

Abstract
Brome mosaic virus (BMV) is a model positive-strand RNA virus whose replication has been studied in a number of surrogate hosts. In transiently transfected human cells, the BMV polymerase 2a activated signaling by the innate immune receptor RIG-I, which recognizes de novo-initiated non-self-RNAs. Active-site mutations in 2a abolished RIG-I activation, and coexpression of the BMV 1a protein stimulated 2a activity. Mutations previously shown to abolish 1a and 2a interaction prevented the 1a-dependent enhancement of 2a activity. New insights into 1a-2a interaction include the findings that helicase active site of 1a is required to enhance 2a polymerase activity and that negatively charged amino acid residues between positions 110 and 120 of 2a contribute to interaction with the 1a helicase-like domain but not to the intrinsic polymerase activity. Confocal fluorescence microscopy revealed that the BMV 1a and 2a colocalized to perinuclear region in human cells. However, no perinuclear spherule-like structures were detected in human cells by immunoelectron microscopy. Sequencing of the RNAs coimmunoprecipitated with RIG-I revealed that the 2a-synthesized short RNAs are derived from the message used to translate 2a. That is, 2a exhibits a strong cis preference for BMV RNA2. Strikingly, the 2a RNA products had initiation sequences (5′-GUAAA-3′) identical to those from the 5′ sequence of the BMV genomic RNA2 and RNA3. These results show that the BMV 2a polymerase does not require other BMV proteins to initiate RNA synthesis but that the 1a helicase domain, and likely helicase activity, can affect RNA synthesis by 2a.
INTRODUCTION
Brome mosaic virus (BMV), a member of the alphavirus-like superfamily of RNA viruses, is a model system for studies of viral RNA replication. In addition to being able to infect monocot and dicot plant hosts, BMV can replicate and transcribe its genome in yeast, Saccharomyces cerevisiae (9, 11, 12, 32). Studies in S. cerevisiae have led to the identification of a number of cellular pathways that impact BMV replication (10) and contributed to our understanding of the membrane-encased BMV RNA replication factories, so-called spherules (38).
The BMV genome is composed of three capped, messenger-sense RNAs. RNA3 encodes two proteins required for cell-to-cell movement and for RNA encapsidation. RNA1 and RNA2 encode nonstructural proteins 1a and 2a, respectively (24, 38). 1a and 2a expressed from mRNAs that lack the cis-acting sequences required for RNA replication can replicate RNA3 (1, 39).
The multifunctional 1a protein containing an N-terminal domain with m7G methyltransferase activity required for viral RNA capping (4, 16) and a C-terminal half that has sequence motifs of DEAD box RNA helicases exhibits ATPase and GTPase activities (17, 47). 1a is necessary and sufficient to form the BMV replication factories in the perinuclear endoplasmic reticulum of barley cells and in the surrogate host, S. cerevisiae (33, 34, 37, 38). It also recruits RNA2 and RNA3 to the replication factory (7, 8). Mapping studies using 1a mutants revealed that the BMV 1a helicase domain interacts with the N-terminal portion of the 2a protein and that this interaction is required for BMV RNA synthesis (7, 8, 13, 41).
The BMV 2a protein is the RNA-dependent RNA polymerase (RdRp) required for BMV RNA replication. In barley protoplasts, and in the yeast two-hybrid assay, 2a interacts with 1a through an ∼200-residue N-terminal domain (26, 27, 45). Mutations within the N-terminal domain abolish BMV RNA replication. However, it is not known whether the 2a has RNA synthesis activity or template specificity in the absence of 1a.
We recently developed an assay, the so-called 5BR assay, to assess RNA synthesis by transiently expressed viral polymerases through innate immune receptor signaling in human cells (31, 42). Briefly, viral RdRps expressed in human cells will produce RNAs that can serve as agonists to activate signaling by the innate immune viral RNA sensors, either RIG-I or MDA5, leading to production of reporter luciferase. In the present study, we adapted the 5BR assay to better understand the activities of the BMV 1a and 2a proteins and their interactions. We found that 2a was sufficient to synthesize RNAs, but RNA synthesis was enhanced by coexpression of the 1a protein. The assay was further used to map the residues required for 1a-2a interaction. Finally, pyrosequencing of the 2a products revealed that their 5′-terminal residues were identical to that of BMV RNA2 and RNA3.
MATERIALS AND METHODS
Constructs for expression in mammalian cells.
The open reading frames (ORFs) coding for BMV 2a (nucleotides [nt] 104 to 2572 in RNA2; GenBank accession number X01678) and 1a (nt 75 to 2960 in RNA1; GenBank accession number X02380) were amplified from BMV virion RNAs using specific forward and reverse primers containing AgeI and NheI restriction enzyme sites. The cDNAs were cloned into the pUNO expression vector. Versions of 1a expressed with or without a FLAG tag at the 1a C terminus and of 2a with or without an HA tag at the 2a C terminus were made. The epitope-tagged constructs had no noticeable effects on the activities of the proteins. Site-directed mutants were made using a QuikChange mutagenesis kit (Agilent Technologies, Santa Clara, CA). All oligonucleotide sequences will be made available upon request. All constructs were confirmed to have the correct sequence by DNA sequencing using the BigDye Terminator v3.1 cycle sequencing kit (Applied Biosystems, Carlsbad, CA).
Plasmids that can express RIG-I (pUNO-hRIG-I) and MDA5 (pUNO-hMDA5) were obtained from InvivoGen (San Diego, CA). The TLR3 plasmid (pcDNA-TLR3) was previously described by Sun et al. (44). The plasmid containing the firefly luciferase reporter gene driven by the beta interferon (IFN-β) promoter (IFNβ-Luc) was kindly provided by R. Lin (Lady Davis Institute for Medical Research, Quebec, Canada) and was previously described by Ranjith-Kumar et al. (30). To monitor and standardize transfection efficiency, plasmid expressing Renilla reniformis luciferase controlled by the herpes simplex virus thymidine kinase (TK) promoter was used (Promega, Madison, WI). Renilla luciferase driven by cytomegalovirus promoter was used for the assay in Huh-7 cells.
Protein expression analysis.
Cells were harvested by gentle scraping in 1× sodium dodecyl sulfate (SDS) sample buffer and analyzed on a continuous NuPAGE Novex Bis-Tris gel using morpholinepropanesulfonic acid-SDS running buffer (Invitrogen, Carlsbad, CA). Proteins resolved in the gels were transferred to polyvinylidene difluoride membranes (Invitrogen). After the membranes were blocked with 5% nonfat milk in Tris-buffered saline containing 0.01% Tween 20 (TBS-T) for 2 h, the membrane was incubated overnight in blocking buffer containing a 1:2,000 dilution of the primary antibody. The membranes were washed in TBS-T, followed by incubation with blocking buffer supplemented with horseradish peroxidase-conjugated secondary antibody and developed using ECL Plus Western blotting detection system (GE Healthcare, Piscataway NJ). Rabbit anti-1a and mouse anti-2a monoclonal antibodies were used as described by Kao et al. (14).
5BR assays.
The dual luciferase reporter assays were performed as described by Ranjith-Kumar et al. (31). Human embryonic kidney (HEK293T) cells were cultured in Dulbecco modified Eagle medium (DMEM) GlutaMax high-glucose medium (Life Technologies, Grand Island NY) containing 10% fetal bovine serum (FBS) at 37°C with 5% CO2. Huh-7 cells were cultured in a DMEM low-glucose medium containing glutamine (Life Technologies) supplemented with 10% FBS and 1× nonessential amino acids (Life Technologies). Plasmids expressing BMV 2a were cotransfected along with plasmids expressing RIG-I or MDA5, as well as two luciferases: the firefly luciferase driven from the IFN-β promoter and the Renilla luciferase driven from a TK promoter. The latter can serve to report on transfection efficiency and cytotoxicity.
For Huh-7 cells, the Renilla luciferase used a cytomegalovirus promoter. TLR3 assays were performed with ISRE-Luc as the reporter plasmid. Where an exogenous agonist for TLR3 was used, poly(I:C) was added to the medium of the cultured cells to a final concentration of 500 ng/ml. Equal amounts of total plasmid DNA were transfected in every experiment by adding vector plasmid (pUNO-MCS), as necessary. At 36 h after transfection, luciferase activity was measured using a Dual-Glo luciferase assay system (Promega, Madison, WI) in a Synergy 2 microplate reader (BioTek, Winooski, VT). The ratios of the firefly luciferase to the Renilla luciferase are shown in our results unless otherwise stated.
Confocal immunofluorescence microscopy.
HEK293T cells were grown for about 12 h on coverslips in six-well plates (BD Biosciences) at 2 × 106 cells/well and transfected with 100 ng of each plasmid coexpressing HA-tagged wild-type (WT) 1a (pUNO-BMV 1a) with FLAG-tagged 2a, HA-tagged 1a with a methyltransferase active-site mutant (pUNO-BMV 1a D106A) with FLAG-tagged 2a, HA-tagged 1a with a helicase active-site mutant (pUNO-BMV 1a K691A) with FLAG-tagged 2a, and empty vector as control using Lipofectamine 2000. Cells were grown for an additional 24 h after transfection and then washed three times with cold 1× phosphate-buffered saline (PBS). The cells were fixed in 4% paraformaldehyde for 10 min at room temperature, followed by two washes with PBS. The cells were rehydrated for 30 min using PBS containing 0.01% Triton X-100. Cells were incubated in a 1:300 dilution of goat anti-HA antibody and mouse anti-FLAG antibodies and then incubated overnight at 4°C. The cells were washed three times with PBS containing 0.01% Triton X-100 and incubated in 1:300 dilutions of Texas Red-conjugated bovine anti-goat and Alexa Fluor 488-conjugated anti-mouse antibodies (Invitrogen) for 2 h at room temperature. The cells were washed three times with 1× PBS containing 0.01% Triton X-100, mounted using ProLong Gold antifade mounting medium containing the DNA dye DAPI (4′,6′-diamidino-2-phenylindole; Invitrogen), and imaged under a scanning confocal microscope (Leica TCS SP5).
Immunoelectron microscopy.
HEK293T cells were grown in six-well collagen-coated plates (BD Biosciences) at 2 × 106 cells/well and transfected with the empty vector and either 0.5 μg of pEGFP or 0.5 μg of pEGFP, along with 1 μg of pUNO-BMV 1a. At 24 h after transfection, the green fluorescent protein (GFP)-positive cells were enriched using a FACSCalibur machine (BD Biosciences). The cells used for this analysis were washed twice with ice-cold fluorescence-activated cell sorting (FACS) buffer (1× PBS containing 10 mM phosphate, 150 mM NaCl [pH 7.4], and 3% FBS) and resuspended in same buffer at 2.0 × 107 cells/ml.
The cells were fixed in 0.25% glutaraldehyde–4% paraformaldehyde in PBS at 4°C for 1 h. The samples were pelleted by centrifugation at 1,000 × g for 5 min, and the pellets were then rinsed three times with PBS, dehydrated in a graded ethanol series to 95%, and infiltrated with L.R. White Resin (Electron Microscopy Sciences, Hatfield, PA) at a dilution of 1:1 with resin dissolved in 95% ethanol and in three changes of 100% L.R. White resin. The samples were then polymerized at 55°C in size 00 gelatin capsules filled with L.R White Resin. Ultrathin sections were produced from the polymerized resin blocks using a Porter-Blum MT-2 ultramicrotome (Sorvall, Inc.). Silver-gold sections were placed on Formvar-coated 300-mesh nickel grids (Electron Microscopy Sciences). The grids were floated on drops of TBS (pH 7.5) containing 1% Tween 20 (TBS-1%T) for 30 min. The grids were then floated on a solution of TBS-1%T containing 1% bovine serum albumin (BSA) for 10 min, followed by a solution of TBS-1%T containing 1% normal goat serum for 10 min. The grids were then incubated with a 1:100 dilution of rabbit anti-1a antibody at room temperature for 1 h before being washed for 15 min in TBS-1%T amended with 1% BSA. Finally, the grids were placed for 30 min in 1:10 dilution of 10-nm colloidal gold conjugated with goat anti-rabbit antibodies (Electron Microscopy Sciences) in a solution of 1% BSA in TBS-T. The grids were washed, stained with 3% aqueous uranyl acetate, and then imaged using a JEM-1010 transmission electron microscope (JEOL, Inc., Tokyo, Japan).
Sequencing of products synthesized by BMV 2a polymerase.
The products of the 2a polymerase were identified by using a method adopted from Baum et al. (5). Briefly, HEK293T cells (106) were transfected with 500 ng of plasmid that expressed FLAG-tagged RIG-I and with 1 μg of either the recombinant plasmid expressing WT BMV polymerase (2a) or the active-site mutant (2aGAA). At 24 h after transfection, the cells were washed once with PBS and lysed in buffer (20 mM Tris [pH 7.5], 150 mM NaCl, 10 mM EDTA, 1% NP-40, 10% glycerol, 0.1 mM sodium vanadate) containing a 1:100 dilution of mammalian protease inhibitor cocktail (Sigma). The anti-FLAG tag polyclonal antibody was covalently linked to Dynabeads M-270 Epoxy resin according to the manufacturer's protocol.
The immunoprecipitated material was treated with RNase-free DNase I to remove any possible DNA contamination, and RNAs were purified by phenol-chloroform extraction, followed by ethanol precipitation in the presence of glycogen. Ribosomal RNAs were removed with the Ribominus kit (Invitrogen). An RNA adaptor (5′-5dsp-GTTCrAGAGUUCUACAGUCCGACGAUC-3′) was ligated to the 5′ termini of purified RNAs by using RNA ligase (New England Biolabs) and, after overnight incubation at 4°C, an RNA adaptor (5′-5phos-AUCGUAUGCCGUCUUCUGCUUGU-3ddC-3′) was then ligated to the 3′ ends of the RNAs. First-strand cDNA synthesis used a reverse primer, 5′-CCTATCCCCTGTGTGCCTTGGCAGTCTCAGCAAGCAGAAGACGGCATAC-3′, specific to the 3′-adaptor sequence and SuperScript III reverse transcriptase (Invitrogen; the underlined sequence can anneal to the PCR primer). A cDNA library was constructed for pyrosequencing by PCR using the 5′-adaptor-specific primer, 5′-CCATCTCATCCCTGCGTGTCTCCGACTCAGACGAGTGCGTGTTCAGAGTTCTACAGTCCGACGATC-3′ (the underlined sequence serves as a barcode; the sequence in boldface can anneal to the 5′ primer for PCR) and the 3′-adaptor-specific primer, 5′-CCTATCCCCTGTGTGCCTTGGCAGTCTCAGCAAGCAGAAGACGGCATAC-3′ (the sequence in boldface is the 3′-adaptor-specific sequence). The PCR products were purified by elution from a 6% PAGE-TBE gel and analyzed by using a Bioanalyzer (Agilent Technologies). The amplicons were pooled at equimolar ratios for sequencing using the 454 Genome Sequencer FLX Titanium (Roche) at the Center for Genomics and Bioinformatics, Indiana University. The sequence reads were sorted based on the barcodes added in the adaptors using Galaxy web service tools (6). These were compared to those in databases using Megablast (48).
Statistical analysis.
Statistical analyses of the data were performed using GraphPad Prism software. The results are shown as means and standard deviations (SD). Groups are compared by the Student t test.
RESULTS
Enzymatically active BMV 2a protein can activate innate immune receptor signaling.
The 5BR assay offers a convenient system to examine viral RNA-dependent RNA synthesis and the interaction between viral proteins. Therefore, we examined whether the WT BMV 2a polymerase could function in the 5BR assay format (31, 42). As a negative control, we used a construct that will express 2a that had the divalent metal binding site (the active site) changed from GDD to GAA (2aGAA). Western blot analysis showed that the 2aGAA mutant was expressed at a level comparable to that of the WT 2a polymerase (Fig. 1A).
Fig 1.
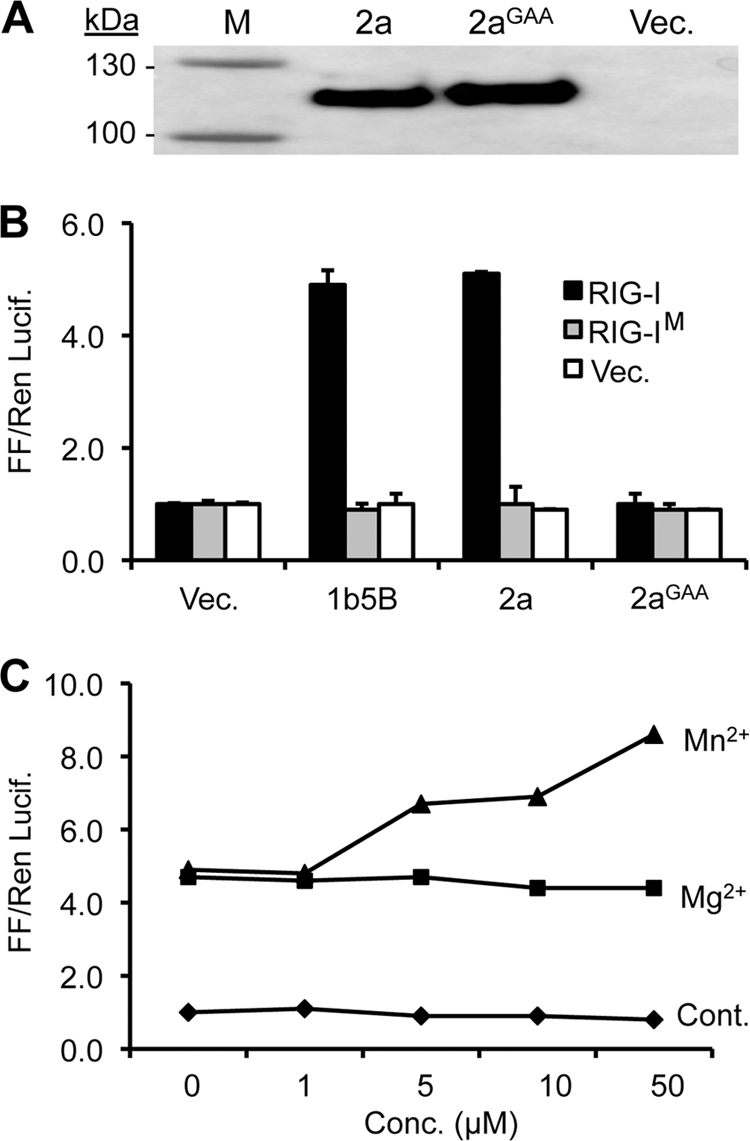
The BMV 2a polymerase can activate innate immunoreceptor-mediated signaling. (A) Western blot showing the expression of WT or the 2aGAA mutant protein from the HEK 293T cells used in the reporter experiment. The empty vector (Vec.) denotes a negative control. (B) Comparison of luciferase reporter levels induced by the BMV and HCV polymerases in transiently transfected HEK 293T cells. 2AGAA denotes an active-site mutant of the BMV 2a and RIG-IM that contains a K888E substitution that renders RIG-I defective for ligand binding. The ratios of the firefly to the Renilla luciferase levels were quantified as described in Materials and Methods and are shown on the y axis. The standard errors shown above the bars are from a minimum of three independent assays. (C) Mn2+, but not Mg2+, affects the 2a polymerase activity. The divalent metals were added directly to the medium of the cultured HEK 293T cells. The control reactions are from cells expressing RIG-I and the two luciferases but not the 2a polymerase. Each data point represents the mean of three independent assays.
The WT 2a reproducibly increased the firefly luciferase level driven by the IFN-β promoter relative to the Renilla luciferase (FF/Ren) by 4- to 6-fold compared to cells where the plasmid expressing 2a was replaced with an empty vector or the 2a active-site mutant (Fig. 1B). The increase in the FF/Ren levels was dependent on the amount of the 2a-expressing plasmid transfected into cells, with ∼60 ng of plasmid being optimal (data not shown). This amount was used throughout the 5BR assay in this work. Mutant 2aGAA did not activate RIG-I signaling (Fig. 1B). The level of RIG-I signaling in the presence of BMV 2a was comparable to that of 1b HCV NS5B (Fig. 1B). Furthermore, firefly luciferase was not induced in cells that lacked RIG-I or that expressed the RNA-binding mutant of RIG-I (RIG-IM; Fig. 1B). Comparable results were observed when the 5BR assay was performed with human hepatocytes, Huh-7 (data not shown). These results suggest that transiently expressed BMV 2a RdRp can synthesize RNAs in multiple human cell lines and induce RIG-I signaling.
The divalent metal ion Mn2+ has been reported to enhance replication initiation of BMV, likely through induced changes in the RdRp conformation (15). We observed that supplementing the culture medium with up to 50 μM MnCl2 enhanced luciferase levels by 4-fold without an effect on Renilla luciferase levels. MgCl2, however, had no effect on the luciferase readout (Fig. 1C). Concentrations of Mn2+ and Mg2+ greater than 50 μM caused cytotoxicity, which is consistent with previous reports (31, 42). The effect of Mn2+ but not Mg2+ is consistent with RNA synthesis by 2a using a de novo initiation mechanism.
We seek to use innate immune receptors to characterize the products of the BMV 2a. RIG-I preferentially recognizes de novo-initiated RNAs with 5′ triphosphate (5, 21). In contrast, MDA5 recognizes blunt-ended double-stranded RNAs (dsRNAs) that are longer than 100-bp (19). Both RIG-I and MDA5 recognize ligands that are present in the cytoplasm of cells. The Toll-like receptor 3 (TLR3) also recognizes dsRNAs, but within acidic endosomes (18, 20). BMV 2a activated MDA5 signaling to levels comparable to that by the HCV NS5B, but did not activate TLR3 signaling (Fig. 2). In the TLR3 assay, exogenously provided poly(I:C) activated TLR3 signaling, whereas mutant TLR3 that is defective for ligand (H539E, TLR3M [18]) did not, showing that the assay is working. These observations reveal that the RNA products synthesized by BMV 2a polymerase could be several hundred base pairs in size and are likely present in the cytoplasm and not in endosomes.
Fig 2.
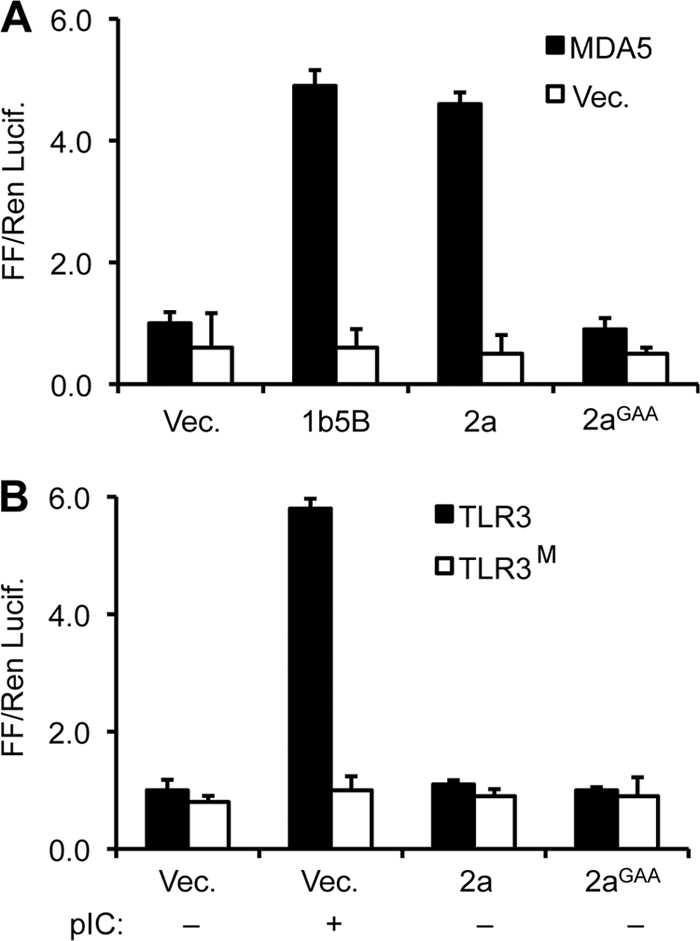
The BMV 2a protein can activate signaling by MDA5, but not TLR3. (A) Assay for the BMV 2a polymerase in HEK 293T cells with MDA5 as the innate immunoreceptor. (B) TLR3 signaling was not activated by the coexpression of the BMV 2a polymerase. TLR3M contains a H539E mutation that abrogates ligand binding and serves to demonstrate that signaling is dependent on the WT TLR3. Polyinosinic and poly(C) (pI:C), a TLR3 agonist, was added to the growth medium of cells to a final concentration of 1 μg/ml. All data for reporter assays in the present study are shown as means and the standard errors of at least three replicates in the experiment. Each result has been reproduced a minimum of twice, with consistent results.
1a-2a coexpression can stimulate 2a-dependent RNA synthesis.
The 5BR assay has been used previously to study the interaction of RdRps with other coexpressed viral nonstructural and structural proteins (31, 42). Therefore, we examined whether 2a activity in 293T cells is affected by the coexpression of 1a. Western blot analysis demonstrated that the proteins used in this set of experiments were all expressed to comparable levels (Fig. 3A). Coexpression of the WT 1a protein with 2a resulted in an approximately 50 to 60% increase in the FF/Ren levels relative to cells expressing only the 2a protein (P = 0.006, Fig. 3B). No stimulation was observed with the 2aGAA. 1a also did not affect RIG-I signaling in the absence of 2a (data not shown). Coexpression of 1a with the RdRps from the HCV 1b genotype and the human norovirus (NoV) increased signaling by 15 to 20% (P = 0.059; Fig. 3B and data not shown). These results suggest that at least some of the effects of the 1a protein may not be directed at 2a. Nonetheless, the 2- to 3-fold higher enhancement of the BMV 2a activity by 1a relative to the effect on NS5B led us to examine whether the interaction between 1a and 2a in human cells will mimic requirements previously determined in plants.
Fig 3.

BMV 1a protein interacts with and enhances 2a polymerase activity. (A) At the top is a schematic of BMV 1a protein showing the locations of active-site residues in the methyltransferase (D106) and helicase (K691) domains. The middle and bottom panels are images from Western blots that show comparable expression of WT 1a and different 1a mutants in HEK 293T cells. (B) Effects of coexpressed WT or mutant 1a proteins on the activities of the 2a polymerase. (C) Effects of the active-site mutations in the truncated methyltransferase or helicase-like domains of the 1a protein on 2a polymerase activity. (D) Several mutations in 1a previously shown to affect BMV RNA replication in barley protoplasts also affect the ability to enhance 2a signaling. The results of statistically significant differences, as determined by the Student t test, with the P values are shown above the bars denoting the means of the results.
Features in 1a required for 1a-2a interaction.
Several 1a mutants have been characterized for BMV RNA replication and interaction with 2a (3, 4, 13, 14, 16, 17, 25, 27, 47). To identify which part of 1a was required to interact with 2a, we expressed the WT 1a, the 1a methyltransferase domain (1aM, residues 1 to 513) and the 1a helicase domain (1aH, residues 516 to 961) in the assay (Fig. 3A). 1aH enhanced 2a's activity to a level similar to that of the WT 1a protein, while 1aM did not (Fig. 3B). To determine whether the ATPase active site of the helicase domain was required for the enhancement, both full-length 1a and 1aH with a K691A substitution were tested. Neither ATPase mutant enhanced 2a's activity (Fig. 3B). Furthermore, 1aK691A was found to have lost the ability to modestly stimulate the activity of the HCV 1b NS5B (Fig. 3B and data not shown). A substitution in the active site of the methyltransferase domain (D106A) of full-length 1a retained the ability to enhance 2a polymerase activity (Fig. 3B and C). These results demonstrate that the 1a helicase domain is required to interact with and enhance 2a polymerase activity. Furthermore, the ATPase motif of 1a helicase domain is important for enhancing 2a's activity.
Three insertion mutants in the 1a helicase domain were previously characterized for interaction with 2a (17). Mutant G-STS has an insertion between amino acids Ser-670 and Ala-671 was temperature sensitive for 1a replication in plants and retained interaction with 1a (14, 27) Mutants D-P869 and D-P905 were unable to replicate BMV RNAs and did not interact with 2a (Fig. 3D) (14, 17, 27). In human cells, only mutant G-STS could enhance 2a's ability to activate signaling by RIG-I to comparable level to WT 1a (Fig. 3D). These results suggest that BMV RNA replication proteins expressed in human cells mimic at least some of the requirements for BMV RNA replication in plant cells.
Features in 2a required for 1a-2a interaction.
Deletions within the N-terminal ca. 200 residues of 2a could abolish BMV RNA replication in barley (13, 45). To determine whether the 2a N-terminal region is required for 1a-2a interaction in human cells, we made and tested a series of N-terminal truncations that deleted of 2a amino acid residues 2 to 25, 2 to 50, 2 to 75, 2 to 100, 2 to 110, 2 to 120 and 2 to 130 for activation of RIG-I signaling in the absence or presence of 1a. All of these truncated proteins were expressed to levels comparable to that of WT 2a (Fig. 4A). Expression of the truncations up to and including residue 110 along with WT 1a, resulted in the enhancement of reporter activities above the levels of 2a alone. None of these truncations were enhanced for reporter activities when coexpressed with mutant 1aK691A (Fig. 4B). Deletion up to residue 120 retained 2a activity in the absence of 1a but was not as responsive to the 1a-dependent enhancement (Fig. 4B). Mutant 2a with deletion up to residue 130 lacked basal RdRp activity. These results further demonstrate that the 2a polymerase alone is sufficient for its RNA synthesis activity and that polymerase activity does not require the N-terminal 120 residues. However, the residues between positions 110 and 120 in the 2a N-terminal region are required to interact with 1a to enhance RNA synthesis.
Fig 4.

BMV 2a residues required for polymerase activity and for interaction with 1a. (A) Western blot of the expression of the WT BMV 2a and various N-terminal deletions and point mutants. The primary antibodies were described in the work of Kao et al. (14), and the secondary antibody was a horseradish peroxidase-linked goat anti-rabbit IgG. (B) Effects of deletions in 2a on activity and the enhancement of polymerase activity by the WT BMV 1a protein. All results were from transiently transfected HEK293T cells. Notably, Δ120 retained 2a polymerase activity, but the activity was not enhanced by coexpression with the BMV 1a protein. (C) Effects of the deletions and point mutations between residues 110 and 120 on 2a polymerase activity and interaction with BMV 1a. The names of the mutant constructs are in parentheses. The statistical analyses were performed using the Student t test, and the P values of key comparisons are shown. All of the single amino acid substitutions had P values of <0.01 compared to cells that do not express 1a.
A stretch of negatively charged residues (sequence EDEIDD) is present between residues 114 to 119 of 2a (Fig. 4C). To determine whether these charged residues contribute to interaction with 1a, four single-residue substitutions that retained polar side chains but lacked the negative charges (E114S, D115Q, D118Q, and D119Q) were made along with a fifth construct named S4 that had substitutions at all four positions (Fig. 4C). Coexpression of 1a enhanced the activities of the four mutants with single residue substitutions, but S4 did not (Fig. 4C). These results suggest that multiple acidic residues between the N-terminal 110 and 120 positions of the 2a protein contribute to a functional interaction with the 1a protein.
Localization of 1a and 2a in human cells.
We attempted to determine whether 1a and 2a could form a coimmunoprecipitable complex in transfected 293T cells but failed despite many attempts that used either existing protocols or protocols modified with the intention of retaining protein-protein interactions (7; data not shown). Therefore, we used immune-confocal microscopy to examine whether 1a and 2a are colocalized in cells. HEK293T cells expressing the empty vector had minimal background in the confocal images with the antibodies used, but expression of 1a resulted in a number of punctate signals at the periphery of the nuclei (Fig. 5B). Similar results were observed for 1aD106A and 1aK691A proteins (Fig. 5C and D). Cells expressing both the 1a and 2a proteins did not noticeably alter the localization of 1a. However, 2a appeared to extensively colocalize in the perinuclear region with 1a (Fig. 5B to D). Analysis of the data using ImageJ-JACoP software revealed that 55, 47, and 39% of the 2a proteins were colocalized with WT 1a, 1aD106A, and 1aK691A, respectively. The fact that 1aK691A could not enhance the activity of 2a but did colocalize with 2a suggests that 1a has additional role(s) in enhancing 2a polymerase activity in addition to trafficking 2a to the replication factory (Fig. 3B and C).
Fig 5.

Colocalization of the BMV 1a and 2a proteins in 293T cells. Alexa Flour 488-conjugated anti-mouse and Texas Red-conjugated anti-goat antibodies were used as antibodies to recognize 1a and 2a antibodies, respectively. Cell nuclei were stained with DAPI and viewed using a DeltaVision personal DV fluorescence microscope (Applied Precision, Issaquah, WA) and a scanning confocal microscope (Leica TCS SP5). All images were collected at ×63 magnification. (A) Cells transfected with empty vector as control (Cont.). (B) Cells expressing both WT BMV 1a and 2a proteins. (C) Cells expressing 1aD106A mutant and WT 2a protein. (D) Cells expressing 1aK691A mutant and WT 2a protein.
1a forms spherules within the nuclear membrane in yeasts cells (38). Since the 1a helicase domain that lacks the membrane-intercalating helix was sufficient to enhance 2a activity, the membrane reconfiguration activity of 1a is not required to enhance 2a activity. Nonetheless, we used immunoelectron microscopy to examine whether 1a formed membrane-associated structures in human cells. In order to enrich cells transfected to express 1a, the cells were cotransfected with GFP and sorted by flow cytometry for the preparation of thin sections. Samples transfected to express only GFP had only a low abundance of gold particles that were randomly distributed (Fig. 6A). However, clusters of gold particles were observed in cells expressing 1a. Approximately half of the gold clusters were in areas that displayed lower electron density (Fig. 6B), but none were surrounded by membranes. These observations were consistent in three independently prepared samples, as well as with the active-site mutants (Fig. 6C and D). These results suggest that 1a's can enhance 2a activity, in a manner independent of the formation of spherule-like structures.
Fig 6.

Immunoelectron microscopy of BMV 1a proteins. Sections of HEK 293T cells transfected with empty vector as a control were viewed under JEF-1010 transmission electron microscope (JEOL, Inc., Tokyo, Japan). The sections are stained with anti-1a antibody, followed by goat anti-rabbit antibodies conjugated to 10-nm colloidal gold. Representative sections where colloidal gold was detected are shown. In all images the nuclei are positioned to the left and labeled as “Nuc”. The arrows identify the clusters of colloidal gold. (A) Thin section of a HEK293T cell expressing WT BMV 1a protein. (B) Section of a cell expressing the 1aD106A mutant. (C) Section of a cell expressing 1aD106A mutant. (D) Section of a cell expressing 1aK691A mutant.
2a RNA products exhibit initiation requirements of the BMV replicase.
The BMV 2a protein could use either cellular RNA or the RNA expressed in the 5BR assay as templates for RNA synthesis. To identify the RNA products synthesized by BMV 2a polymerase, we adapted the protocol of Baum et al. (5), wherein a FLAG-tagged RIG-I was coexpressed with WT 2a polymerase or with the 2aGAA. RIG-I was immunoprecipitated, and the associated RNAs were extracted and either characterized for function or processed for pyrosequencing. The HCV NS5B from genotype 4a and its active-site mutant were also processed in parallel to allow comparison of the results. Immunoprecipitation revealed comparable amounts of precipitated RIG-I and 2a from the two treatments (Fig. 7A).
Fig 7.

Analysis of the BMV 2a products made in 293T cells. (A) The top panel shows a Western blot analysis of the FLAG-tagged RIG-I protein immunoprecipitated from cells coexpressing the WT 2a or the 2aGAA mutant. The bottom panel shows an image from a Western blot of the total lysate from the cells, detecting 2a expression. “M” denotes protein molecular mass markers from Invitrogen, Inc. (B) A comparison of the immunostimulatory activity of RNAs coimmunoprecipitated with RIG-I cells expressing either WT 2a or the 2aGAA. The RNAs were quantified using a Nanodrop spectrometer and known amounts either mock-treated (□) or treated (■) with Antarctic phosphatase treatment (+AP) and then transfected into 293T cells expressing WT RIG-I. shR9 is triphosphorylated short hairpin RNA that is a known agonist of RIG-I signaling. Total RNA was extracted from 293T cells and used as a control. (C) cDNA products of RNAs that coimmunoprecipitated with the RIG-I proteins. The RNAs were ligated to adaptors and synthesized cDNAs by RT-PCR; the products were electrophoresed on an agarose gel and stained with ethidium bromide.
To demonstrate that the RNAs were RIG-I ligands, aliquots of eluted RNAs were either treated or mock treated with shrimp alkaline phosphatase and then transfected into 293T cells expressing WT RIG-I. The signals from 5 and 10 ng of the RNAs eluted from the 2a-expressing cells were at least 10-fold higher than that of total RNAs extracted from the 293T cells harboring the empty plasmid vector (Fig. 7B). These results confirm that immunoprecipitation of RIG-I from 2a-expression cells enriched for RIG-I ligands.
Aliquots of the RNAs precipitated with RIG-I from cells expressing 2a, 2aGAA, the HCV NS5B, or NS5BGAA were then ligated with 5′ and 3′ adapters, followed by reverse transcription and PCR to amplify the cDNA. Products of ca. 200 to 400 bp were observed with cells expressing the BMV and HCV NS5B but not with cells expressing the catalytically inactive RdRps (Fig. 7C).
The cDNAs were then subjected to pyrosequencing, and the results were compared to the nucleic acid sequences in GenBank to identify the templates used by the BMV RdRps. Of the 901 sequences from the BMV 2a sample that had perfect adaptor sequences, all were derived from BMV RNA2. Furthermore, 897 of the 901 of the RNAs had a 5′ nucleotide that matched either residue 2261 or residue 2262 of BMV RNA2 (Fig. 8A). The longest sequence is shown in Fig. 8B. Notably, the first 2 nt starting with residue 2262 are GU, a sequence identical to that at the 5′ termini of all of the BMV genomic RNAs in GenBank and that of several members of the Bromoviridae (Fig. 8C). In fact, the first 5 nt, GUAAA, is a perfect match to the 5′-terminal 5 nt of BMV RNA2 and RNA3. The 3′ termini of the RNAs were more heterogeneous and were located at the various underlined nucleotides in the sequence shown in Fig. 8B. Notably, ∼70% of the terminal or penultimate nucleotides at the 3′ termini of the 901 reads contained either one or two cytidylates, suggesting that the complementary strand predominantly initiated with a guanylate.
Fig 8.

Features of RNAs coimmunoprecipitated with RIG-I and processed by pyrosequencing. (A) Summary of the 901 reads obtained from cells expressing the WT 2a polymerase. A schematic of the cDNA for the BMV 2a protein and the location of the reads from pyrosequencing within the 2a message are shown. A total of 855 of the reads initiated at either nt 2261 or nt 2262 within the BMV RNA2 sequence, the alignments of which are shown below and to the left of the schematic. The 3′ termini of the reads varied in location, but the terminal 2 nt are shown, along with the number of reads with these nucleotides. (B) The sequence of the longest of the 901 reads generated from cells expressing the BMV 2a protein. The preferred initiation nucleotide from all of the reads is identified with an arrow, and the 3′-most nucleotides in the reads are underlined. (C) Sequences present at the 5′ termini of the genomic RNAs of three members of the Bromoviridae family. CMV, cucumber mosaic virus; AMV, alfalfa mosaic virus. (D) A summary of the 5′-terminal nucleotides in the RNAs precipitated with the genotype 4A HCV NS5B polymerase. The arrow denotes the predominant 5′ nucleotide within the 265 sequences.
We have previously demonstrated that initiation by the BMV replicase was most efficient when the template could direct the synthesis of GUA as the first 3 nt (1, 39). The deep-sequencing results demonstrate that the BMV 2a protein alone is sufficient to exhibit the initiation requirements for BMV RNA replication in human cells.
To determine whether the initiation requirement exhibited by 2a applies to another viral RdRp, we examined the HCV RdRp products that were processed and sequenced in the same set of reactions. A total of 265 sequences with perfect 5′ and 3′ adaptors were identified, and 208 of these had 56 consecutive residues that aligned with the human solute carrier family 33 (acetyl-CoA) transporter, member 1 (SLC33A1) (data not shown). The remaining reads appear to be a collection of RNA sequences, including that from the acetyl-CoA transporter, which could have resulted from active template switch. However, the lengths of the homologies are generally <20 nt in length, thus precluding firm assignments of the identities of the RNAs. It is unambiguous, however, that multiple cellular RNAs served as templates for the HCV RdRp. Notably, 263 of the 265 RNAs contained the identical 6 nt at their 5′ termini: 5′-GAUGAA (Fig. 8D). Therefore, the HCV NS5B, like the BMV 2a, also exhibited a preferred initiation sequence even in the absence of other viral replication-associated proteins, similar to their initiation requirements in vitro (14, 22, 29).
DISCUSSION
Using transiently transfected human cells that could sense non-self-RNAs, we determined that (i) the BMV 2a polymerase can synthesize RNAs in the absence of additional BMV proteins, (ii) coexpression of BMV 1a enhanced 2a polymerase activity, (iii) mutations previously characterized to affect BMV RNA replication and 1a-2a interaction affected 1a's ability to enhance 2a activity, (iv) mutations in the 1a ATPase motif, but not the methyltransferase motif, prevented enhancement of 2a activity, (v) several acidic residues between amino acids 110 and 120 in the 2a protein were required to interact with 1a, (vi) BMV 1a and 2a can colocalize in human cells and that colocalization does not require the 1a ATPase motif, (vii) the BMV 2a preferentially used the RNAs from which it was translated as a template for RNA synthesis, (viii) the products made by 2a contains the initiation sequence found in BMV genomic RNAs, and (ix) finally, like 2a, the HCV RdRp also exhibits a strongly preferred initiation sequence.
De novo-initiated RNA synthesis allows copying of the entire viral RNA genome and serves as the substrate for the capping of the mRNAs. De novo-initiated RNA can also serve as a molecular pattern detected by innate immunoreceptors to trigger antiviral responses. Although the BMV replicase has been demonstrated to initiate by a de novo mechanism (43), whether both 1a and 2a are required was not known. Since both BMV RNA1 and RNA2 were required to detect BMV RNA replication in yeasts and plant cells, it is not known whether 2a has polymerase activity independent of the BMV replicase. Using the 5BR assay, we demonstrated that 2a alone is sufficient for RNA synthesis, likely by a de novo-initiated mechanism since RIG-I preferentially recognizes RNAs with 5′ triphosphates (5, 21) (Fig. 7B). In addition, adding Mn2+ to the media of the cultured cells increased the 2a activity, a finding consistent with Mn2+ stimulating de novo-initiated RNA synthesis in vitro (15) (Fig. 1C).
Pyrosequencing revealed that the products made by 2a exhibit features previously determined to be preferred for efficient RNA synthesis by the BMV replicase (1, 39). In fact, 765 of the 901 sequences were identical to the 5′ termini of the BMV genomic RNA. Interestingly, the newly synthesized RNAs were derived mostly from RNA2, reflecting a strong cis preference for BMV RNA2. Another 132 of the sequences contained an additional cytidylate 5′ of the genomic sequence. We speculate that the additional cytidylate was added in a nontemplated manner, perhaps during the processing of the samples for DNA sequencing. More importantly, highly enriched BMV replicase appears to be programmed to preferentially produce RNAs with the initiation sequence found at the termini of the genomic and subgenomic RNAs (1, 36, 39). Since our deep sequencing was from cells expressing only 2a, these results show that 2a is sufficient to select the template sequence for de novo initiation. Since the HCV RdRp also used one predominant initiation sequence (5′-GAUGAA-3′), a selection of the proper initiation sequence may be a general property of viral RdRps.
It is currently unclear why 2a initiated from nt 2262 in the 5BR assay. The sequence for the first five residues (5′ GUAAA) was also present at two other positions in the message used to translate the 2a protein (starting at nt 1274 and 1341, using the numbering for BMV RNA2 nucleotides in GenBank entry NC_002027). Also, there were eight sequences that matched the first 4 nt of the product (GUAA) within the 2a cDNA that we expressed. The strong preference for initiation at position 2262 suggests that there may be additional requirements, perhaps RNA secondary structure, which facilitates initiation or that the 2a protein must be translated past this sequence to bind RNA.
The preference of 2a to use its own RNA as the template was unexpected. The norovirus RdRp, as well as the genotype 4a HCV RdRp, used cellular RNA as templates (42). BMV RNA2, unlike BMV RNA3, is not known to contain a replicase assembly sequence (28). It is possible that the cis preference of 2a to recognize RNA2 provides a mechanism for the specific replication of RNA2 and the assembly of the BMV RNA replicase. Should this be the case, replicase assembly on RNA2 will be mechanistically different than on RNA3.
It is also worth noting that all 901 reads were of positive-sense RNA. This identification is possible since we used adaptor sequence ligated to the termini of the RNAs extracted from immunoprecipitated RIG-I. We did not observe a minus-strand RNA that could serve as the template in the 901 reads. Whether this is due to sequence-specific binding by RIG-I or an insufficient number of reads during sequencing remains to be determined. RIG-I has been reported to prefer A-U rich sequences, but this has not been supported in follow-up studies (35, 46), and the RNAs made by the BMV 2a have a comparable amounts of A-U residues (54%) for both the positive and the negative strands. Furthermore, positive-strand bromovirus RNAs accumulate to >100-fold greater levels than those observed for negative-strand RNAs (23, 40), potentially resulting in our reads having only positive-strand RNAs. We have tried to coexpress BMV RNA3 in cells transfected to express 1a and 2a but were unable to detect either BMV RNA4 by Northern blots or minus-strand RNAs by reverse transcription-PCR (RT-PCR). We do note that there is a preference for termination of the 901 reads of positive-sense RNAs with a cytidylate at the terminal or penultimate position. This suggests that the minus-strand RNAs were preferentially initiated at a guanylate.
BMV 1a-2a interaction.
BMV RNA replication occurs within membrane-encased structures. In human cells, we do not believe that the membrane-encased structures are responsible for the enhanced 2a activity. First, spherule formation requires an α-helix from the 1a methyltransferase domain to insert into one face of the membrane bilayer to mediate membrane curvature (2). In our system, the methyltransferase domain was not required to enhance 2a activity (Fig. 3B and C). Second, the helicase domain could enhance the 2a polymerase activity, while the K691A mutation in the ATPase domain prevents the enhancement of 2a activity (Fig. 3B and C). Third, immunoelectron microscopy did not reveal reconfigured membranous structures in association with 1a in human cells. Together, these observations suggest that 1a can affect RNA synthesis in addition to formation of spherules. Furthermore, the helicase activity could be involved in enhancing RNA synthesis by 2a polymerase. The NTPase activity of 1a is required for RNA synthesis, but not for spherule formation, localization of 1a to perinuclear endoplasmic reticulum membranes, and recruitment of 2a polymerase (47).
An important implication of 2a being able to direct RNA synthesis in the absence of 1a is that viral RNA synthesis does not have to be restricted to spherules. In plants, Seo et al. (40) have demonstrated that the cucumber mosaic virus 2a protein could synthesize positive-strand RNAs from negative-strand templates with similar activity compared to the presence of 1a. Whether spherule-independent RNA synthesis can influence bromovirus infection remains to be determined.
Finally, we note that while model systems such as BMV have provided useful information concerning the basic requirements for viral infection, a large number of viruses cannot infect cultured cells, and this has hindered the study of the requirements for these viruses as well as the interventions. The 5BR assay has now been used to study the requirements for RNA synthesis for six major genotypes of HCV, two noroviruses and, in the present study, BMV (31, 42). Furthermore, for all three classes of viruses, RdRp activities were modified by interactions with other viral proteins, and it may be a useful system for analyzing the requirements for RNA synthesis by other viruses that cannot be cultured.
ACKNOWLEDGMENTS
We thank members of the IU and TAMU Cereal Killers, former and present, for wonderful discussions through the years and for being willing to do Friday afternoon experiments.
This study was supported by NIH grant 1R01AI090280 to C.C.K.
Footnotes
Published ahead of print 8 February 2012
REFERENCES
- 1. Adkins S, Kao C. 1998. Subgenomic RNA promoters dictate the mode of recognition by bromoviral RNA-dependent RNA polymerases. Virology 252:1–8 [DOI] [PubMed] [Google Scholar]
- 2. Ahlquist P. 2006. Parallels among positive-strand RNA viruses, reverse-transcribing viruses and double-stranded RNA viruses. Nat. Rev. Microbiol. 4:371–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ahola T, den Boon JA, Ahlquist P. 2000. Helicase and capping enzyme active site mutations in brome mosaic virus protein 1a cause defects in template recruitment, negative-strand RNA synthesis, and viral RNA capping. J. Virol. 74:8803–8811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ahola T, Ahlquist P. 1999. Putative RNA capping activities encoded by brome mosaic virus: methylation and covalent binding of guanylate by replicase protein 1a. J. Virol. 73:10061–10069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Baum A, Sachidnaandam R, Garcia-Sastre A. 2010. Preference of RIG-I for short viral RNA molecules in infected cells revealed by next-generation sequencing. Proc. Natl. Acad. Sci. U. S. A. 107:16303–16308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Blankenberg D, et al. 2010. Galaxy: a web-based genome analysis tool for experimentalists. Curr. Protoc. Mol. Biol. Chapter 19:Unit 19.10.1–19.10.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen J, Ahlquist P. 2000. Brome mosaic virus polymerase-like protein 2a is directed to the endoplasmic reticulum by helicase-like viral protein 1a. J. Virol. 74:4310–4318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen J, Noueiry A, Ahlquist P. 2001. Brome mosaic virus protein 1a recruits viral RNA2 to RNA replication through a 5′ proximal RNA2 signal. J. Virol. 75:3207–3219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dzianott AM, Bujarski J. 1991. Generation and analysis of nonhomologous RNA-RNA recombinants in brome mosaic virus: sequence complementarities at crossover sites. J. Virol. 65:4153–4159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gancarz BL, Hao L, He Q, Newton MA, Ahlquist P. 2011. Systematic identification of novel, essential host genes affecting bromovirus RNA replication. PLoS One 6:e23988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gopinath K, Dragnea B, Kao CC. 2005. Interaction between brome mosaic virus proteins and RNAs: effects on RNA replication, protein expression, and RNA stability. J. Virol. 79:14222–14234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Janda M, Ahlquist P. 1993. RNA-dependent replication, transcription, and persistence of brome mosaic virus RNA replicons in Saccharomyces cerevisiae. Cell 72:961–970 [DOI] [PubMed] [Google Scholar]
- 13. Kao CC, Ahlquist P. 1992. Identification of the domains required for the direct interaction of the helicase-like and polymerase-like RNA replication proteins of brome mosaic virus. J. Virol. 66:7293–7302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kao CC, Quadt R, Hershberger RP, Ahlquist P. 1992. Brome mosaic virus RNA replication protein 1a and 2a form a complex in vitro. J. Virol. 66:6322–6329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kao CC, Singh P, Eckert D. 2001. De novo initiation of viral RNA-dependent RNA synthesis. Virology 287:251–260 [DOI] [PubMed] [Google Scholar]
- 16. Kong F, Sivakumaran K, Kao CC. 1999. The N-terminal half of the brome mosaic virus 1a protein has RNA capping-associated activities: specificity for GTP and S-adenosylmethionine. Virology 259:200–210 [DOI] [PubMed] [Google Scholar]
- 17. Kroner PA, Young B, Ahlquist P. 1990. Analysis of the role of brome mosaic virus la protein domains in RNA replication, using linker insertion mutagenesis. J. Virol. 64:6110–6120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Leonard JN, et al. 2008. The TLR3 signaling complex forms by cooperative receptor dimerization. Proc. Natl. Acad. Sci. U. S. A. 105:258–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li X, et al. 2009. Structural basis of double-stranded RNA recognition by the RIG-I-like receptor MDA5. Arch. Biochem. Biophys. 488:23–33 [DOI] [PubMed] [Google Scholar]
- 20. Liu L, et al. 2008. Structural basis of Toll-like receptor 3 signaling with double-stranded RNA. Science 320:379–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lu C, et al. 2010. The structural basis of 5′-triphosphate double-stranded RNA recognition by RIG-I C-terminal domain. Structure 18:1032–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Luo G, et al. 2000. De novo initiation of RNA synthesis by the RNA-dependent RNA polymerase (NS5B) of hepatitis C virus. J. Virol. 74:851–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Marsh LE, Huntley CC, Pogue GP, Connell JP, Hall TC. 1991. Regulation of (+):(−)-strand asymmetry in replication of brome mosaic virus RNA. Virology 182:76–83 [DOI] [PubMed] [Google Scholar]
- 24. Noueiry AO, Ahlquist P. 2003. Brome mosaic virus RNA replication: revealing the role of the host in RNA virus replication. Annu. Rev. Phytopathol. 41:77–98 [DOI] [PubMed] [Google Scholar]
- 25. O'Reilly EK, Tang N, Ahlquist P, Kao CC. 1995. Biochemical and genetic analyses of the interaction between the helicase-like and polymerase-like proteins of the brome mosaic virus. Virology 214:59–71 [DOI] [PubMed] [Google Scholar]
- 26. O'Reilly E, Paul J, Kao CC. 1997. Analysis of the interaction of viral RNA replication proteins by using the yeast two-hybrid assay. J. Virol. 71:7526–7532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. O'Reilly E, Wang Z, French R, Kao CC. 1998. Interaction between the structural domains of the RNA replication proteins of plant-infecting RNA viruses. J. Virol. 72:7160–7169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Quadt R, Ishikawa M, Janda M, Ahlquist P. 1995. Formation of brome mosaic virus RNA-dependent RNA polymerase in yeast requires coexpression of viral proteins and viral RNA. Proc. Natl. Acad. Sci. U. S. A. 92:4892–4896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ranjith-Kumar CT, et al. 2002. Mechanism of de novo initiation by the hepatitis C virus RNA-dependent RNA polymerase: role of divalent metals. J. Virol. 76:12513–12525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ranjith-Kumar CT, et al. 2009. Agonists and antagonists recognition by RIG-I, a cytoplasmic innate immunity receptor. J. Biol. Chem. 284:1155–1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ranjith-Kumar CT, Wen Y, Baxter N, Bhardwaj K, Kao CC. 2011. A cell-based assay for RNA synthesis by the HCV polymerase reveals new insights on mechanism of polymerase inhibitors and modulation by NS5A. PLoS One 6:e22575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rao AL. 2006. Genome packaging by spherical plant RNA viruses. Annu. Rev. Phytopathol. 44:61–87 [DOI] [PubMed] [Google Scholar]
- 33. Restrepo-Hartwig M, Ahlquist P. 1996. Brome mosaic virus helicase and polymerase-like proteins colocalize on the endoplasmic reticulum at sites of viral RNA synthesis. J. Virol. 70:8908–8916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Restrepo-Hartwig M, Ahlquist P. 1999. Brome mosaic virus RNA replication proteins 1a and 2a colocalize and 1a independently localizes on the yeast endoplasmic reticulum. J. Virol. 73:10303–10309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Saito T, Owen DM, Jiang F, Marcotrigiano J, Gale M., Jr 2008. Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature 454:523–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stawicki S, Kao CC. 1999. Spatial requirements for promoter recognition by a viral RNA-dependent RNA polymerase. J. Virol. 73:198–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schwartz M, et al. 2002. A positive-strand RNA virus replication complex parallels form and function of retrovirus capsids. Mol. Cell 9:505–514 [DOI] [PubMed] [Google Scholar]
- 38. Schwartz M, Chen J, Lee WM, Janda M, Ahlquist P. 2004. Alternate, virus-induced membrane rearrangements support positive-strand RNA virus genome replication. Proc. Natl. Acad. Sci. U. S. A. 101:11263–11268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sivakumaran K, Kim CH, Tayon R, Kao C. 1999. RNA sequence and structural determinants for the recognition and efficiency of RNA synthesis by a viral RNA replicase. J. Mol. Biol. 294:667–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Seo J-K, Kwon S-J, Choi H-S, Kim K-H. 2009. Evidence for alternate states of cucumber mosaic virus replicase assembly in positive- and negative-strand RNA synthesis. Virology 383:284–290 [DOI] [PubMed] [Google Scholar]
- 41. Smirnyagina E, Lin N, Ahlquist P. 1996. The polymerase-like core of brome mosaic virus 2a protein, lacking a region interacting with viral 1a in vitro, maintains activity and 1a selectivity in RNA replication. J. Virol. 70:4729–4736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Subba-Reddy CV, Goodfellow I, Kao CC. 2011. VPg-primed RNA synthesis of norovirus RNA-dependent RNA polymerases by using a novel cell-based assay. J. Virol. 85:13027–13037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sun J, Adkins S, Faurote G, Kao CC. 1996. Initiation of negative-strand RNA synthesis catalyzed by the brome mosaic virus RNA-dependent RNA polymerase: synthesis of oligonucleotides. Virology 226:1–12 [DOI] [PubMed] [Google Scholar]
- 44. Sun J, et al. 2006. Structural and functional analyses of the human Toll-like receptor 3. Role of glycosylation. J. Biol. Chem. 281:11144–11151 [DOI] [PubMed] [Google Scholar]
- 45. Traynor P, Young BM, Ahlquist P. 1991. Deletion analysis of brome mosaic virus 2a protein: effects on RNA replication and systemic spread. J. Virol. 65:2807–2815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Uzri D, Gehrke L. 2009. Nucleotide sequences and modifications that determine RIG-I/RNA binding and signaling activities. J. Virol. 83:4174–4184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wang X, et al. 2005. Brome mosaic virus 1a nucleoside triphosphatase/helicase domain plays crucial roles in recruiting RNA replication templates. J. Virol. 79:13747–13758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhang Z, Schwartz S, Wagner L, Miller WW. 2000. A greedy algorithm for aligning DNA sequences. J. Comput. Biol. 7:203–214 [DOI] [PubMed] [Google Scholar]