disease (MCD). KSHV replicates actively, via a controlled gene expression program, but can also remain latent. It had been thought that the transition from latent to lytic replication was controlled exclusively by the replication and transcription activator protein RTA (open reading frame 50 [ORF50] gene product). A dominant-negative (DN) ORF50 mutant, ORF50ΔSTAD, blocks gene expression and replication. We produced a PEL cell line derivative containing both latent KSHV genomes and an inducible ORF50ΔSTAD. We unexpectedly found that induction of apoptosis triggered high-level viral replication, even when DN ORF50ΔSTAD was present, suggesting that apoptosis triggers KSHV replication through a distinct RTA-independent pathway. We verified that apoptosis triggers KSHV replication independent of RTA using ORF50 small interfering RNA (siRNA) and also showed that caspase activity is required to trigger KSHV replication. We showed that when apoptosis triggers KSHV replication, the kinetics of late gene expression is accelerated by 12 to 24 h and that virus produced following apoptosis has reduced infectivity. KSHV therefore appears to replicate via two distinct pathways, a conventional pathway requiring RTA, with slower replication kinetics, producing virus with higher infectivity, and an alternative apoptosis-triggered pathway that does not require RTA, has faster replication kinetics, and produces virus with lower infectivity. The existence of a distinct apoptosis-triggered, accelerated replication pathway may have evolutionary advantages for the virus and clinical significance for the treatment of KSHV-associated neoplasms. It also provides further evidence that KSHV can sense and react to its environment.
INTRODUCTION
Kaposi's sarcoma-associated herpesvirus (KSHV) (also called human herpesvirus 8 [HHV-8]) (10) is a member of the gammaherpesvirus subfamily of the genus Rhadinovirus. KSHV is closely related to Herpesvirus saimiri of squirrel monkeys and Epstein-Barr virus (EBV). KSHV been causally linked to Kaposi's sarcoma (KS), primary effusion lymphoma (PEL), a B cell lymphoproliferative disorder (8), and a plasmablastic subset of multicentric Castleman's disease (42). KSHV-related neoplasms sometimes respond clinically to restoration of immune function, for example when AIDS patients with KS experience immune reconstitution following treatment with combination antiretroviral therapy. KSHV-related neoplasms often require cytotoxic chemotherapy, as do neoplasms causally linked with other herpesviruses, such as Epstein-Barr virus (EBV).
KSHV has an ∼145-kb genome, organized into >87 open reading frames (ORFs), flanked by ∼5 kb of terminal repeat sequence. KSHV has many genes typical of other herpesviruses (28, 37). These typical herpesvirus genes include genes that encode capsid, tegument, envelope, replication proteins, and regulatory proteins (transcriptional activators) (28, 37). However, KSHV also has a large complement of genes homologous to cellular genes and others of unknown function (1, 2, 4, 7, 9, 29, 32, 33). The accessory cellular gene homologs, sometimes called “pirated” cellular genes, encode proteins that have effects on the host cell: viral homologs of host cell cytokines and chemokines, chemokine receptors, anti-apoptotic factors, cyclins, and transactivators. Several of these viral proteins can transform cells (12, 13).
KSHV not only has pirated cellular genes within its genome, which have apparently evolved to alter the physiology of the host cell and the host, the virus also has demonstrated the abilities to sense and respond to the host and host cell signals. For example, KSHV's genome has promoter regulatory sites homologous to known cellular regulatory sequences or that bind host cell factors, e.g., AP-1, CCAAT/enhancer binding protein alpha (C/EBPα), NF-κB, NFAT, OCT, and recombination signal binding protein Jκ (RBP-Jκ) (17, 22, 38, 39, 46, 47). Treatment of PEL cells with agents targeting host cell regulatory mechanisms, like diacylglycerol homologs (phorbol myristyl acetate [PMA]/tetradecanoylphorbol-13-acetate [TPA]) and agents that affect chromatin and histone acetylation, such as butyrate, triggers viral replication (36). There are additional examples of KSHV responding to host cell signaling cascades: alpha interferon (IFN-α) directly activates the KSHV viral interleukin-6 (IL-6) gene, so the virus can sense the levels of host IFN-α and modify the cellular environment accordingly by inhibiting IFN signaling (11). KSHV therefore has a complicated, nuanced relationship with the host and the host cell. The virus has genes that can have significant effects on the host cell and the host as a whole and can sense host and host cell signals.
Like other herpesviruses, KSHV can both replicate lytically and remain latent. During latency, KHSV expresses only a few viral genes, such as LANA-1, LANA-2, ORF72, ORFK12, ORFK13, and ORFK15. The expression of these genes does not lead to the initiation of a full replication cycle resulting in the production of virus. LANA-1, the major latency-associated KSHV nuclear antigen (19), plays a critical role in latency and tethers viral DNA to host DNA via sequence-specific interactions between viral DNA and mitotic host cell chromosomes (3), via interactions with RBP-Jκ, the downstream effector of the Notch signaling pathway (21).
Several agents, such as proinflammatory cytokines, phorbol esters, and histone deacetylase inhibitors can trigger an end to latency, initiating lytic replication. Activation initiates a carefully controlled, temporally determined program of viral gene expression involving the full complement of viral genes, culminating in the production of infectious virus (25, 34).
The initiation of lytic replication is believed to proceed through the activities of the replication and transcription activator protein (RTA) (or ORF50 protein), a 691-amino-acid (aa) product of the KSHV open reading frame 50 (ORF50) (27). RTA lies at the apex of the expression regulation hierarchy (27, 44). RTA directly activates eight KSHV genes (nut-1/PAN, ORF57/Mta, ORF56/primase, K2/viral interleukin-6 [vIL-6], ORF37/SOX, K14/vOX, K9/viral interferon regulatory factor [vIRF1], and ORF52), with the rest of the KSHV genes activated indirectly through these genes alone or in combination with RTA or other cellular factors and further downstream KSHV genes (6). RTA binds directly to the promoters of several KSHV genes, including genes expressed early in the replication cycle, like ORF57 and K8 (26). RTA acts together with cellular proteins, such as Oct1 and HMGB1 (15), SWI/SNF and TRAP/Mediator (14), and recombination signal binding protein Jκ RBPj-κ (or CBF-1 or CSL) (23, 48) to transactivate other genes. RTA interactions with RBP-Jκ help mediate RTA binding to the promoters of ORF57, ORF6, the KSHV basic leucine zipper protein (K-bZIP), the KSHV viral G-protein-coupled receptor protein (vGPCR), and binding to its own promoter (23). Other cellular factors involved in mediating RTA binding to viral promoters include octamer-1, AP-1, and C/EBPα) (38, 48). RTA appears to function as a tetramer (5).
RTA alone can initiate KSHV replication and has a powerful positive autoregulatory activity that forms an important component of the latent-to-lytic switch, which eventually leads to the upregulation of the expression of all lytic cycle KSHV genes (27, 31). This is true when KSHV replication is triggered with PMA, or with other inducers, such as butyrate or ionomycin (27).
Several studies employing both wild-type and mutant RTA led to the widely accepted model that RTA is the single factor that is both necessary and sufficient for the initiation of the KSHV replication cycle and gene expression program (27, 43). The production of a truncation mutant of ORF50, ORF50ΔSTAD, lent strong support to this model. The ORF50ΔSTAD truncation mutant maintains the RTA DNA binding domain and the multimerization function involving a proline-rich, N-terminal leucine heptapeptide repeat (LR) spanning amino acids 244 to 275, together with a central region of RTA (amino acids 245 to 414). The ORF50ΔSTAD truncation mutant, however, lacks the 161 C-terminal amino acids, which include the protein's activation domain. The activation domain by itself can function as a powerful transactivator when fused to a heterologous DNA binding domain (27, 40, 45). ORF50ΔSTAD binds and multimerizes with wild-type RTA, via interactions with the RTA LR (5), but lacking the activation domain, functions as a powerful dominant-negative (DN) mutant. When ORF50ΔSTAD is present, even in small amounts, KSHV replication is completely blocked (27).
We produced a derivative of the BCBL-1 PEL cell line, ORF50ΔSTAD BCBL-1, which contains both latent copies of KSHV and a tetracycline-inducible version of the ORF50ΔSTAD DN mutant. Our initial aim was to determine which viral genes were expressed when cells expressing the ORF50ΔSTAD DN mutant were treated with inducing agents. However, in the course of initially characterizing the cell line, we made the unexpected observation that treatment of the cells with agents that induce apoptosis appeared to trigger KSHV replication, even in the presence of the ORF50ΔSTAD DN mutant, suggesting that KSHV has an alternative replication program triggered by host cell death that proceeds without a requirement for RTA. Our subsequent experiments further showed that this apoptosis-triggered pathway requires the activity of certain caspases, has accelerated late gene expression kinetics, and produces virus with decreased, but still present infectivity. These observations suggest that KSHV can sense and respond to changes in its host cell in previously unappreciated ways that should confer a strong advantage to a virus. The observations also have implications for therapeutics directed against neoplasms caused by KSHV or other herpesviruses.
MATERIALS AND METHODS
Cell culture.
BCBL-1 cells and ORF50ΔSTAD BCBL-1 cells (see below) were grown in RPMI 1640 medium enriched with 10% fetal bovine serum (FBS), glutamine, penicillin-streptomycin, and β-mercaptoethanol (standard growth medium) as previously described (25, 34). The cells were maintained between densities of 0.25 × 106 to 0.5 × 106 cell/ml. Cell density and viability were routinely assessed by using a hemocytometer and trypan blue staining and through flow cytometric assays for apoptotic markers (see below).
Human umbilical vein epithelial cells (HUVEC) (umbilical vein product catalog no. cc-2519; Lonza, Allendale, NJ) were grown in the EBM basal medium provided by Lonza and supplemented with the EGM Single Quot kit provided. The cells were maintained at a confluence of 70 to 80% between densities of 0.18 × 106 to 0.37 × 106 cells/75 cm2. Adherent HUVEC were removed using trypsin-EDTA (Lonza) after washes with HEPES-buffered saline, followed by treatment with trypsin neutralizing solution (Clonetics umbilical vein endothelial cell systems; Lonza). The cells were maintained at 37°C, 5% CO2 and 80% humidity.
KSHV lytic replication was induced by adding tetradecanoylphorbol-13-acetate (TPA) (Sigma-Aldrich, St. Louis, MO) at a final concentration of 20 ng/ml. The cells were incubated with TPA for 1 h at 37°C. The cells were then washed with medium, and if the cells were treated with a low concentration of doxycycline (DOX), they were replenished with DOX at a final concentration of 0.1 μg/ml (unless otherwise indicated).
Construction and characterization of ORF50ΔSTAD BCBL-1 cells.
The dominant-negative ORF50ΔSTAD (C-terminal truncated dominant-negative mutant of ORF50 (27) was provided in pv5-50ΔSTAD. The ORF50ΔSTAD segment integrated into the pv50 vector has an EcoRI site and an XhoI site at either end. pv5-50ΔSTAD was first cut with EcoRI and converted to a blunt end using T4 DNA polymerase. pv5-50ΔSTAD was then cut with XhoI and cloned into pcDNA5/FRT/TO (Invitrogen, Carlsbad, CA) using its EcoRV and XhoI sites. Electroporation of pcDNA5/FRT/TO-50ΔSTAD into Flp-In BCBL-1 cells (31), kindly provided by Jae Jung (University of Southern California), was performed using a Gene Pulser II (Bio-Rad, Hercules, CA) using a capacitance of 975 μF and 250 V. Transfected cells were selected as previously described (31). After final selection, successfully transformed cells were grown in standard growth medium without selection. Cell viability and cell density were monitored every 48 h.
The ORF50ΔSTAD BCBL-1 cells were treated with doxycycline (DOX) (BD Biosciences, San Jose, CA) at the stated concentrations, and the cells were harvested at the indicated times. Subsequently, for experiments in which ORF50ΔSTAD expression was induced to block KSHV replication, we used DOX at a final concentration of 0.1 μg/ml.
RT-PCR assays.
Real-time reverse transcription-PCR (RT-PCR) assays were used to quantitate full-length wild-type ORF50 and the truncated ORF50ΔSTAD RNAs and to quantitate RNAs encoding an early gene that is normally activated by RTA, ORF57, and a late gene, ORF17, which encodes a minor capsid protein.
Total DNA from cells was isolated using the FlexiGene DNA kit (Qiagen, Valencia, CA). Total RNA was isolated using the Qiagen RNeasy Plus minikit. Nucleic acids were quantified using a Nanodrop ND-1000 spectrophotometer (Nanodrop, Wilmington, DE). cDNA was prepared from 800 ng of the total RNA isolated from individual samples. Random hexamers (50 μM) and deoxynucleoside triphosphates (dNTPs) (10 mM) were mixed in a 1.7-ml microcentrifuge tube and 2 μl of hexamer/dNTP mixture was distributed into PCR tubes containing RNA. The RNA was then denatured at 65°C for 5 min and snap chilled on ice. A cocktail of 5× buffer (SSII RT buffer; Invitrogen), 0.1 M dithiothreitol (DTT), Superasin RNase inhibitor (Ambion), and SSII reverse transcriptase were mixed, and 8 μl of this cocktail mix was added to the denatured RNA/hexamer/dNTP mix. PCR was performed using the following protocol: 25°C for 10 min, 42°C for 50 min, and 70°C for 15 min.
ORF50 and ORF50ΔSTAD RT-PCR assays.
Full-length (FL) ORF50 is comprised of 686 amino acids, while ORF50ΔSTAD, a C-terminus-deleted dominant-negative mutant of ORF50, is comprised of 530 amino acids (26). To detect expression of both the FL ORF50 and ORF50ΔSTAD, TaqMan real-time PCR assays were designed to detect only FL ORF50 RNA by assaying for 3′ sequence not included in the truncated ORF50ΔSTAD RNA and total ORF50 RNA by assaying for 5′ sequences included in both FL ORF50 RNA and ORF50ΔSTAD RNA. To detect FL ORF50, we used ORF50FL TaqMan TAMRA probe (5′-6FAM-ACCACCCCCCAGGATGTGCAC-TAMRA-3′) and primers (forward: 5′-ACGCCATCGACCACGACACCT-3′; reverse: 5′-TGAGGGACCATCGGAGGC-3′). To detect both the full-length ORF50 RNA and the truncated ORF50ΔSTAD RNA, we used the ORF50 TaqMan 6-carboxytetramethylrhodamine (TAMRA) probe (5′-6-carboxyfluorescein [6FAM]-ACCTGTGCGCCCTCTTCGACACCT-TAMRA-3′) and primers (forward, 5′-CGCAATGCGTTACGTTGTTG-3′; reverse, 5′-GCCCGGACTGTTGAATCG-3′). The results were normalized to a real-time RT-PCR assay for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (internal standard) using a commercially available assay, Hu GAPDH assay kit (Applied Biosystems). Reverse transcription real-time PCR assays were performed by employing the relative quantitative ΔΔC[r]T method (CT stands for threshold cycle) in an Applied Biosystems 7900 HT fast real-time PCR system. Assays were performed in triplicate. In presenting the data, we plotted the relative expression of ORF50ΔSTAD versus DOX concentration, using both a log scale to better visualize the low concentrations and a linear scale to better visualize the higher concentrations. A Gompertz four-component function was fitted to the values using Stata 11 (Stata Corporation).
Total DNA and protected KSHV DNA isolation.
Protected KSHV DNA was quantitated using a previously described method (25). Briefly, supernatant from each condition was centrifuged at 5,000 × g for 5 min to pellet cellular debris. The clear supernatant was then treated for 10 min with RQ DNase I (Promega, Madison, WI) at a final concentration of 100 U/ml and incubated at 37°C for 1 h to digest free DNA. The samples were then treated with 0.5 M EDTA (final concentration of 10 mM) and incubated at 65°C for 30 min to inactivate DNase. The samples were further treated with 10% SDS (final concentration of 0.5%) and proteinase K (final concentration of 200 μg/ml) and incubated at 65°C for 2 h to disrupt the envelope and digest protein capsid. These samples were extracted with phenol-chloroform-isoamyl alcohol (25:24:1) (Invitrogen), precipitated with ethanol, dried, and dissolved in Tris-EDTA (TE).
Quantitation of total and protected KSHV DNA.
Total and protected KSHV DNAs, isolated as described above, were quantitated using a TaqMan real-time PCR assay for sequences in ORF57, as previously described (25). KSHV ORF57 TaqMan probe 57TM (5′-6FAM-AGAAACCGCAGCCGCCGGAG-TAMRA-3′) forward primer 57RT-F5 (5′-TTTGTGACCAGCCGCCATAATCAAG-3′), and reverse primer 57RT-B5 (5′-TCATTTGTTCCTCCACGAAAGCCCC-3′) were used. Real-time PCR assays were performed using the Applied Biosystems 7900 HT fast real-Time PCR system. Assays were performed in triplicate.
Induction and determination of apoptosis.
BCBL-1 or ORF50ΔSTAD BCBL-1 cells were seeded overnight at a concentration of 0.25 × 106 cells/ml. Apoptosis was induced by adding 2[[3-(2,3-dichlorophenoxy)propyl]amino]ethanol (DCPE) (51, 53) at a final concentration of 50 nM. In some experiments, KSHV replication was also induced using TPA, and/or ORF50ΔSTAD expression was induced with DOX at a low concentration.
The cells were harvested at the indicated times by centrifugation (∼150 × g), after assessment of cell number and viability. Supernatants were flash frozen and stored at −80°C for additional assays, including assays for released virions. Cell pellets were washed with cold phosphate-buffered saline (PBS) (without calcium and magnesium) at pH 7.4 and either analyzed using flow cytometry or flash frozen and stored at −80°C and subsequently assayed for KSHV viral RNA and total KSHV viral DNA.
Apoptosis was assessed using the annexin V01 kit (Caltag, Invitrogen) to determine cell surface phosphatidylserine with flow cytometry. In these experiments, 106 cells from each treatment condition were resuspended in 100 μl of 1× binding buffer, followed by the addition of fluorescein-conjugated annexin V and propidium iodide (Annex-PI [50 μg/ml] in 1× PBS). The cells were incubated for 15 min at room temperature (RT) and analyzed by flow cytometry (Becton Dickinson FACSCalibur; BD Biosciences). Untreated cells were used to establish forward- and side-scatter gates for compensation baselines. Data were analyzed using FlowJo 8.5.2 software (FlowJo, Ashland, OR).
Immunofluorescence assays for the KSHV late gene ORF K8.1A protein.
Detection of KSHV late gene ORF K8.1A protein expression was performed by immunofluorescence using a confocal Zeiss LSM 510 microscope. Coverslips in 24-well plates were coated with poly-l-lysine and incubated at room temperature overnight. Cells from each treatment condition (∼0.5 × 106 to 1.0 × 106 cells) were pelleted and then resuspended in RPMI 1640 medium. The cells were transferred to coated coverslips in 24-well plates and allowed to settle for 15 min, washed with PBS, and fixed with 4% paraformaldehyde at 4°C for 30 min. The cells were washed twice with PBS and permeabilized with PBS containing 0.1% Triton X-100 (PBS–0.1% Triton X-100) at room temperature for 10 min. The cells were washed twice with PBS, blocked with PBS containing 5% bovine serum albumin (PBS–5% BSA), and then incubated at room temperature for 1 h. The cells were washed twice with PBS and incubated with mouse monoclonal anti-KSHV ORF K8.1A antibody (Advanced Biotechnologies Inc.) at a 1:400 dilution in PBS and incubated for 2 h at 4°C. The cells were washed twice with PBS and incubated with goat anti-mouse secondary antibody conjugated to Alexa Fluor 647 (Invitrogen) at a 1:1,000 dilution in PBS. The coverslips were mounted on slides with Prolong Gold antifade reagent with 4′,6′-diamidino-2-phenylindole (DAPI) (Invitrogen). A magnification of ×40 (oil) was used to acquire all images. Excitation wavelengths of 400 nm and 650 nm were used.
Inhibition of apoptosis and KSHV replication by caspase inhibitors.
ORF50ΔSTAD BCBL-1 cells were treated with different concentrations of caspase inhibitors to examine their inhibitory effects on apoptosis and KSHV replication. The general caspase inhibitor Z-Val-Ala-Asp-fluoromethylketone (Z-VAD-FMK) (41) (Enzo Life Sciences), caspase-3 inhibitor acetyl (Ac)-AAVALLPAVLLAPDGVD-CHO, caspase-6 inhibitor Ac-VEID-CHO, caspase-9 inhibitor Ac-LEHD-CHO were reconstituted individually in dimethyl sulfoxide (DMSO) (Fisher Scientific) to a stock concentration of 10 mM. The cells were seeded in 6-well plates at a density of 0.25 × 106 cells/ml. The cells were treated with caspase inhibitors at concentrations ranging from 0 μM to 150 μM and incubated for 12 to 16 h. This was followed by induction of apoptosis with DCPE (50 nM) and/or treatment with TPA (20 ng/ml) to induce KSHV replication conventionally. The cells were harvested after 24 h. Supernatants were assayed for protected KSHV DNA. Cell pellets were washed with PBS and stained to detect annexin V- and PI-positive cells (apoptosis kit; BD Biosciences) and assayed by flow cytometry.
Transfection of ORF50 ΔSTAD BCBL-1 cells with a plasmid expressing a caspase-3–GFP fusion protein and assays for apoptosis and KSHV late gene K8.1 using confocal microscopy.
We transfected ORF50ΔSTAD BCBL-1 cells with a plasmid, pcasp3-Wt-GFP, that expresses functional wild-type caspase-3 fused to green fluorescent protein (GFP), and pUC19 as a negative control. The pcasp3-Wt-GFP plasmid was a kind gift from Shinji Kamada, Biosignal Research Center, Kobe University (18). APC Annexin V (BD Pharminogen) was used to detect apoptosis. Monoclonal mouse anti-KSHV ORF K8.1 and goat anti-mouse antibody labeled with peridinin chlorophyll protein (goat anti-mouse-PerCP) (Advanced Biotechnologies Inc.) were used to detect KSHV late gene expression. ORF50ΔSTAD BCBL-1 cells were maintained at a density of 0.25 × 106 cells/ml and seeded in RPMI 1640 medium without FBS in 24-well plates the night prior to transfection. Doxycycline (0.1 μg/ml) was added to selected wells at the time of seeding. Two hours prior to transfection, the medium was replaced by RPMI 1640 medium with no FBS and no antibiotics. Lipofectamine 2000 was mixed with Opt Mem I medium (Invitrogen) at a 1:25 dilution and kept for 10 min at room temperature. Lipofectamine 2000 complexed with plasmid DNA was prepared by taking 500 ng plasmid DNA and mixing it with diluted Lipofectamine 2000 at a 1:1 ratio, and the mixture was kept at room temperature for 30 min. Two hundred microliters of Lipofectamine 2000–plasmid DNA complex was added to each well. The plate was gently rocked at 37°C for 5 h. TPA (final concentration of 20 ng/ml) was added to selected wells and incubated for 1 h, followed by careful removal of supernatant from wells and replenishing with fresh medium without FBS and antibiotics. DCPE (final concentration of 50 nM) was added to indicated samples.
Coverslips coated with poly-l-lysine (Sigma) were prepared 2 days prior to confocal imaging. After 24 h of incubation posttransfection, cells were washed and resuspended in RPMI 1640 medium and transferred to dried poly-l-lysine coverslips in 24-well plates. The cells were allowed to settle for 15 min at room temperature, followed by two gentle washes with 1× PBS. The cells on the coverslips were fixed with 400 μl of 4% paraformaldehyde (Sigma) and incubated at 4°C for 30 min, followed by two washes with PBS. Cell permeabilization was accomplished by adding PBS–0.1% Triton X-100 and incubating at RT for 10 min, followed by a wash with 1× PBS. The cells were blocked with PBS–5% BSA with incubation for 1 h, followed by a wash with 1× PBS. Staining was done with monoclonal mouse anti-KSHV ORF K8.1 (1:500 in PBS–1.5% BSA) followed by incubation at 4°C for 5 h. This was followed by washing with 1× PBS and staining with goat anti-mouse-PerCP (1:1,000 in PBS–1.5% BSA) for 1.5 h at room temperature in the dark. The supernatants were carefully removed, and 200 μl of 1× binding buffer was added to each well, followed by 10 μl of APC Annexin V in each well and incubated in the dark for 15 min. Individual coverslips were mounted by adding a drop of ProLong Gold antifade reagent with DAPI (Invitrogen) to individual glass slides. The mounted coverslips on individual glass slides were kept at room temperature in the dark overnight and sealed with a thin coat of clear nail polish around the edges. A Zeiss LSM510 single photon laser confocal microscope was used for taking the images. All images were taken at an (oil) objective lens magnification of ×40, 1-arbitrary-unit (AU) pinhole, and a gain set at 572 nm. The excitation and emission wavelengths for GFP were 490 nm and 510 nm, respectively, while for DAPI, they were 360 nm and 460 nm, respectively. The excitation and emission wavelengths for APC were 650 nm and 660 nm, respectively, and for PerCP, they were 482 nm and 678 nm, respectively.
Knockdown of KSHV ORF50 expression.
siRNA against KSHV ORF50 (5′-GCAGAGAACACCGGAGATT-3′) was designed using Block-iT RNAi Designer (Invitrogen). The sequence starts at position 1180. The cells were transfected with siRNAs into 1 × 106 to 2 × 106 cells and maintained in six-well plates. For each transfection, 1 × 106 to 2 ×106 cells/well were resuspended in 100 μl of Nucleofector solution V using protocols for nucleofection of BCBL-1 cells provided by Lonza to which siRNA was added (2.5 μM). As a control, SiGenome nontargeting siRNA pool 1 (Thermo Scientific Dharmacon, Lafayette, CO) was used. The cells and siRNA were transferred to individual Amaxa transfector cuvettes and transfected using Amaxa program T-001 in the Amaxa nucleofector (Amaxa, Lonza). Transfected cells were placed in individual wells for 15 min. The cells were collected and resuspended in RPMI 1640 medium containing 10% FBS for 3 h at 37°C. In some experiments, cells were treated with TPA (10 ng/ml) and incubated for 15 min. In some experiments, cells were treated with DCPE. The cells were harvested after 24 h and assayed for viral RNA using the TaqMan real-time RT-PCR assay and protein by immunoblotting.
Determination of ORF50/RTA and ORF50ΔSTAD protein expression.
Total protein was quantitated using a bicinchoninic acid (BCA) protein assay (Pierce, Thermo Fisher Scientific). Specific detection of ORF50 and ORF50ΔSTAD proteins was performed by immunoblotting. Approximately 30 μg of protein per sample was loaded on 4 to 12% Bis-Tris gel (NuPAGE, Invitrogen). Samples were prepared using Bio-Rad Laemmli buffer (with β-mercaptoethanol) and heated at 90°C for 10 min. The gels were run using NuPAGE morpholinepropanesulfonic acid (MOPS) SDS (1×) running buffer at a constant voltage of 80 V for 4 h. SeeBlue Plus2 prestained standards (Invitrogen) were used as markers. Transfer to nitrocellulose membranes (Bio-Rad Laboratories) was performed using Tris-glycine-SDS buffer at 90 V for 1 h at 4°C with constant stirring of the buffer. The quality of the transfer was tested by staining the nitrocellulose membrane with Ponceau S stain. The membranes were rinsed with deionized water and blocked in 1× PBS–0.1% Tween 20–5% nonfat dry milk overnight. The membranes were incubated with rabbit polyclonal antipeptide antibody produced against an N-terminal ORF50 sequence, MAQDDKGKKLRRSC (ProSci, Poway, CA) that recognizes both full-length ORF50 and the C-terminus-deleted ORF50ΔSTAD mutant overnight at 4°C. The membranes were washed with blocking buffer and incubated in goat anti-rabbit antibody conjugated to horseradish peroxidase (goat anti-rabbit-HRP) secondary antibody (Abcam) at 1:2,000 dilution (in PBS–0.1% Tween 20–1.5% nonfat dry milk) for 1 h at room temperature with constant shaking. The membranes were washed three times with blocking buffer, followed by washing with 1× PBS–0.1% Tween 20 twice for 15 min. Enhanced chemiluminescence (ECL) detection was performed using Western Lightning Plus-ECL (Perkin Elmer, Waltham, MA). The membranes were reprobed for actin using goat anti-actin (1:1,000) and donkey anti-goat-HRP (1:2,000) (Abcam) following the procedure described above. The films were scanned, and the resulting digital images were analyzed and quantitated using ImageJ (http://rsbweb.nih.gov/ij/).
Quantitative RT-PCR assays for a KSHV early gene (ORF57) and late gene (ORF17).
TaqMan real-time RT-PCR assays for ORF57 and ORF17 were used as previously described (25). The following primers and probes were used to detect ORF57 and ORF17, following the reverse transcription-PCR assay described above. To detect KSHV ORF57, the KSHV ORF57 TaqMan probe 57TM (5′-6FAM-AGAAACCGCAGCCGCCGGAG-TAMRA-3′), forward primer 57RT-F5 (5′-TTTGTGACCAGCCGCCATAATCAAG-3′), and reverse primer 57RT-B5 (5′-TCATTTGTTCCTCCACGAAAGCCCC-3′) were used as previously described (25). To detect KSHV ORF17, the TaqMan TAMRA probe (5′-6FAM-TTCTGCACCGGAGCCATCACGTC) and forward and reverse primers (forward, 5′-TGGGCTGGACACTGGGTCTATTTC-3′; reverse, 5′-AGATTTTTCACGGGGGCTCTGG-3′) were used. Results were normalized to the results of a real-time RT-PCR assay for GAPDH using a commercially available assay, the Hu GAPDH assay kit (Applied Biosystems). Assays were performed, analyzed, and normalized as described above.
Determination of KSHV virion infectivity.
HUVEC were maintained at 70 to 80% confluence and seeded at ∼50,000 cells/well for the assay. Prior to inoculation, HUVEC were treated with TPA (final concentration of 20 ng/ml) and incubated for 30 min, after which they were washed with prewarmed EBM medium (Lonza) and replenished with fresh medium. HUVEC were then inoculated with KSHV. Approximately 200 μl of medium containing 106 copies of protected KSHV DNA was added to the cells. After 10 h, the cells were washed three times with EBM medium. Washes after the first wash did not contain any detectable KSHV as assayed using PCR. After infection, the HUVEC were incubated for a total of 48 h, after which supernatants and cells were harvested. The cells showed no evidence of toxicity following exposure to the viral inocula during the course of the experiment. Supernatants from each condition were then assayed for protected KSHV DNA as described above.
RESULTS
Production and characterization of a cell line expressing the dominant-negative RTA mutant ORF50ΔSTAD.
To better understand RTA's contribution to KSHV replication, we produced, using the well-described TREx BCBL-1 cell and the Invitrogen Flp-In T-REx system (31), a derivative of the BCBL-1 PEL cell line, ORF50ΔSTAD BCBCL-1, which contains both latent copies of KSHV and a tetracycline-inducible version of the ORF50ΔSTAD DN mutant.
After we produced the ORF50ΔSTAD BCBCL-1 cell line, first we assessed the induction of expression of ORF50ΔSTAD RNA. We conducted a doxycycline (DOX) titration, with concentrations ranging from 0.001 μg/ml to 5.0 μg/ml, and used real-time RT-PCR to assay for ORF50ΔSTAD (Fig. 1). We found that as the DOX concentration increased, from 0.001 to 1.0 μg/ml (Fig. 1A and B), ORF50ΔSTAD RNA expression steadily increased. At higher DOX concentrations, beyond 1.0 μg/ml, ORF50ΔSTAD RNA expression plateaued (Fig. 1A). Thus, DOX induced ORF50ΔSTAD RNA expression in ORF50ΔSTAD BCBL-1 cells, but concentrations beyond ∼1.0 μg/ml did not lead to further increases in ORF50ΔSTAD expression. This observation is consistent with the stated performance characteristics of the Invitrogen Flp-In T-REx system (16). We also determined the expression of ORF50ΔSTAD RNA after treatment with TPA. Additional direct comparisons of the expression of ORF50ΔSTAD and FL ORF50 RNA and protein following several different treatments are also described below and shown below in Fig. 4. We found that in the presence of TPA, the expression of ORF50ΔSTAD increased and plateaued after DOX concentrations of 0.01 μg/ml. The results show that DOX induces ORF50ΔSTAD expression in both uninduced and TPA-induced ORF50ΔSTAD BCBCL-1 cells, that quite low concentrations of DOX induce high levels of ORF50ΔSTAD RNA expression, and that higher concentrations of DOX (beyond 0.1 to 0.5 μg/ml) do not induce further increases in ORF50ΔSTAD RNA expression. In the presence of TPA, even the lowest levels of DOX induction of DN ORF50ΔSTAD block the expression of wild-type full-length ORF50 (Fig. 1C, squares), as expected.
Fig 1.

Expression of ORF50ΔSTAD in ORF50ΔSTAD BCBL-1 cells treated with DOX. (A) Expression of ORF50ΔSTAD RNA as a function of the concentration of DOX in ORF50ΔSTAD BCBL-1 cells. The ORF50ΔSTAD BCBL-1 cells have both latent copies of KSHV, including a wild-type ORF50, and a DOX-inducible ORF50ΔSTAD gene engineered into the cell line using the Invitrogen Flp-In T-REx system. The DOX concentrations are shown in micrograms per milliliter. In panel A, the data are plotted on a linear scale for DOX concentration. In panel B, the data are plotted on a logarithmic scale for DOX concentration to enhance visibility of the lower concentration data points. As the DOX concentration increases, ORF50ΔSTAD RNA expression reaches a plateau at a DOX concentration of about 0.5 μg/ml, with no subsequent increases in ORF50ΔSTAD RNA expression at higher DOX concentrations. ORF50ΔSTAD was determined using a real-time RT-PCR assay, normalized to an internal GAPDH standard. (C) Expression of ORF50ΔSTAD RNA ORF50ΔSTAD BCBL-1 cells treated with TPA. ORF50ΔSTAD expression (circles) reaches a plateau between 0.01 μg/ml and 0.1 μg/ml. Further increases in DOX concentration do not lead to increases in expression of ORF50ΔSTAD RNA. Expression of full-length wild-type ORF50 RNA (squares) does not increase in the TPA-treated cells when the DN ORF50ΔSTAD is induced with even the lowest tested concentrations of DOX, confirming that the DN ORF50ΔSTAD can block ORF50 expression.
Fig 4.

Expression of wild-type full-length (FL) ORF50 and truncated DN ORF50ΔSTAD after treatment with inducers of ORF50 and ORF50ΔSTAD expression and inducers of apoptosis. ORF50ΔSTAD BCBL-1 cells were treated with TPA to induce ORF50 expression and viral replication via the normal pathway, with a low concentration of DOX (0.1 μg/ml) to induce ORF50ΔSTAD expression, with DCPE to induce apoptosis, and with various combinations of the agents. (A) Expression of full-length wild-type ORF50 and ORF50ΔSTAD RNA, measured by real-time RT-PCR assays. (B) Expression of ORF50 and ORF50ΔSTAD proteins, assessed using immunoblots employing an antipeptide antibody made against an amino-terminal ORF50 peptide. TPA induces expression of FL ORF50 RNA and protein. DOX induces expression of ORF50ΔSTAD RNA and protein. When ORF50ΔSTAD is present, the expression of FL ORF50 is blocked at both the RNA and protein level. The immunoblot in panel B was deliberately overexposed to show that there is a very small amount of residual FL ORF50 expression in the cells prior to induction, slightly visible in the untreated and DCPE-only lanes. BCBL-1 cells spontaneously experience a small amount of lytic KSHV replication even without treatment with an inducing agent, which is suppressed when ORF50ΔSTAD expression is induced with DOX. (Bottom) The blot was reprobed with an antiactin monoclonal antibody (the antiactin-probed blot is a much shorter exposure).
Lukac et al. have shown that expression of the DN ORF50ΔSTAD completely blocks KSHV replication in other experimental systems (27). We assessed the effect of ORF50ΔSTAD on KSHV replication in the ORF50ΔSTAD BCBCL-1 cells with a real-time PCR assay (25) to quantitate virions (Fig. 2). We treated the ORF50ΔSTAD BCBL-1 cells with TPA to induce KSHV replication and with increasing amounts of DOX (0.001 to 5 μg/ml) to induce ORF50ΔSTAD expression (Fig. 2A). Induction of DN ORF50ΔSTAD by low DOX concentrations completely blocked viral replication triggered by TPA, as expected. However, at higher concentrations of DOX (5 μg/ml), the cells produced large amounts of virus, even though the DN ORF50ΔSTAD was induced with DOX, an unexpected result. The amount of ORF50ΔSTAD present at 5 μg/ml was equivalent to those seen with lower levels of DOX, at which KSHV replication was blocked (Fig. 1). When we examined the cells, we noted that at higher DOX concentrations, the cells showed significant cytotoxicity, as judged by trypan blue exclusion (Fig. 2B). The cells also expressed markers of apoptosis (Fig. 2C), staining positive for annexin V and propidium iodide. Previous studies (24) showed that high concentrations of tetracyclines trigger apoptosis through a Fas-FasL-like pathway. These results indicated that in our cells, high DOX concentrations triggered apoptosis, with the production of large amounts of KSHV virions, even in the presence of DN ORF50ΔSTAD sufficient to block KSHV replication after treatment with TPA. The results suggested that host cell apoptosis was associated with induction of KSHV replication via a process that did not require RTA, a factor previously considered essential for KSHV replication.
Fig 2.

KSHV production and cell viability in ORF50ΔSTAD BCBL-1 cells treated with increasing DOX. (A) KSHV virions, assayed as protected KSHV DNA. TPA induces KSHV virion production in the absence of DOX. With DOX at low concentrations, induction of ORF50ΔSTAD blocks KSHV virion production. However, at high DOX concentrations (≥5 μg/ml), DOX not only fails to block KSHV virion production triggered by TPA treatment but also is associated with the induction of high levels of KSHV virion production. (B) High DOX concentrations (≥5 μg/ml) are associated with a decrease in cell viability as measured by trypan blue exclusion. (C) The cells were examined with flow cytometry for apoptotic markers using propidium iodide (PI) and annexin V (Ann). At high DOX (≥5 μg/ml), most cells scored positive for PI and Ann staining, indicating apoptosis. Ann-/PI-, no staining for either annexin V or propidium iodide; Ann+, positive staining for annexin V alone; PI+, positive staining for propidium iodide alone; Ann+/PI+, positive staining for both annexin V and propidium iodide.
Apoptosis induction of KSHV replication.
To assess whether ORF50-independent KSHV replication was triggered by general features of apoptosis or by some process unique to DOX treatment or to the extrinsic apoptotic pathway and to conduct conceptually simpler experiments that employed different agents to trigger apoptosis and induce expression of the DN ORF50ΔSTAD, we used a different apoptosis inducer, 2[[3-(2,3-dichlorophenoxy) propyl] amino] ethanol (DCPE), which acts via the intrinsic pathway (51, 53). We first assessed DCPE's ability to induce apoptosis with flow cytometry (Fig. 3). We found that DCPE triggered apoptosis, with increasing numbers of apoptotic cells observed over time. TPA also had slight cytotoxic effects, presumably due to induction of KSHV replication. Expression of ORF50ΔSTAD, induced by low concentrations of DOX, blunted TPA's cytotoxic effect in the ORF50ΔSTAD BCBL-1 cells, but not in the normal BCBL-1 cells, presumably because a substantial portion of the cytotoxic effect resulted from KSHV replication, which was prevented when the DN ORF50ΔSTAD was induced by DOX in the ORF50ΔSTAD BCBL-1 cells. DOX-induced expression of ORF50ΔSTAD did not protect the cells against the apoptotic effects of DCPE. We studied both the ORF50ΔSTAD BCBL-1 cell line and the parental BCBL-1 cell line, and both cell types showed similar patterns of apoptosis following treatment with TPA, DCPE, and a low concentration of DOX with the exception of the slight protective effect observed in the ORF50ΔSTAD BCBL-1 cells when the cells were treated with TPA and DOX.
Fig 3.

Induction of apoptosis in BCBL-1 and ORF50ΔSTAD BCBL-1 cells by DCPE. BCBL-1 and ORF50ΔSTAD BCBL-1 cells were examined at 0 h and at 24 h and 48 h after treatment with DOX (a low concentration [0.1 μg/ml]), TPA, and DCPE, and combinations of the agents, as indicated. DCPE had significant cytotoxic effects in both cell lines. TPA treatment was associated with mildly cytotoxic effects in both cell lines. However, in the ORF50ΔSTAD BCBL-1 cells, the toxic effects of TPA, which was likely due to induction of viral replication, was inhibited by the presence of ORF50ΔSTAD induced by DOX, which blocks viral replication.
Since DCPE appeared to effectively induce apoptosis in both the parental BCBL-1 and ORF50ΔSTAD BCBL-1 cell lines, we sought to confirm that the DN ORF50ΔSTAD was expressed upon treatment with DOX when apoptosis was induced with DCPE (Fig. 4). We performed real-time RT-PCR assays for wild-type full-length (FL) and truncated ORF50ΔSTAD RNA (Fig. 4A) and immunoblots employing a rabbit polyclonal antipeptide antiserum made against a peptide from the N-terminal region of ORF50, which can detect both the FL wild-type ORF50 and truncated ORF50ΔSTAD proteins (Fig. 4B). The observations confirmed the following. (i) TPA induces FL ORF50 expression in both normal BCBL-1 cells and ORF50ΔSTAD BCBL-1 cells. (ii) A low concentration of DOX induces expression of ORF50ΔSTAD, but not FL ORF50. (iii) ORF50ΔSTAD expression induced by a low concentration of DOX blocks FL ORF50 expression after TPA treatment. (iv) DCPE treatment induces neither FL ORF50 nor DN ORF50ΔSTAD expression at either the RNA or protein level.
To determine whether the apoptotic inducer DCPE triggers KSHV replication in the parental BCBL-1 cells and in the ORF50ΔSTAD BCBL-1 cells, we treated the cells with DCPE, with and without a low concentration of DOX to induce expression of DN ORF50ΔSTAD, and assayed for KSHV total DNA (Fig. 5A and B) and DNA protected in virions (Fig. 5C and D). We assayed for both total KSHV DNA and KSHV DNA protected in virions to obviate any concerns that increases in KSHV virions observed following treatment with apoptosis inducers might just be due to enhanced release of KSHV virions by dying cells. We observed that TPA and DCPE triggered KSHV replication in both BCBL-1 cells and ORF50ΔSTAD BCBL-1 cells. Observing increases in both total (Fig. 5A and C) and protected (Fig. 5B and D) KSHV DNA makes it unlikely that the increases seen with DCPE result merely from cells undergoing apoptosis releasing more viruses. The steady, time-dependent increase in both total and protected KSHV DNA suggests ongoing viral replication. Induction of ORF50ΔSTAD expression by a low concentration of DOX blocked total and protected KSHV DNA production in ORF50ΔSTAD BCBL-1 cells treated with TPA alone. DOX had no effect on KSHV total and protected KSHV DNA in normal BCBL-1 cells treated with TPA, since those cells cannot express the DN ORF50ΔSTAD. When the BCBL-1 cells and ORF50ΔSTAD BCBL-1 cells were treated with DCPE, they also produced large amounts of KSHV total DNA and DNA in virions, with the ORF50ΔSTAD BCBL-1 cells producing large amounts of KSHV total DNA and DNA in virions upon DCPE treatment, even when the cells were treated with a low concentration of DOX to induce ORF50ΔSTAD expression. This suggested that DN ORF50ΔSTAD could not block KSHV replication when KSHV replication was induced in association with apoptosis.
Fig 5.

Effects of TPA, apoptosis triggered by DCPE, and induction of the ORF50ΔSTAD expression by a low concentration of DOX on KSHV DNA and virion production in BCBL-1 cells and ORF50ΔSTAD BCBL-1 cells. BCBL-1 and ORF50ΔSTAD BCBL-1 cells were subjected to several treatments: TPA to induce FL ORF50 expression and viral replication via the normal pathway, DOX (a low concentration of 0.1 μg/ml) to induce ORF50ΔSTAD expression, and DCPE to induce apoptosis, and combinations of the agents. Production of total and protected KSHV DNA was measured using a real-time PCR assay (25). (A) KSHV total DNA in BCBL-1 cells. (B) KSHV total DNA in ORF50ΔSTAD BCBL-1 cells. (C) KSHV virion protected DNA in BCBL-1 cells. (D) KSHV virion protected DNA in ORF50ΔSTAD BCBL-1 cells. (E) Effects of low concentrations of DOX on KSHV replication in ORF50ΔSTAD BCBL-1 cells. The levels of ORF50ΔSTAD induced by very low concentrations of DOX (0.001 μg/ml) completely suppress KSHV replication induced by TPA. (The levels of KSHV replication are below the baseline level for samples from cells treated with a low concentration of DOX, because ORF50ΔSTAD blocks the small amount of spontaneous KSHV replication that occurs in the cells.) TPA induces KSHV replication, and TPA-induced KSHV replication is blocked by ORF50ΔSTAD. DCPE also induces KSHV replication, but DCPE-induced KSHV replication is not blocked by ORF50ΔSTAD.
Even low ORF50ΔSTAD expression, induced by DOX concentrations as low as 0.001 μg/ml (Fig. 1 and 5E) was enough to block viral replication induced by TPA. Therefore, the approximately 4-fold-more ORF50ΔSTAD expression induced by 0.1 μg/ml DOX, the standard low concentration of DOX used to induce the ORF50ΔSTAD expression in these experiments, was far in excess of that required to block KSHV replication initiated via DCPE-triggered apoptosis. Since as much or more virus was produced after DCPE treatment as after TPA treatment, it is unlikely that virus made after apoptotic induction was produced in only a small number of cells, since there is a limit on the per cell production of virions. These results further validated our basic observation that KSHV has a new replication pathway, which is triggered in association with host cell and does not require RTA, which is required for normal lytic replication.
Uniformity of induction of KSHV replication associated with apoptosis.
While it is clear that DCPE induces both apoptosis and KSHV replication and gene expression (Fig. 3, 4, 5, 8, and 10), we wanted to clearly show that the virus produced following induction of apoptosis was not derived from just a small minority of the latently infected cells, but instead from the great majority of the cells. To determine whether apoptosis induced KSHV replication in most of the latently infected BCBL-1 cells or just a few cells after treatment with an apoptosis inducer, we treated BCBL-1 cells either with TPA as a positive control for induction of KSHV replication or with DCPE to induce apoptosis and then examined the cells for the expression of the KSHV late protein ORF K8.1A (50, 52) using anti-ORF K8.1A monoclonal antibodies and immunofluorescence confocal microscopy (Fig. 6). We found that both TPA and DCPE powerfully induced the expression of ORF K8.1A in most of the cells in the culture. Hence, the induction of KSHV replication by apoptosis did not occur in only a small minority of the cells.
Fig 8.

Effects of the caspase inhibitors on apoptosis induced by DCPE and KSHV replication induced by treatment with DCPE. The effects of increasing concentrations of the general caspase inhibitor Z-VAD-FMK, the caspase-9 inhibitor Ac-LEHD-CHO, the caspase-6 and caspase-8 inhibitor Ac-VEID-CHO, and the caspase-3 inhibitor Ac-AAVALLPAVLLALLAPDGVD-CHO on cell apoptosis, as measured by flow cytometric assays for Annexin V and propidium iodide and the production of KSHV virions as protected KSHV DNA. Control treatments included cells not treated with any inducer (Untreated), in which the cells were treated with DMSO, the vehicle used to dissolve DCPE, or cells treated with TPA (as a positive control for the induction of viral replication). Four-component Gompertz plots were fitted to the dose-response data (Stata 11), except for the data obtained when the cells were treated with the caspase-6 inhibitor Ac-VEID-CHO, since no activity was observed for this inhibitor. The general Z-VAD-FMK and specific caspase-9 and caspase-3 inhibitors had similar dose-response curves for inhibition of apoptosis and viral replication. Inhibition of the end effector caspase-3 appears to be sufficient to block KSHV replication triggered in association with apoptosis.
Fig 10.

Effects of silencing ORF50 on KSHV replication. (A) ORF50 silencing in BCBL-1 cells treated with TPA and DCPE to induce KSHV replication. BCBL-1 cells were transfected with an ORF50 siRNA and control GAPDH siRNA and then treated with TPA or DCPE to induce KSHV replication. RNAs were assayed for ORF50 RNA at 24 h via a TaqMan real-time RT-PCR assay (results normalized to actin control and a nontargeting siRNA pool). Transfection of ORF50 siRNA resulted in a decrease in ORF50 RNA expression in both cells treated with TPA (86% decrease) and DCPE (93% decrease). (B) ORF50 protein levels in normal BCBL-1 cells after induction by TPA and DCPE and ORF50 silencing. ORF50 siRNA knocks down expression of ORF50 when cells are treated with both TPA and DCPE. (Note that the DCPE lanes were exposed longer, because TPA is a potent inducer of ORF50 expression, while DCPE is not). (C) Effects on KSHV replication of silencing ORF50 in BCBL-1 cells induced with TPA or DCPE. Cells were transfected with siRNA and induced with either TPA or DCPE and assayed for KSHV virions using a real-time PCR assay for protected KSHV DNA after 48 h. Silencing ORF50 led to a statistically significant decrease in virus production when cells were induced with TPA (P = 0.004 [indicated by an asterisk in the figure]), but no statistically significant decrease when ORF50 was silenced in DCPE-treated cells.
Fig 6.

KSHV protein expression in cells treated with TPA or the apoptosis inducer DCPE. ORF50ΔSTAD BCBL-1 cells were treated either with TPA to induce KSHV replication via the conventional pathway or with DCPE to induce apoptosis or left untreated. The cells were studied with confocal immunofluorescence microscopy using as the primary antibody a monoclonal antibody against KSHV ORF K8.1A, a well-characterized KSHV late gene whose product is associated with the plasma membrane (red – ORF K8.1 detected using an anti-ORF K8.1A mouse monoclonal antibody and an Alexa Fluor 687-conjugated goat anti-mouse secondary antibody). Cells were counterstained with DAPI (blue). Both positive-control TPA treatment and treatment with the apoptosis inducer DCPE resulted in high levels of expression of ORF K8.1A in essentially all of the treated cells.
Caspase inhibitor inhibition of KSHV replication following treatment with apoptosis inducers.
Since the observation that apoptosis appeared to trigger KSHV replication was unanticipated, we wanted to confirm the association between apoptosis and the induction of viral replication using an additional approach. Furthermore, since one plausible mechanism for the virus to sense host cell apoptosis could be cleavage of a viral or host cell protein by a caspase, we performed experiments using general and specific caspase inhibitors to determine whether there was an association between caspase activity and the induction of KSHV replication. We employed the general caspase inhibitor Z-VAD-FMK (41), the caspase-9 inhibitor Ac-LEHD-CHO, the caspase-6 and caspase-8 inhibitor Ac-VEID-CHO, and the caspase-3 inhibitor Ac-AAVALLPAVLLALLAPDGVD-CHO. See Fig. 7 for a diagram of the apoptotic pathways and the inhibitors used in this study. We induced apoptosis with DCPE, as described above, in the presence of different concentrations of the caspase inhibitors. We assessed apoptosis using flow cytometric assays for annexin V and propidium iodide and real-time PCR assays for KSHV virions (Fig. 8). We found that there was a clear dose-response relationship between the concentration of the general caspase inhibitor Z-VAD-FMK and both the inhibition of apoptosis and inhibition of KSHV replication. Both processes showed half-maximal inhibition at about 75 μM, suggesting that these processes may be closely linked.
Fig 7.

Schematic diagram of the apoptotic cascade and the sites of action of the general and specific caspase inhibitors used in this study. The figure illustrates some key features of the apoptotic cascade, highlighting sites of action of the caspase inhibitors used in this study (⊣). Abbreviations: TRAIL, tumor necrosis factor-related apoptosis-inducing ligand; TRAIL-R, TRAIL receptor; TNFα, tumor necrosis factor alpha; TNF-R1, tumor necrosis factor receptor 1; IL-1, interleukin-1; IL-1R, IL-1 receptor; FADD, Fas-associated death domain protein; TRADD, TNF receptor-associated death domain; CASP8, caspase-8; PARP, poly(ADP) ribose polymerase; EndoG, endonuclease G; CytC, cytochrome c; ICE, mammalian interleukin-1 beta-converting enzyme; IRAK, interleukin-1 receptor-associated kinase; FLICE, FADD-like ICE; FLIP, FLICE-inhibitory protein; AIF, apoptosis-inducing factor; DFF40, DNA fragmentation factor, 40 kDa; CAD, caspase-activated DNase; IAP, inhibitor of apoptosis; TRAF2, TNF receptor-associated factor 2; MyD88, myeloid differentiation primary response gene (88); tBID, truncated BID; BH3, interactive domain death agonist.
When we examined more-specific caspase inhibitors, we found that the caspase-9 inhibitor Ac-LEHD-CHO and the caspase-3 inhibitor Ac-AAVALLPAVLLALLAPDGVD-CHO produced both an inhibition of apoptosis and KSHV replication, with similar activities. In contrast, the caspase-6 and -8 inhibitor Ac-VEID-CHO inhibited neither apoptosis nor KSHV replication, even at high concentration (150 μM). This finding was as expected, since caspase-6 and -8 are not in the intrinsic pathway activated by DCPE, but the result does provide further evidence of specificity. Overall, the data suggest that the activation of caspase-3, the end effector caspase, is sufficient for the activation of KSHV replication in association with apoptosis.
Direct activation of KSHV replication by caspase-3.
Since the experiments employing caspase inhibitors suggested that caspase-3 was necessary for the induction of KSHV replication in association with apoptosis, we conducted additional experiments to determine whether caspase-3 was sufficient to activate KSHV and induce apoptosis in the PEL cells. We transfected ORF50ΔSTAD BCBL-1 cells with pUC19 as a negative control, together with the inducer of viral replication, TPA, DOX to induce expression of the DN ORF50ΔSTAD, and DCPE to directly induce apoptosis in the cells in various combinations, and a plasmid, pcasp3-Wt-GFP, produced by Kamada et al. (18), which expresses wild-type, functional caspase-3 as a fusion protein with GFP, and used confocal microscopy to assess apoptosis using staining for annexin V and antibodies to the KSHV late gene ORF K8.1, as well as imaging the GFP signal to confirm transfection with the pcasp3-Wt-GFP (Fig. 9). In these experiments, the cell nuclei were stained with DAPI and are shown as blue, GFP is shown as green, the signal due to annexin V is shown as magenta, and staining with anti-ORF K8.1 is shown as red. We found that pUC19 alone (Fig. 9A) showed no signal except the nuclear DAPI signal, as did cells transfected with pUC19 treated with DOX (Fig. 9B). When the ORF50ΔSTAD BCLB-1 cells were transfected with pUC19 and treated with TPA, the expression of the KSHV ORFK8.1 is apparent (Fig. 9C). However, when the ORF50ΔSTAD BCBL-1 cells were transfected with pUC19 and treated with TPA plus DOX (Fig. 9D), no KSHV ORFK8.1 expression is apparent, since the DN ORF50ΔSTAD shuts off viral replication induced by TPA. When the cells were transfected with pUC19 and treated with TPA plus DOX plus DCPE to induce apoptosis, the cells express both annexin V and KSHV ORFK8.1, since DOX induction of the DN ORF50ΔSTAD cannot shut off KSHV replication induced by apoptosis (Fig. 9E). (These results were obtained after approximately 24 h, so KSHV ORFK8.1 late gene expression is not fully developed.) When the cells were transfected with the plasmid expressing the caspase-3–GFP fusion protein, pcasp3-Wt-GFP (Fig. 9F), the expression of GFP is clearly evident within the great majority of the cells, the cells are undergoing apoptosis as shown by the magenta signal due to annexin V, and the KSHV ORFK8.1 late gene is also expressed in the cells, as shown by the red stain. Treating the cells with DOX to induce expression of ORF50ΔSTAD did not block induction of viral replication triggered by caspase-3 (not shown). Caspase-3, by itself, can thus induce apoptosis and KSHV gene expression in the ORF50ΔSTAD BCBL-1 cells. Caspase-3 is therefore both necessary and sufficient to induce KSHV replication in the context of host cell apoptosis.
Fig 9.
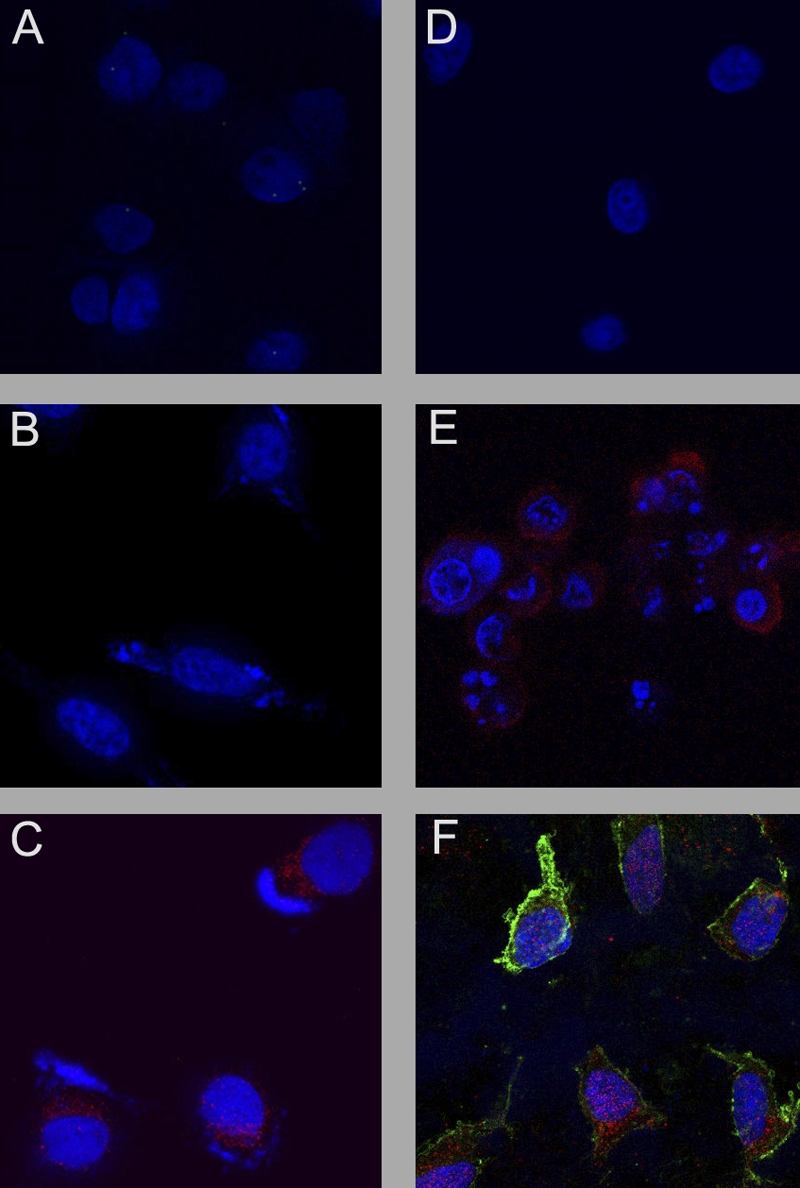
The effects of transfecting the caspase-3-expressing plasmid pcasp3-Wt-GFP into ORF50ΔSTAD BCBL-1 cells were assessed using confocal microscopy. ORF50ΔSTAD BCBL-1 cells were transfected with either the negative-control plasmid pUC19 or pcasp3-Wt-GFP, which expresses functional wild-type caspase-3 as a fusion protein with GFP, and various chemical inducers. Nuclei are stained blue with DAPI, the KSHV late gene ORF K8.1 is stained red, Annexin V is stained magenta, and GFP is shown as green. TPA was given to induce KSHV replication via the orthodox pathway, a low concentration of DOX was given to induce expression of the DN ORF50DSTAD, and DCPE was given to induce apoptosis in the following combinations. The cells were treated with pUC19 alone (A), pUC19 plus DOX (B), pUC19 plus TPA (C), pUC19 plus TPA plus DOX (D), pUC19 plus TPA plus DOX plus DCPE (E), and pcasp3-Wt-GFP (F), The pcasp3-Wt-GFP lead induces both apoptosis and KSHV gene expression, indicating that the end effector caspase-3 is sufficient to induce KSHV replication.
Inhibition of TPA-induced, but not DCPE-induced, KSHV replication by ORF50 siRNA.
To ensure that the inability of DN ORF50ΔSTAD to block KSHV replication triggered by DCPE did not result from some artifact unique to ORF50ΔSTAD BCBL-1 cells, we performed transfection experiments with ORF50 siRNA in BCBL-1 cells. We performed real-time RT-PCR assays to assess knockdown of ORF50 expression at the RNA level (Fig. 10A) and immunoblotting to assess knockdown of ORF50 expression at the protein level (Fig. 10B). At the RNA level, we were able to achieve 86% knockdown of ORF50 expression in the presence of TPA and 93% knockdown of ORF50 expression in the presence of DCPE (Fig. 8). siRNAs knocked down ORF50 protein expression by 93% for TPA-treated cells and 88% for DCPE-treated cells. Transfection with the siRNA was associated with a statistically significant (P < 0.04 by t test) inhibition of protected and total KSHV DNA when KSHV replication was triggered by normal replication pathway, but not when replication was triggered by apoptosis (Fig. 10C). These results from a second, independent approach further confirmed that KSHV replication triggered by apoptosis does not require ORF50.
Early and late gene expression kinetics following induction of KSHV replication by apoptosis.
KSHV replication triggered by apoptosis appeared to have at least one characteristic that is fundamentally different from the normal replication pathway—the lack of a requirement for ORF50. We therefore undertook studies of other key characteristics of KSHV replication to determine whether KSHV replication triggered by apoptosis differed in other ways from KSHV replication occurring via the normal pathway. We began with a study of KSHV gene expression kinetics (Fig. 11). We used real-time RT-PCR to examine the expression pattern of a prototypical KSHV early gene, ORF57, and a prototypical late gene, ORF17, following treatment of ORF50ΔSTAD BCBL-1 cells with low and high concentrations of DOX. We found (Fig. 11A) that when we treated cells with high concentrations of DOX, which triggers apoptosis and KSHV replication, both the early gene ORF57 (triangles) and the late gene ORF17 (circles) were expressed, with the expression of the late gene following expression of the early gene (a high concentration of DOX [shown in red] compared to no treatment [green] or a low concentration of DOX [orange] [0.1 μg/ml]). Interestingly, the expression of the late gene was accelerated by ∼12 to 24 h compared to what is typically seen when KSHV replication is induced with TPA (25, 34). The data indicate that when KSHV replication that occurs after cells are exposed to high DOX concentrations, which produce host cell apoptosis, the virus maintains a clear temporal ordering of gene expression, with an early, regulatory gene expressed prior to a late structural gene, but the expression of the late gene ORF17 occurred sooner than when KSHV replication is induced conventionally.
Fig 11.

KSHV gene expression kinetics during viral replication induced by high concentrations of DOX, by TPA, and by the apoptosis inducer DCPE. Expression of an early KSHV gene, ORF57 (triangles) and a late gene, ORF17 (circles), was assayed using TaqMan real-time RT-PCR assays at serial times after treatment. (A) ORF50ΔSTAD BCBL-1 cells were left untreated (green) or treated with a low concentration (0.1 μg/ml) of DOX (orange) or treated with a high concentration of DOX (red). Exposure to a high concentration (but not a low concentration) of DOX induced expression of the early gene ORF57 followed by expression of the late gene ORF17, but the kinetics of ORF17 expression was accelerated by ∼12 to 24 h over that previously described (25, 34). (B) ORF50ΔSTAD BCBL-1 cells were treated with TPA to induce viral replication via the normal pathway, with a low concentration of DOX to induce ORF50ΔSTAD expression, with DCPE to induce apoptosis, and with various combinations of the agents as follows: untreated (gray), low concentration of DOX (0.1 μg/ml) (lavender), TPA (blue), low concentration of DOX plus TPA (green), DCPE (yellow), low concentration of DOX plus DCPE (orange), and TPA plus DCPE (red). Real-time RT-PCR was used to assay for ORF57 RNA (triangles) and ORF17 RNA (circles). The key comparison is between ORF17 after induction with TPA (blue circles) versus ORF17 after induction with DCPE (yellow circles) or DCPE plus DOX (orange circles) or TPA plus DCPE (red symbols). ORF17 expression kinetics are accelerated after induction with the proapoptotic agent DCPE compared to kinetics observed following induction with TPA whether or not DN ORF50ΔSTAD is induced with DOX. Curves fitted were Gompertz four-component functions. The difference between ORF17 expression at 12 h following TPA induction (blue circles) compared to ORF17 expression following DCPE/apoptosis induction (red circles) was highly significant (P = 0.0003 by t test).
Since a high concentration of DOX appeared to be associated with accelerated KSHV late gene expression, we conducted a more extensive study of KSHV gene expression kinetics following induction of KSHV replication by the conventional inducing agent TPA and the apoptosis inducer DCPE. We performed real-time RT-PCR assays for the early ORF57 KSHV gene and the late ORF17 KSHV gene at serial times after initiation of KSHV replication by TPA or DCPE (Fig. 11B). In Fig. 11B, expression of the early gene ORF57 is indicated with triangles, while expression of the late gene ORF17 is indicated with circles. Untreated cells are shown in gray, DOX-treated cells (a low concentration of DOX alone to induce the DN ORF50ΔSTAD) are shown in lavender, TPA-treated cells are shown in blue, DOX-plus-TPA-treated cells are shown in green, DCPE-treated cells are shown in yellow, DOX-plus-DCPE-treated cells are shown in orange, and TPA-plus-DCPE-treated cells are shown in red. We found that, as expected, TPA induces expression of the early gene ORF57, which reached maximal levels of expression at 6 to 12 h. With TPA treatment, the expression of the ORF17 late gene increased, as expected, reaching maximal levels of expression after 24 h. Treatment with the apoptosis inducer DCPE led to maximal ORF57 early gene expression by 6 h, similar to that observed with TPA, but in contrast to the expression kinetics observed after induction with TPA, ORF17 expression reached maximal expression much earlier, by shortly after 12 h. The differences in expression of ORF17 induced by TPA and DCPE at 12 h were highly significant (P = 0.0003 by t test). As expected, when the cells were treated with a low concentration of DOX to induce expression of the DN ORF50ΔSTAD, the DN ORF50ΔSTAD blocked both early gene and late gene expression induced when KSHV replication was induced with TPA, but not when KSHV replication was induced with DCPE. This result was consistent with the results of the viral replication studies shown in Fig. 5. However, when replication was induced with the DCPE apoptosis inducer, expression of the late gene ORF17 was accelerated by ∼12 h compared to the kinetics observed when KSHV replication was triggered by TPA, consistent with the results observed in association with apoptosis when the cells were treated with a high concentration of DOX.
KSHV virion infectivity following induction of replication associated with apoptosis.
Since KSHV replication associated with apoptosis appeared to involve accelerated late gene expression, we hypothesized that virions produced in association with apoptosis would have impaired infectivity, since accelerated late gene expression may not lead to the well-ordered assembly of maximally infectious virions. To assess KSHV virion infectivity, we produced virions from ORF50ΔSTAD BCBL-1 cells treated with TPA to induce viral replication conventionally and with DCPE to induce viral replication via the apoptosis-associated pathway in the presence of a low concentration (0.1 μg/ml) of DOX to block replication via the normal pathway. We assayed for virions produced in the two pathways using assays for protected KSHV DNA and exposed TPA-treated HUVEC, which can support limited KSHV replication (3, 35) to equal numbers of virions produced in the two different pathways. After inoculation, we washed the HUVEC three times. There was no KSHV detectable by PCR in the supernatant after the first wash (not shown). After 48 h, we assayed for the KSHV virions produced by the HUVEC that had been infected with the two kinds of virions made following treatment with either TPA or DCPE (Fig. 12). We found that virions produced in cells in which KSHV replication was triggered by apoptosis, while still infectious, had significantly decreased infectivity compared with the infectivity of KSHV virions made when cells were induced by TPA to produce virions via the normal replication pathway. The infectivity of the virions made following DCPE treatment was only about 20% of that of the normal virions (P < 0.0001 by t test). While the alternative KSHV replication cycle triggered by host cell apoptosis still results in the production of infectious virions, the more rapid viral replication cycle induced in association with host cell apoptosis exacts a cost upon virion infectivity.
Fig 12.

Infectivity of KSV virions made in the normal and apoptosis-associated pathways. Equal numbers of KSHV virions produced by cells in which KSHV replication was induced with TPA and by cells in which KSHV replication was induced in association with apoptosis induced by DCPE were added to activated HUVEC, followed by extensive washing after inoculation (no KSHV DNA was detectable by PCR in the last wash). After 48 h, supernatants from the HUVEC were assayed for KSHV virions using an assay for protected KSHV DNA (25). The virions produced by the normal, TPA-induced replication pathway displayed infectivity more than 5-fold greater than virions made following induction of host cell apoptosis. Values are means plus standard deviations (SD) (error bars). (P < 0.0001 by t test).
DISCUSSION
Many viruses, including KSHV, have genes that specifically inhibit apoptosis (reviewed in reference 20). Our study suggests that KSHV also has an emergency “escape” replication pathway that it utilizes when its efforts to block host cell apoptosis fail. Our data further suggest that KSHV has two distinct viral replication pathways: an orthodox replication pathway and an alternate, emergency replication pathway. This alternate replication pathway appears to have several important characteristics. (i) The pathway is triggered in association with host cell apoptosis. (ii) The pathway does not require the activity of RTA, a protein previously thought essential for KSHV replication. (iii) The pathway requires the activity of caspases. (iv) The pathway exhibits late gene expression kinetics that are accelerated compared to the conventional pathway. (v) The pathway produces large numbers of virions. (vi) The virions made in the alternative replication pathway have reduced infectivity. While the existence of an alternative apoptosis-associated KSHV replication pathway is unconventional, it would have several evolutionary advantages for the virus. If the virus could sense when the host cell is about to undergo apoptosis and accelerate the pace of viral replication in response to the impending death of the host cell, it would aid in the survival of the virus. Even the production of virus with diminished infectivity would be advantageous—if the other option were an inability to produce any virus at all before the host cell died.
While our observations suggest that KSHV has an alternative replication pathway, triggered by apoptosis, we do not yet know what regulates this pathway. We hypothesize that there is a viral target that initiates replication pathway that is altered by a cellular factor involved in apoptosis. An example from another herpesvirus may be instructive. Caspase-3 can cleave the herpes simplex virus (HSV) ICP22, which has regulatory activity for the UL38 and US11 late genes (30), so there is precedent for a herpesvirus having a component that can sense a key component of the activated apoptotic cascade. The ability to detect when host cell apoptosis threatens may therefore be a general feature of herpesviruses. Our data suggest that caspase-3 activity is required for the KSHV alternative apoptosis-associated replication pathway, so a plausible mechanism might involve a KSHV or cellular protein that is cleaved by caspase-3 to form an active transcription factor that triggers the alternative replication pathway, but many more studies are required before the mechanisms that regulate the apoptosis-associated KSHV gene expression program can be established.
Since the virions made in the alternative replication pathway have decreased infectivity, we also hypothesize that the virus produced in the alternative pathway has altered biophysical characteristics, and potentially an altered ultrastructure and protein and/or lipid composition, although the composition of the icosahedral virion capsid may not differ due to geometric and stoichiometric constraints.
Many examples exist of organisms that change reproductive strategies depending on environmental variables (reviewed in reference 49). Such strategies are known in complex organisms, but the notion that viruses replicate differently when death threatens the host cell appears to be an unknown, but potentially important, aspect of herpesvirus biology. The ability of KSHV to respond to critical challenges by making major changes in its replication strategy may also have important implications for therapy for KSHV-associated neoplasms and potentially for other herpesvirus-associated neoplasms. For example, it may be clinically advantageous to treat patients with such neoplasms with antiviral agents when they are being treated with cytotoxic chemotherapy to block potentially clinically adverse production of apoptosis-induced virus. Finally, since two of the key attributes of life, beyond the mere ability to reproduce, are the abilities to sense and respond in a meaningful way to changes in the external environment, the observations that KSHV has two distinct replication programs, a normal program and an emergency program triggered by host cell apoptosis, lend additional support to the notion that KSHV can sense changes in its environment and respond to those changes in ways that make an important contribution to its survival.
ACKNOWLEDGMENTS
We thank Michael Bukrinsky, Claire Fraser-Liggett, Allen Herre, Fatah Kashanchi, and Sam Simmens for helpful advice and discussions. We thank Charles Wood for helpful advice and discussions and the gift of polyclonal anti-KSHV ORF50 antisera that was used in initial stages of the work. We thank Hiroyuki Nakamura and Jae Jung for the kind gift of Flp-In BCBL-1 cells. We thank Shinji Kamada, Kobe University, for the kind gift of pcasp3-Wt-GFP. We thank Bhargavi Ragan for help and advice with the flow cytometric assays and Jiang-Chang Wang of the CRI core imaging facility for help and advice with the confocal microscopic studies.
This work was supported in part by a Collaborative Faculty Award for AIDS Research from the George Washington University AIDS Institute, a Children's Research Institute Research Advisory Committee Award, a gift from the Conner Family Foundation, and the District of Columbia Developmental Center for AIDS Research (P30AI087714 from NIAID, NIH).
Footnotes
Published ahead of print 15 February 2012
REFERENCES
- 1. Arvanitakis L, Geras-Raaka E, Varma A, Gershengorn MC, Cesarman E. 1997. Human herpesvirus KSHV encodes a constitutively active G-protein-coupled receptor linked to cell proliferation. Nature 385:347–350 [DOI] [PubMed] [Google Scholar]
- 2. Bais C, et al. 1998. G-protein-coupled receptor of Kaposi's sarcoma-associated herpesvirus is a viral oncogene and angiogenesis activator. Nature 391:86–89 [DOI] [PubMed] [Google Scholar]
- 3. Ballestas ME, Chatis PA, Kaye KM. 1999. Efficient persistence of extrachromosomal KSHV DNA mediated by latency-associated nuclear antigen. Science 284:641–644 [DOI] [PubMed] [Google Scholar]
- 4. Boshoff C, et al. 1997. Angiogenic and HIV-inhibitory functions of KSHV-encoded chemokines. Science 278:290–295 [DOI] [PubMed] [Google Scholar]
- 5. Bu W, Carroll KD, Palmeri D, Lukac DM. 2007. Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 ORF50/Rta lytic switch protein functions as a tetramer. J. Virol. 81:5788–5806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bu W, et al. 2008. Identification of direct transcriptional targets of the Kaposi's sarcoma-associated herpesvirus Rta lytic switch protein by conditional nuclear localization. J. Virol. 82:10709–10723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Burysek L, et al. 1999. Functional analysis of human herpesvirus 8-encoded viral interferon regulatory factor 1 and its association with cellular interferon regulatory factors and p300. J. Virol. 73:7334–7342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 332:1186–1191 [DOI] [PubMed] [Google Scholar]
- 9. Cesarman E, et al. 1996. Kaposi's sarcoma-associated herpesvirus contains G protein-coupled receptor and cyclin D homologs which are expressed in Kaposi's sarcoma and malignant lymphoma. J. Virol. 70:8218–8223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chang Y, et al. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865–1869 [DOI] [PubMed] [Google Scholar]
- 11. Chatterjee M, Osborne J, Bestetti G, Chang Y, Moore PS. 2002. Viral IL-6-induced cell proliferation and immune evasion of interferon activity. Science 298:1432–1435 [DOI] [PubMed] [Google Scholar]
- 12. Damania B. 2004. Oncogenic gamma-herpesviruses: comparison of viral proteins involved in tumorigenesis. Nat. Rev. Microbiol. 2:656–668 [DOI] [PubMed] [Google Scholar]
- 13. Guo HG, Pati S, Sadowska M, Charurat M, Reitz M. 2004. Tumorigenesis by human herpesvirus 8 vGPCR is accelerated by human immunodeficiency virus type 1 Tat. J. Virol. 78:9336–9342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gwack Y, et al. 2003. Principal role of TRAP/mediator and SWI/SNF complexes in Kaposi's sarcoma-associated herpesvirus RTA-mediated lytic reactivation. Mol. Cell. Biol. 23:2055–2067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Harrison SM, Whitehouse A. 2008. Kaposi's sarcoma-associated herpesvirus (KSHV) Rta and cellular HMGB1 proteins synergistically transactivate the KSHV ORF50 promoter. FEBS Lett. 582:3080–3084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Invitrogen Life Technologies Corporation 2003. Flp-In T-REx core kit manual. Catalog number K6500-01. Invitrogen Life Technologies Corporation, Grand Island, NY [Google Scholar]
- 17. Izumiya Y, et al. 2003. Kaposi's sarcoma-associated herpesvirus K-bZIP is a coregulator of K-Rta: physical association and promoter-dependent transcriptional repression. J. Virol. 77:1441–1451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kamada S, Kikkawa U, Tsujimoto Y, Hunter T. 2005. Nuclear translocation of caspase-3 is dependent on its proteolytic activation and recognition of a substrate-like protein(s). J. Biol. Chem. 280:857–860 [DOI] [PubMed] [Google Scholar]
- 19. Kedes DH, Lagunoff M, Renne R, Ganem D. 1997. Identification of the gene encoding the major latency-associated nuclear antigen of the Kaposi's sarcoma-associated herpesvirus. J. Clin. Invest. 100:2606–2610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lamkanfi M, Dixit VM. 2010. Manipulation of host cell death pathways during microbial infections. Cell Host Microbe 8:44–54 [DOI] [PubMed] [Google Scholar]
- 21. Lan K, et al. 2005. Induction of Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen by the lytic transactivator RTA: a novel mechanism for establishment of latency. J. Virol. 79:7453–7465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liang Y, Chang J, Lynch SJ, Lukac DM, Ganem D. 2002. The lytic switch protein of KSHV activates gene expression via functional interaction with RBP-Jkappa (CSL), the target of the Notch signaling pathway. Genes Dev. 16:1977–1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liang Y, Ganem D. 2004. RBP-J (CSL) is essential for activation of the K14/vGPCR promoter of Kaposi's sarcoma-associated herpesvirus by the lytic switch protein RTA. J. Virol. 78:6818–6826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu J, Kuszynski CA, Baxter BT. 1999. Doxycycline induces Fas/Fas ligand-mediated apoptosis in Jurkat T lymphocytes. Biochem. Biophys. Res. Commun. 260:562–567 [DOI] [PubMed] [Google Scholar]
- 25. Lu M, et al. 2004. Dissection of the Kaposi's sarcoma-associated herpesvirus gene expression program by using the viral DNA replication inhibitor cidofovir. J. Virol. 78:13637–13652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lukac DM, Garibyan L, Kirshner JR, Palmeri D, Ganem D. 2001. DNA binding by Kaposi's sarcoma-associated herpesvirus lytic switch protein is necessary for transcriptional activation of two viral delayed early promoters. J. Virol. 75:6786–6799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lukac DM, Kirshner JR, Ganem D. 1999. Transcriptional activation by the product of open reading frame 50 of Kaposi's sarcoma-associated herpesvirus is required for lytic viral reactivation in B cells. J. Virol. 73:9348–9361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Moore P, Chang Y. 2001. Kaposi's sarcoma-associated herpesvirus, p 2803–2833 In Knipe D, Howley P, Griffin D, Lamb R, Martin M, Roizman B, Straus S. (ed), Fields virology, 4th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 29. Moore PS, Boshoff C, Weiss RA, Chang Y. 1996. Molecular mimicry of human cytokine and cytokine response pathway genes by KSHV. Science 274:1739–1744 [DOI] [PubMed] [Google Scholar]
- 30. Munger J, Hagglund R, Roizman B. 2003. Infected cell protein no. 22 is subject to proteolytic cleavage by caspases activated by a mutant that induces apoptosis. Virology 305:364–370 [DOI] [PubMed] [Google Scholar]
- 31. Nakamura H, et al. 2003. Global changes in Kaposi's sarcoma-associated virus gene expression patterns following expression of a tetracycline-inducible Rta transactivator. J. Virol. 77:4205–4220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nicholas J, et al. 1997. A single 13-kilobase divergent locus in the Kaposi sarcoma-associated herpesvirus (human herpesvirus 8) genome contains nine open reading frames that are homologous to or related to cellular proteins. J. Virol. 71:1963–1974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nicholas J, et al. 1997. Kaposi's sarcoma-associated human herpesvirus-8 encodes homologues of macrophage inflammatory protein-1 and interleukin-6. Nat. Med. 3:287–292 [DOI] [PubMed] [Google Scholar]
- 34. Paulose-Murphy M, et al. 2001. Transcription program of human herpesvirus 8 (Kaposi's sarcoma-associated herpesvirus). J. Virol. 75:4843–4853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Raghu H, Sharma-Walia N, Veettil MV, Sadagopan S, Chandran B. 2009. Kaposi's sarcoma-associated herpesvirus utilizes an actin polymerization-dependent macropinocytic pathway to enter human dermal microvascular endothelial and human umbilical vein endothelial cells. J. Virol. 83:4895–4911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Renne R, et al. 1996. Lytic growth of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat. Med. 2:342–346 [DOI] [PubMed] [Google Scholar]
- 37. Russo JJ, et al. 1996. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc. Natl. Acad. Sci. U. S. A. 93:14862–14867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sakakibara S, Ueda K, Chen J, Okuno T, Yamanishi K. 2001. Octamer-binding sequence is a key element for the autoregulation of Kaposi's sarcoma-associated herpesvirus ORF50/Lyta gene expression. J. Virol. 75:6894–6900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sarid R, Wiezorek JS, Moore PS, Chang Y. 1999. Characterization and cell cycle regulation of the major Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) latent genes and their promoter. J. Virol. 73:1438–1446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Seaman WT, et al. 1999. Gene expression from the ORF50/K8 region of Kaposi's sarcoma-associated herpesvirus. Virology 263:436–449 [DOI] [PubMed] [Google Scholar]
- 41. Slee EA, et al. 1996. Benzyloxycarbonyl-Val-Ala-Asp (OMe) fluoromethylketone (Z-VAD.FMK) inhibits apoptosis by blocking the processing of CPP32. Biochem. J. 315(Part 1):21–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Soulier J, et al. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood 86:1276–1280 [PubMed] [Google Scholar]
- 43. Sun R, et al. 1998. A viral gene that activates lytic cycle expression of Kaposi's sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. U. S. A. 95:10866–10871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sun R, et al. 1999. Kinetics of Kaposi's sarcoma-associated herpesvirus gene expression. J. Virol. 73:2232–2242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang S, Liu S, Wu M, Geng Y, Wood C. 2001. Kaposi's sarcoma-associated herpesvirus/human herpesvirus-8 ORF50 gene product contains a potent C-terminal activation domain which activates gene expression via a specific target sequence. Arch. Virol. 146:1415–1426 [DOI] [PubMed] [Google Scholar]
- 46. Wang SE, et al. 2003. Role of CCAAT/enhancer-binding protein alpha (C/EBPalpha) in activation of the Kaposi's sarcoma-associated herpesvirus (KSHV) lytic-cycle replication-associated protein (RAP) promoter in cooperation with the KSHV replication and transcription activator (RTA) and RAP. J. Virol. 77:600–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wang XP, et al. 2001. Characterization of the promoter region of the viral interferon regulatory factor encoded by Kaposi's sarcoma-associated herpesvirus. Oncogene 20:523–530 [DOI] [PubMed] [Google Scholar]
- 48. Wang Y, Yuan Y. 2007. Essential role of RBP-Jkappa in activation of the K8 delayed-early promoter of Kaposi's sarcoma-associated herpesvirus by ORF50/RTA. Virology 359:19–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. West-Eberhard MJ. 2003. Developmental plasticity and evolution, 1st ed Oxford University Press, New York, NY [Google Scholar]
- 50. Wu L, et al. 2000. Three-dimensional structure of the human herpesvirus 8 capsid. J. Virol. 74:9646–9654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wu S, et al. 2004. Induction of apoptosis and down-regulation of Bcl-XL in cancer cells by a novel small molecule, 2[[3-(2,3-dichlorophenoxy)propyl]amino]ethanol. Cancer Res. 64:1110–1113 [DOI] [PubMed] [Google Scholar]
- 52. Zhu FX, Cusano T, Yuan Y. 1999. Identification of the immediate-early transcripts of Kaposi's sarcoma-associated herpesvirus. J. Virol. 73:5556–5567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhu H, et al. 2004. Induction of S-phase arrest and p21 overexpression by a small molecule 2[[3-(2,3-dichlorophenoxy)propyl] amino]ethanol in correlation with activation of ERK. Oncogene 23:4984–4992 [DOI] [PubMed] [Google Scholar]