

Background: Doxorubicin produces genotoxic stress and p53 activation in both carcinoma and non-carcinoma cells.
Results: AMP-activated protein kinase (AMPK) inhibition by doxorubicin causes p53 accumulation and SIRT1 dysfunction.
Conclusion: AMPK regulates p53.
Significance: Pharmacological activation of AMPK might alleviate the side effects of doxorubicin.
Keywords: AMP Kinase, AMP-activated Kinase (AMPK), Oxidative Stress, p53, Sirt1
Abstract
Doxorubicin, an anthracycline antibiotic, is widely used in cancer treatment. Doxorubicin produces genotoxic stress and p53 activation in both carcinoma and non-carcinoma cells. Although its side effects in non-carcinoma cells, especially in heart tissue, are well known, the molecular targets of doxorubicin are poorly characterized. Here, we report that doxorubicin inhibits AMP-activated protein kinase (AMPK) resulting in SIRT1 dysfunction and p53 accumulation. Spontaneously immortalized mouse embryonic fibroblasts (MEFs) or H9C2 cardiomyocyte were exposed to doxorubicin at different doses and durations. Cell death and p53, SIRT1, and AMPK levels were examined by Western blot. In MEFs, doxorubicin inhibited AMPK activation, increased cell death, and induced robust p53 accumulation. Genetic deletion of AMPKα1 reduced NAD+ levels and SIRT1 activity and significantly increased the levels of p53 and cell death. Pre-activation of AMPK by 5-aminoimidazole-4-carboxamide ribonucleoside or transfection with an adenovirus encoding a constitutively active AMPK (AMPK-CA) markedly reduced the effects of doxorubicin in MEFs from Ampkα1 knock-out mice. Conversely, pre-inhibition of Ampk further sensitized MEFs to doxorubicin-induced cell death. Genetic knockdown of p53 protected both wild-type and Ampkα1−/− MEFs from doxorubicin-induced cell death. p53 accumulation in Ampkα1−/− MEFs was reversed by SIRT1 activation by resveratrol. Taken together, these data suggest that AMPK inhibition by doxorubicin causes p53 accumulation and SIRT1 dysfunction in MEFs and further suggest that pharmacological activation of AMPK might alleviate the side effects of doxorubicin.
Introduction
Doxorubicin is an anthracycline antibiotic that is used widely in treatment of cancers, including leukemia, prostate cancer, and breast cancer. The significant side effects to non-carcinoma cells, especially cardiomyocytes, limit its clinical utilization (1). The pathophysiological mechanism of doxorubicin-induced tissue injury is poorly understood. Reactive oxygen species have been reported to play an important role in doxorubicin-induced cardiomyopathy (2, 3), and several antioxidants have been shown to reduce doxorubicin-induced cardiotoxicity in animal experiments (2, 4). However, antioxidant supplementation has only a very limited protective effect on doxorubicin-induced cardiotoxicity in clinically relevant chronic animal models or clinical trials (5, 6). These results suggest that other mechanisms may be involved in doxorubicin-induced cardiotoxicity, and recently, researchers suggest that toxicity associated with doxorubicin is mediated by the p53 protein (7, 8).
p53 is well known as a tumor suppressor that acts as a sequence-specific transcription factor to control cellular proliferation or apoptosis by regulating wide variety of genes, including p21/WAF1, BAX, PUMA, and NOXA (9, 10). p53 also plays a key role in regulating cellular stress responses (11) and in cell survival under stressful conditions. p53 facilitates the transient adaptation of cells to multiple stresses, including UV-induced DNA damage, by increasing DNA repair on cell cycle arrest (12, 13). In the cytoplasm, p53 has a transcript-independent role in regulating cellular apoptosis and metabolism (14). The p53 protein has a fast turnover rate and is maintained at low levels at normal physiological conditions. Under conditions of cellular stress, such as genotoxic stress, p53 is stabilized and accumulates. Consistent with its broad and sometimes contradictory functions, the regulation of p53 activation and stabilization is complex. In addition to protein stability, p53 regulatory mechanisms include phosphorylation, methylation, and acetylation (15). Multiple modifications of p53 are thought to be required for its transcript regulation function (16). Recent research has highlighted the importance of acetylation modification on p53 function, stabilization, and subcellular localization, and the class III NAD+-dependent deacetylase SIRT1 has been reported to play a key role in p53 acetylation levels (17). The outcomes of p53 induction or stabilization are determined by the cell type, intensity of stress, and the genetic background (18).
AMP-activated protein kinase (AMPK)2 is a heterotrimeric serine/threonine protein kinase complex that includes a catalytic α-subunit and regulatory β- and γ-subunits (19). AMPK is a well known energy sensor whose activity is regulated by energy status (20, 21). Several upstream kinases, including LKB1, CaMKKβ, and TAK1, activate AMPK by phosphorylation of a threonine residue (Thr172) in the kinase domain of the α-subunit (22). Beside its role in maintaining cellular or whole body energy homeostasis, AMPK is reported to play important roles in the genotoxic stress response and in the regulation of apoptosis (23). Activated AMPK is reported to directly phosphorylate p53 at Ser15 in response to glucose deprivation (24). However, the role of AMPK activation in the induced genotoxic stress response and its underlying mechanisms are not clear.
We previously reported that AMPKα1 is required for erythrocyte survival (25) and that AMPK is important for the survival of endothelial cells under conditions of hypoxia and energy deprivation (26). Here, we use a genetically modified cellular model to investigate the roles of AMPK in the genotoxic stress response. We report that doxorubicin inhibited AMPK activation, resulting in accumulation of p53. We find that AMPK activation protects non-carcinoma cells from genotoxic stress-induced apoptosis by regulating the function and stability of p53 through direct phosphorylation and SIRT1-dependent acetylation.
EXPERIMENTAL PROCEDURES
Reagents and Animals
Rabbit anti-AMPKα, rabbit anti-phospho-acetyl-CoA carboxylase, rabbit anti-caspase-3, rabbit anti-acetyl-p53 (Lys382 in human, and Lys379 in mouse), and rabbit anti-phospho-p53 (Ser15) antibodies were obtained from Cell Signaling Technology (Beverly, MA). Goat anti-AMPKα1, goat anti-AMPKα2, mouse anti-p53, and mouse anti-p21 antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Mouse anti-GAPDH antibody was from Abcam, Inc. (Cambridge, MA). Other chemicals and organic solvents of the highest available grade were obtained from Sigma-Aldrich. The H9C2 cell line was obtained from ATCC and cultured in DMEM with 10% FBS. Ampkα1−/− mice and Ampkα2−/− mice were described elsewhere (27, 28). Mice were handled in accordance with study protocols approved by the Institutional Animal Care and Use Committee of the University of Oklahoma Health Science Center (Oklahoma City, OK).
Isolation, Spontaneous Immortalization, and Culture of Mouse Embryonic Fibroblasts
Mouse embryonic fibroblasts (MEFs) were isolated from 13.5-days post-coitus embryos from Ampkα1-deficient (AMPKα1−/−), Ampkα2-deficient (Ampkα2−/−), or WT mice and cells were immortalized using the 3T3 protocol as described previously (29). Briefly, 13.5-day mice embryos were decapitated, thoroughly minced, and trypsinized. The dissociated cells were resuspended. To immortalize MEFs, cells were passaged according to the 3T3 protocol (3 × 105 cells were plated per 60-mm dish every 3 days) continuously until growth rates in culture stabilized. Cells were then cultured for an additional 15 passages (to about passage 35) and at that point were considered immortalized and used for experiments. MEFs were cultured in DMEM (Invitrogen) containing 10% fetal bovine serum (Invitrogen) and 50 μg/ml penicillin/streptomycin.
Western Blot
Cells were washed with ice-cold PBS two times and then lysed in 25 mm Tris·HCl, pH 7.6, 150 mm NaCl, 1% Nonidet P-40, 1% sodium deoxycholate, 0.1% SDS, supplemented with 0.25 mg/ml phenylmethylsulfonylfluoride, 1× protease inhibitor, and 1× phosphatase inhibitor mixtures (Calbiochem), using 0.15 ml of lysis solution per 1 × 107 cells. An equal volume of 2× sample buffer (62.5 mm Tris-Hcl, 25% glycerol, 2% SDS, 0.02% bromphenolblue,5% β-mercaptoethanol, pH 6.8) was added, and cleared extracts were frozen at −80 °C. Protein concentration was determined by the Bradford method. Equal protein amounts were denatured, electrophoresed, and blotted. For chemiluminescence, HRP-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA) were applied, and SuperSignal West Dura reagents (Pierce) were used for detection.
NAD+-NADH Quantification
Intracellular NAD+, NADH content, and NAD+/NADH ratio were determined using BioVision's NAD+/NADH quantification kit (Mountain View, CA) according to the manufacturer's instructions.
Mitochondria Labeling and Immunocytochemical Staining
Mitochondria were labeled with BacMam 2.0 CellLight Mito-RFP (Invitrogen) according to manufacturer's instructions. The day after Mito-RFP labeling, cells were trypsinized, seeded on sterilized glass coverslips, and fixed by 4% (v/v) paraformaldehyde in PBS. After rinsing, cells were permeabilized in 0.1% Triton X-100. After blocking the coverslips with 10% goat-serum, cells were incubated with mouse-anti p53 primary antibody at 4 °C overnight. Cells were then incubated with Dylight 488 anti-mouse IgG (Vector Laboratories, Burlingame, CA), and the nucleus was stained with DAPI. The coverslips were mounted and images were captured using an Olympus BX51 microscope and DP-2BSW software.
mRNA Isolation and Semiquantitative RT-PCR
Total mRNA was isolated and purified using the RNeasy mini kit from Qiagen (Valencia, CA) according to the manufacturer's instructions. cDNA was synthesized from isolated mRNA using the iScript cDNA synthesis kit (Bio-Rad), as described by the manufacturer. RT-PCR was performed using 1 μl of first-strand cDNA as template with specific primers for p53 (5′-GGA AAT TTG TAT CCC GAG TAT CTG-3′, 5′-GTC TTC CAG TGT GAT GAT GGT AA-3′) and β-actin (5′-CCT GAA CCC TAA GGC CAA CC-3′, 5′-GCA ATG CCT GGG TAC ATG GT-3′). Amplification was performed using the Mastercycler Thermal Cycler (Eppendorf) in a 20-μl mixture under following condition: 95 °C for 5 min, followed by 30 cycles at 95 °C for 30 s, 60 °C for 30 s, and 72 °C for 30 s, and a final extension at 72 °C for 2 min. Expression of the β-actin housekeeping gene was used as a control. PCR products were analyzed by 1.5% agarose gel electrophoresis.
Adenoviral Infection
A replication-defective adenoviral vector expressing green fluorescent protein (Ad-GFP) served as a control for all adenoviral experiments. AMPK-CA adenoviral vector was generated as described previously (30). MEFs were infected with Ad-GFP or Ad-AMPK-CA overnight in medium supplemented with 10% FBS. These conditions typically produced an infection efficiency of at least 80% as determined by GFP expression.
siRNA Transfection in MEFs
Mouse p53 siRNA and control siRNA were obtained from Santz Cruz Biotechnology, and transient siRNA transfection was carried out according to the Santa Cruz Biotechnology protocol. Briefly, p53 siRNA and control siRNA were dissolved in double distilled H2O to prepare a 10 μmol/liter stock solution. Wild-type and Ampkα1−/− MEFs grown in six-well plates were transfected with siRNA in Opti-MEM (Invitrogen) containing RNAiMax (Invitrogen). For each transfection, 250 μl of transfection medium containing 5 μg of siRNA was gently mixed with 250 μl of transfection medium containing 5 μl of transfection reagent. After a 20-min incubation at room temperature, the mixture was added to cells in 2 ml of culture medium and cultured for 48 h before treatment.
p53 Stable Knockdown Cell Line Set Up
Mouse-specific p53 shRNA and control shRNA lentiviral particles were obtained from Santa Cruz Biotechnology. The lentivirus vector-based RNAi approach was used to knockdown p53 in wild-type and Ampkα1−/− MEFs according to the manufacturer's instructions. Briefly, MEFs seeded in 24-well plates were infected with p53-shRNA lentiviral particles and control-shRNA lentiviral particles. Multiple independent clones were isolated and expanded in the complete growth medium containing puromycin (2.5 μg/ml). shRNA expression efficiency was analyzed by Western blot.
Statistical Analysis
The two-tailed Student's t test or two-way analysis of variance followed by Bonferroni post hoc analysis was used to determine statistical differences between various experimental and control groups. A p value < 0.05 was considered statistically significant.
RESULTS
AMPKα1 Is Predominant Isoform in Mouse Embryonic Fibroblast Cells
AMPKα1 is reported to be the predominant isoform of AMPKα in major cell types and tissues, whereas AMPKα2 is reported to be the predominant isoform expressed in skeletal muscle and cardiomyocytes (31). The expression of AMPKα1 and AMPKα2 in MEFs has not been reported. Here, we report that AMPKα1 is the predominant isoform of AMPKα in murine embryonic fibroblast cells, as demonstrated by Western blot. Using antibodies specific for either AMPKα1 or AMPK α2, we confirmed that both AMPKα1 and AMPKα2 are expressed in MEFs (Fig. 1A). We proceeded to use an antibody that recognizes both AMPKα1 and AMPKα2 isoforms. As shown in Fig. 1A, AMPKα was largely absent, as expected, in MEFs from Ampkα1−/− mice, whereas there was no significant difference in the expression of total AMPKα between Ampkα2−/− and wild-type MEFs. Quantitative analysis (Fig. 1B) indicated that AMPKα2 and AMPKα1 account for ∼20 and 80% of AMPKα MEFs, respectively. These results indicate that AMPKα2 is a minor AMPKα isoform in MEFs. Our subsequent experiments were mainly focused on the Ampkα1−/− deletion in MEFs.
FIGURE 1.

AMPKα1 is the predominant isoform of AMPK expressed in mouse embryonic fibroblasts, and long term treatment of doxorubicin inhibits AMPK activation. A, AMPKα1, AMPKα2, and total AMPKα protein expression in MEFs from wild-type, Ampkα1−/−, and Ampkα2−/− mice were detected by immunoblotting. B, qualification of the expression profile of AMPKα1 and AMPKα2 in MEFs. C and D, doxorubicin treated at 0.5, 1.0 μm for 16 h inhibits AMPK function in wild-type MEFs (#, *, p < 0.05 versus control; n = 3). E, 2 mm 5-aminoimidazole-4-carboxamide ribonucleoside (AICAR) inhibited and 1 μm compound C sensitized wild-type MEFs from doxorubicin (1.0 μm for 16 h)-induced cellular apoptosis. DMSO, dimethyl sulfoxide; Dox, doxorubicin; Ctrl, control; p-ACC, phospho-acetyl-CoA carboxylase.
Prolonged Doxorubicin Treatment Inhibits AMPK in MEF
Next, we determined the effects of doxorubicin on AMPK in MEFs. As depicted in Fig. 1, C and D, doxorubicin inhibited AMPK phosphorylation at Thr172 after 16 h of treatment. The Ser79 phosphorylation level of acetyl-CoA carboxylase, a well characterized downstream target of AMPK, was concomitantly reduced (Fig. 1, C and D).
AMPK Activation Protects from Doxorubicin-induced Apoptosis
As expected, doxorubicin induced cellular apoptosis in wild-type MEFs (Fig. 1E). Pre-activation of AMPK by the pharmacological activator 5-aminoimidazole-4-carboxamide ribonucleoside protected wild-type MEFs from doxorubicin-induced apoptosis, whereas inhibition of AMPK by compound C, a specific pharmacological inhibitor, further enhanced doxorubicin-induced apoptosis (Fig. 1E).
Ampkα1 Deletion Sensitizes MEFs to Genotoxic Stress-induced Cell Death and Increased p53 Levels
Consistent with the above observation, genetic deletion of Ampkα1 led to higher cell apoptosis even under normal growth conditions (Fig. 2A). Ampkα1-deficient cells were much more sensitive to doxorubicin-induced apoptosis as indicated by increased levels of the apoptosis marker caspase-3 in Ampkα1-deficient MEFs (Fig. 2B). The morphology changes after doxorubicin treatment (Fig. 2C) further confirmed that Ampkα1−/− MEFs were much more sensitive to doxorubicin-induced genotoxic stress than wild-type MEFs.
FIGURE 2.

Genetic deletion of AMPKα1 sensitizes MEFs to doxorubicin-induced apoptosis. A, Ampkα1-deficient MEFs show increased DNA damage and apoptosis compared with wild-type MEFs. B, genetic deletion of Ampkα1 increased doxorubicin-induced apoptosis in MEFs. C, morphology changes in wild-type and Ampkα1−/− MEFs after doxorubicin treatment. PARP, poly (ADP-ribose) polymerase.
Activation of AMPK in MEFs Protects Cells from Doxorubicin-induced Cell Death and p53 Induction
Because the p53-p21 pathway plays an important role in controlling cellular proliferation, the genotoxic stress response, and cellular survival (32), we examined the expression of p53 and p21 in MEFs. As expected, p53 protein was detected at very low levels in wild-type MEFs under normal conditions but showed dramatic accumulation, associated with significant induction of p21, when cells were treated with doxorubicin (Fig. 3A). In Ampkα1−/− MEFs, even under normal culture conditions, there was a higher amount of p53 expressed and much lower p21 expression compared with wild-type control cells (Fig. 3A).
FIGURE 3.

AMPK deletion leads to defects in p53 function and increased p53 accumulation. A, low p53 levels in wild-type MEFs under normal conditions and doxorubicin-induced robust accumulation of p53 and p21 induction. B, Ampkα1−/− MEFs showed stronger DNA damage signals but lower Ser15 phosphorylation levels of p53 compared with wild-type MEFs under doxorubicin treatment. C and D, AMPK-CA increased p53 phosphorylation levels and rescued Ampkα1−/− MEFs from doxorubicin-induced apoptosis. Ctrl, control.
To confirm these results, we transfected MEFs with the AMPK-CA adenovirus. Our results confirmed that activation of AMPK increased p53 phosphorylation at Ser15 (Fig. 3C) and that AMPK-CA transfection lowered p53 protein levels and protected both wild-type and Ampkα1−/− MEFs from doxorubicin-induced apoptosis (Fig. 3, C and D). Taken together, these results indicate that AMPK inactivation by doxorubicin might play a causative role in doxorubicin-induced cellular apoptosis in normal, non-carcinoma cells.
p53 Half-life Is Increased in Ampkα1−/− MEFs
Because no significant difference in p53 gene expression levels was found between Ampkα1−/− MEFs and wild-type MEFs (Fig. 4A), we hypothesized that p53 should be stabilized in Ampkα1−/− MEFs. We used cycloheximide to block protein synthesis and measure the half-life of p53. The half-life of p53 in wild-type MEFs was <15 min, which is consistent with previous findings (33). However, the half-life of p53 in Ampkα1−/− MEFs was prolonged dramatically to ∼12 h (Fig. 4, B–E).
FIGURE 4.

Elongated half-life of p53 protein in Ampkα1-deficient MEFs. A, PCR results of mRNA expression levels of p53 in wild-type and Ampkα1−/− MEFs. B, p53 levels in wild-type MEFs treated with cycloheximide (CHX, 50 μg/ml) for the indicated times. C, Ampkα1−/− MEFs were treated with cycloheximide (50 μg/ml) for the indicated times, and p53 levels were detected with Western blot. D and E, quantitative analysis of the half-life of p53 in wild-type and Ampkα1−/− MEFs. Ctrl, control.
p53 Is Stabilized in Ampkα1−/− MEFs Due to Increased Acetylation and Decreased Degradation
The stability of p53 is regulated by multiple post-translational modifications, including phosphorylation and acetylation (34). Phosphorylation of p53 at serine 15 is not only critical for p53 activation but also is reported to disrupt the MDM2 and p53 interaction to stabilize p53 during the DNA damage response (35). The ataxia-telangiectasia-mutated (ATM) protein kinase has a fundamental role in the DNA damage response and is rapidly and specifically activated in response to DNA double-strand breaks (36). As shown in Fig. 3B, when cells were treated with doxorubicin, the phosphorylation level of ATM increased, and the Ser15 phosphorylation level of p53 decreased in Ampkα1−/− MEFs compared with wild-type controls. These findings indicate higher DNA damage levels are associated with defective p53 function in Ampkα1−/− MEFs. However, we have shown that phosphorylation of Ser15 of p53 is actually decreased in Ampkα1−/− MEFs and that p53 was stabilized. Thus, there must be another mechanism(s) leading to the increased stability of p53 when AMPKα1 is absent.
Modification by acetylation is another important mechanism to regulate the stability of p53 (37) and is regulated by NAD+-dependent SIRT1 activation (38, 39). We tested the acetylation levels of p53 using a specific antibody for Lys379 in p53. As shown in Fig. 5A, acetyl-p53 levels were dramatically increased in Ampkα1−/− cells. Inhibition of AMPK by compound C also caused an increase in p53 acetylation levels in wild-type cells treated with doxorubicin (Fig. 5B).
FIGURE 5.

Increased acetylation and decreased degradation of p53 in Ampkα1−/− MEFs due to defective SIRT1 activation. A, increased acetylation of p53 (Ace-p53) and total p53 levels in Ampkα1−/− MEFs compared with wild-type MEFs. B, inhibition of AMPK by compound C (CmpC) increased the doxorubicin (Dox)-induced acetylation of p53 in wild-type MEFs. C, intracellular NAD+ content (**, p < 0.01; n = 6 in each group). D, NADH levels in MEFs. E, calculated NAD+/NADH ratio (**, p < 0.01; n = 6 in each group). F, p53 protein levels after resveratrol treatment in Ampkα1-deficient MEFs for 16 h at the indicated dose. G, p53 protein levels after resveratrol treatment in Ampkα1−/− MEFs at 100 μm for indicated time points. H, MG132 blocks the effect of resveratrol in decreasing p53 levels in Ampkα1−/− MEFs. Ctrl, control; p-ACC, phospho-acetyl-CoA carboxylase; AICAR, 5-aminoimidazole-4-carboxamide ribonucleoside.
Decreased NAD+/NADH Ratio in Ampkα1−/− MEFs
Activation of SIRT1 is dependent on intracellular NAD+ content and NAD+/NADH ratios (40), and AMPK activation is reported to increase NAD+ content and change intracellular NAD+/NADH ratios (41). Our experimental results support this relationship as the NAD+ content and NAD+/NADH ratio were decreased significantly in Ampkα1−/− MEFs compared with wild-type MEFs (Fig. 5, C–E).
SIRT1 Activation with Resveratrol Decreases p53 Levels in Ampkα1−/− MEFs
We also treated cells with the SIRT1 activator resveratrol, which significantly decreased p53 levels in a dose- and time-dependent manner (Fig. 5, F and G). More importantly, pretreatment of cells with the proteasome inhibitor MG132 strongly blocked the resveratrol-induced decrease in p53 levels in Ampkα1-deficient MEFs (Fig. 5H).
Cytoplasmic Localization of p53 in Ampkα1−/− MEFs
Because the cytoplasmic localization of p53 is reported to play an important role in the induction of cellular apoptosis by a mechanism that is independent of its role as a transcription factor, we investigated the subcellular distribution of p53. As shown in Fig. 6A, immunocytochemistry staining indicated that some p53 was localized in cytoplasm even in mitochondria in Ampkα1−/− MEFs, whereas in wild-type MEFs, p53 was not detectable under normal culture condition (data not shown). Western blot analysis of subcellular fractions further confirmed the cytoplasmic localization of p53 in Ampkα1−/− MEFs (Fig. 6B).
FIGURE 6.

Cytoplasmic localization of p53 in Ampkα1-deficient MEFs and genetic silencing of p53 partially protect Ampkα1−/− MEFs from doxorubicin-induced apoptosis. A, Ampkα1−/− MEFs were labeled with mito-RFP (red) and then stained with anti-p53 (green) and DAPI (blue). The yellow shading in the merged image indicates mitochondria-localized p53 (phase contrast image; magnification, 40×). B, an obvious increase in cytoplasmic (Cyto) localization of p53 is seen in Ampkα1−/− MEFs compared with wild-type controls. C and D, transient silencing of p53 by siRNA in wild-type (C) and Ampkα1-deficient (D) MEFs protects cells against doxorubicin (Dox)-induced apoptosis. E and F, stable knockdown of p53 by shRNA lentiviral particles protect MEFs from doxorubicin-induced apoptosis. G, representative phase contrast images of MEFs after doxorubicin (1 μm) treatment in wild-type and Ampkα1−/− MEFs. Ctrl, control; Nucl, nucleus; PARP, poly (ADP-ribose) polymerase; LDH, Lactate dehydrogenase.
Knockdown of p53 in Ampkα1−/− MEFs Protects Cells from Genotoxic Stress-induced Cell Apoptosis
To investigate the role of p53 accumulation in cellular apoptosis, we knocked down p53 expression in MEFs using a specific siRNA. As shown in Fig. 6, C and D, p53 knockdown partially protected Ampkα1−/− MEFs from genotoxic stress-induced apoptosis when compared with control siRNA-transfected cells. We also generated p53 stable knockdown cells using shRNA lentinovirus infection followed by puromycin (2.5 g/ml) selection, and similar results were obtained with silencing of p53 by shRNA (Fig. 6, E and F). Morphology changes in wild-type and Ampkα1−/− MEFs transfected with control shRNA or p53 shRNA provide further support for the role of p53 (Fig. 6G).
Doxorubicin Inhibits Phosphorylation of AMPK and Acetyl-CoA Carboxylase, Increases Levels of p53, and Promotes Apoptosis in Cardiomyocytes
Because the major side effect of doxorubicin is cardiomyopathy, we next determined the effects of doxorubicin on AMPK, p53, and apoptosis in cardiomyocytes. To this end, H9C2 cardiomyocytes were incubated with doxorubicin (1 μm) for 1 to 16 h. As depicted in Fig. 7A, short exposure (1 h) of H9C2 to doxorubicin slightly increased the phosphorylation of AMPK at Thr172 and acetyl-CoA carboxylase at Ser79 without altering the levels of p53, cleaved caspase-3, and cleaved poly (ADP-ribose) polymerase (PARP). However, increased incubation time to 4 h inhibited the phosphorylation of both AMPK and acetyl-CoA carboxylase (Fig. 7A). Prolonged incubation of H9C2 to doxorubicin (>16 h) markedly reduced the levels of p-AMPK and phospho-acetyl-CoA carboxylase (Fig. 7A). Consistently, doxorubicin markedly increased the levels of p53 with increased detection of cleaved caspase-3 after 16 h incubation (Fig. 7A).
FIGURE 7.

Doxorubicin inhibits the phosphorylation of AMPK and acetyl-CoA carboxylase (ACC), increases the levels of p53, and promotes apoptosis in cardiomyocytes. A, H9C2 cardiomyocytes were treated with doxorubicin at 1 μm for the indicated times. Western blots were performed for indicated target proteins. B, H9C2 cardiomyocytes were treated with doxorubicin at indicated doses for 16 h. Proteins were detected in Western blots by using the specific antibodies (* and #, p < 0.05 compared with control group, respectively). Ctrl, control; PARP, poly (ADP-ribose) polymerase.
Next, we examined the dose-dependent effects of doxorubicin on H9C2 cardiomyocytes. H9C2 cells were exposed to doxorubicin (0.25 to 2 μm) for 16 h. Exposure of H9C2 to doxorubicin (0.25 to 2 μm) dose-dependently inhibited both p-AMPK and phospho-acetyl-CoA carboxylase (Fig. 7B). Similarly, doxorubicin (0.25 to 2 μm) markedly increased the detection of p53 in H9C2 (Fig. 7B). This increase of p53 was paralleled with increased detection of cleaved caspase-3 and cleaved poly (ADP-ribose) polymerase in H9C2. Taken together, our results suggest that increased p53 and apoptosis in cardiomyocytes might be linked to doxorubicin-induced AMPK inhibition.
DISCUSSION
In this study, we unveiled a novel mechanism by which AMPKα can modulate p53 accumulation and contribute to doxorubicin-induced apoptosis in non-carcinoma cells. Our results suggest that AMPK activation, via SIRT1 activation, protects non-carcinoma cells from doxorubicin-induced cellular apoptosis by modulation of p53 function and stability.
There are several lines of evidence supporting the key role of AMPK inhibition in the toxicity of doxorubicin in non-carcinoma cells. First, in wild-type MEF cells, we found that doxorubicin had an inhibitory effect on AMPK activation (Fig. 1C), consistent with studies in rat heart tissues (42). Second, the MEFs isolated from Ampkα1−/− mice showed increased apoptosis when exposed to doxorubicin compared with wild-type controls (Fig. 2, B and C), indicating that AMPKα1 is important for a normal genotoxic stress response. Notably, we observed that doxorubicin might activates AMPK at initial stage (<4 h), but it did not induce cell apoptosis under short time treatment under our current conditions (Fig. 7, A and B), similar results were reported by other groups (43, 44), which might be due to a transient increase in reactive oxygen species levels after adding doxorubicin (44). But the detailed mechanism of this transient activation of AMPK by doxorubicin is unknown, which warrants further investigation. The ATM protein kinase has a fundamental role in the DNA damage response and is rapidly and specifically activated in response to DNA double-strand breaks (36). ATM activation induces a two-phase dynamics of p53 in the DNA damage response, which is believed to play a key role in the fate decision between cell survival and death (45). Fig. 3B shows that Ampkα1−/− cells had a higher level of phosphorylated ATM (phosphorylated at serine 428), which is consistent with the increased sensitivity of these cells to doxorubicin-induced DNA damage. Overall, our results demonstrate a protective role of AMPK against doxorubicin-induced cellular apoptosis in MEFs.
p53 plays an essential role in cellular stress response pathways (46). It has a dual role in cellular fate decisions, possessing both anti-apoptotic and proapoptotic functions, which can be dependent or independent on its transcriptional function (47, 48). It is believed that p53 accumulation is essential for doxorubicin-induced cellular death (49) and doxorubicin-induced p53 accumulation might be through ATM-dependent pathway (50). Consistent with its multiple functions, the intracellular protein levels of p53 and its activities are regulated tightly by complicated pathways, including transcriptional regulation and post-translational modifications (16). AMPK is activated under stressful conditions and the function of AMPK in cell fate has been linked with the p53 pathway. However, data supporting a relationship between AMPK activation and p53 function are largely unclear and even contradictory. Jones et al. (24) reported that AMPK helps cells survival under energy stress conditions through direct phosphorylation of p53 at Ser15 to regulate its function. In contrast, Kizaki et al. (51) reported that, under similar glucose depletion conditions, AMPK activation induced transcriptional up-regulation of p53 and increased phosphorylation of p53 at Ser46 leading to cellular apoptosis. These apparently contradictory findings could be due to different cell types and different treatment durations. Here, we found that AMPK was important for p53 Ser15 phosphorylation under doxorubicin-induced genotoxic stress in MEFs. AMPK regulates p53 protein levels largely through effects on p53 stability by post-transcriptional modification. No significant change in p53 transcriptional level was observed under those conditions, which is supported by our observations of p53 stability and p53 mRNA expression levels under doxorubicin treatment in AMPK-integrated cells and Ampkα1-deficient cells (Fig. 4, A–E). More importantly, restoration of AMPK function in Ampkα1−/− MEFs by infection with the AMPK-CA adenovirus resulted in a lowering of the p53 protein level in these cells (Fig. 3C).
p53 was the first non-histone protein reported to be acetylated by histone acetyl transferases (52). Our experimental results show that AMPK activation affects p53 stability and function through p53 acetylation. High p53 acetylation levels were observed in Ampkα1−/− MEFs (Fig. 5A), and inhibition of AMPK by compound C dramatically increased doxorubicin-induced p53 acetylation levels in wild-type MEFs (Fig. 5B). Acetylation of p53 is reversible, and deacetylation is mediated largely by the tightly regulated class III NAD+-dependent deacetylase SIRT1 (53, 54). AMPK is reported to regulate energy metabolism by modulating NAD+/NADH levels and SIRT activity in C2C12 myocytes (41), and similar results were reported by Auwerx et al. (55) in skeletal muscle using an Ampkγ3 knock-out mouse model. In this report, we used genetic knock-out MEFs to demonstrate the regulation of NAD+/NADH levels by AMPK to control SIRT1 function. Overall, our results suggest that AMPK regulates p53 stability and function in MEFs during doxorubicin-induced genotoxic stress conditions by regulation of SIRT1-dependent deacetylation (Fig. 8).
FIGURE 8.
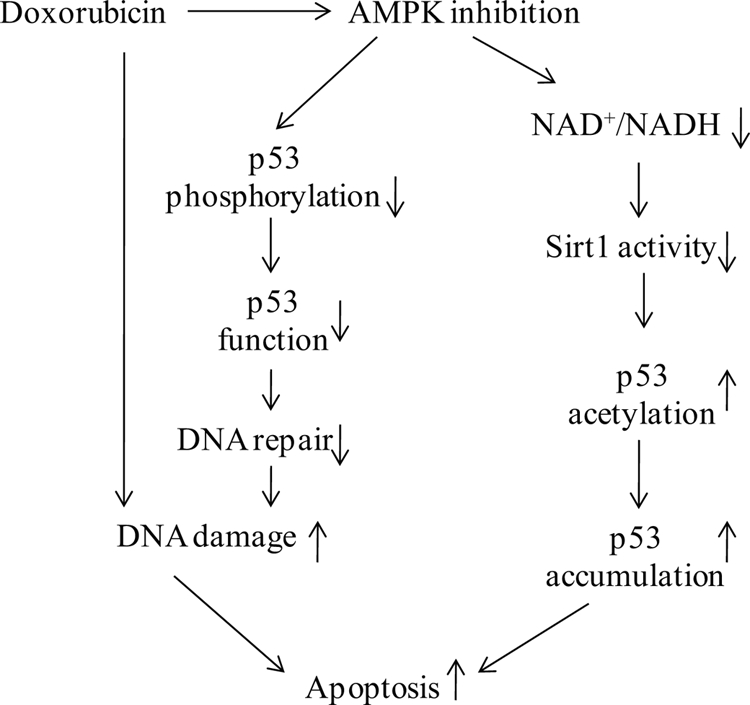
Proposed mechanisms of AMPK regulation of the genotoxic stress response in mouse embryonic fibroblast cells. Genotoxic stress inducers, such as doxorubicin, induce DNA damage and inhibit AMPK activation. AMPK inactivation by doxorubicin leads to p53 dysfunction and an altered NAD+/NADH ratio, resulting in decreased SIRT1 activation, which, in turn, leads to the accumulation of p53 and cellular apoptosis.
In clinical studies, doxorubicin has been proven to be a powerful and efficient genotoxic stress inducer to fight cancer, but at the same time, it leads to significant damage to normal tissues as well (56), especially in cardiomyocytes (57, 58). However, the role of AMPK in doxorubicin-induced apoptosis is a matter of debate. For example, Chen et al. (44) and Ji et al. (43) have described that doxorubicin-induced early AMPK (<6 h) contributes to cellular death (>16 h) in cultured H9C2 cells or in cancer cell lines. Whether or not doxorubicin-induced AMPK activation at early stage (<6 h) is responsible for late stage apoptosis is unknown because prolonged incubation of doxorubicin inhibits AMPK. Thus, we consider prolonged AMPK inhibition but not a transient AMPK activation by doxorubicin was responsible for doxorubicin-induced apoptosis. Indeed, doxorubicin has been reported to inhibit AMPK activation in isolated perfused heart (42) and in MHC-CB7 mouse hearts (8). In support of this notion, several most recent publications (59–61) have provided evidence that activation of AMPK protects cells (HL-1 cardiomyocyte cell line, cultured rat cardiomyocytes, and C57BL/6 mouse cardiomyocytes, respectively) from doxorubicin-induced cellular apoptosis. Here, we also found a strong inhibitory effect of doxorubicin on AMPK activation (Fig. 1, C and D), but the details of the mechanism by which doxorubicin inhibits AMPK activation is unknown. Moreover, how doxorubicin causes a transient induction of AMPK-α at the early stage but inhibits its activity in later stage warrants further investigation. Our results suggest that AMPK inhibition plays an important role in mediating the toxic effects of doxorubicin in non-carcinomal cells via effects on the post-translational modification of p53.
In conclusion, our research indicates that AMPK regulates responses to genotoxic stress response via novel p53-dependent mechanisms and suggests potential strategies to attenuate doxorubicin-induced toxicity in clinical patients.
Acknowledgment
We acknowledge Dr. Benoit Viollet (Institut Cochin, Université Paris Descartes, CNRS UMR 8104, Paris, France) for providing Ampk KO mice.
This work was supported in part by National Institutes of Health Grants HL074399, HL079584, HL080499, HL089920, HL096032, HL105157, and HL110448. This work was also supported by a research award from the American Diabetes Association and by the Warren Chair in Diabetes Research from the University of Oklahoma Health Sciences Center (all to M.-H. Z.).
- AMPK
- AMP-activated protein kinase
- MEF
- mouse embryonic fibroblast cell(s)
- Ad
- adenovirus
- CA
- constitutively active
- ATM
- ataxia-telangiectasia-mutated protein kinase.
REFERENCES
- 1. Kalyanaraman B., Joseph J., Kalivendi S., Wang S., Konorev E., Kotamraju S. (2002) Doxorubicin-induced apoptosis implications in cardiotoxicity. Mol. Cell Biochem. 234–235, 119–124 [PubMed] [Google Scholar]
- 2. Kang Y. J., Chen Y., Epstein P. N. (1996) Suppression of doxorubicin cardiotoxicity by overexpression of catalase in the heart of transgenic mice. J. Biol. Chem. 271, 12610–12616 [DOI] [PubMed] [Google Scholar]
- 3. Ewer M. S., Ewer S. M. (2010) Cardiotoxicity of anticancer treatments: What the cardiologist needs to know. Nat. Rev. Cardiol. 7, 564–575 [DOI] [PubMed] [Google Scholar]
- 4. Kang Y. J., Chen Y., Yu A., Voss-McCowan M., Epstein P. N. (1997) Overexpression of metallothionein in the heart of transgenic mice suppresses doxorubicin cardiotoxicity. J. Clin. Invest. 100, 1501–1506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ferreira A. L., Matsubara L. S., Matsubara B. B. (2008) Anthracycline-induced cardiotoxicity. Cardiovasc. Hematol. Agents Med. Chem. 6, 278–281 [DOI] [PubMed] [Google Scholar]
- 6. Simunek T., Stérba M., Popelová O., Adamcová M., Hrdina R., Gersl V. (2009) Anthracycline-induced cardiotoxicity: Overview of studies examining the roles of oxidative stress and free cellular iron. Pharmacol. Rep. 61, 154–171 [DOI] [PubMed] [Google Scholar]
- 7. Nithipongvanitch R., Ittarat W., Cole M. P., Tangpong J., Clair D. K., Oberley T. D. (2007) Mitochondrial and nuclear p53 localization in cardiomyocytes: Redox modulation by doxorubicin (Adriamycin)? Antioxid. Redox Signal. 9, 1001–1008 [DOI] [PubMed] [Google Scholar]
- 8. Zhu W., Soonpaa M. H., Chen H., Shen W., Payne R. M., Liechty E. A., Caldwell R. L., Shou W., Field L. J. (2009) Acute doxorubicin cardiotoxicity is associated with p53-induced inhibition of the mammalian target of rapamycin pathway. Circulation 119, 99–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vousden K. H., Lu X. (2002) Live or let die: The cell's response to p53. Nat. Rev. Cancer 2, 594–604 [DOI] [PubMed] [Google Scholar]
- 10. Michalak E. M., Villunger A., Adams J. M., Strasser A. (2008) In several cell types tumor suppressor p53 induces apoptosis largely via Puma but Noxa can contribute. Cell Death Differ. 15, 1019–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Levine A. J., Oren M. (2009) The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 9, 749–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ford J. M., Hanawalt P. C. (1997) Expression of wild-type p53 is required for efficient global genomic nucleotide excision repair in UV-irradiated human fibroblasts. J. Biol. Chem. 272, 28073–28080 [DOI] [PubMed] [Google Scholar]
- 13. Rubbi C. P., Milner J. (2003) p53 is a chromatin accessibility factor for nucleotide excision repair of DNA damage. EMBO J. 22, 975–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Green D. R., Kroemer G. (2009) Cytoplasmic functions of the tumor suppressor p53. Nature 458, 1127–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dai C., Gu W. (2010) p53 post-translational modification: Deregulated in tumorigenesis. Trends Mol. Med. 16, 528–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kruse J. P., Gu W. (2009) Modes of p53 regulation. Cell 137, 609–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Brooks C. L., Gu W. (2011) The impact of acetylation and deacetylation on the p53 pathway. Protein Cell 2, 456–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lane D., Levine A. (2010) p53 research: The past thirty years and the next thirty years. Cold Spring Harb. Perspect. Biol. 2, a000893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hardie D. G. (2008) Role of AMP-activated protein kinase in the metabolic syndrome and in heart disease. FEBS Lett. 582, 81–89 [DOI] [PubMed] [Google Scholar]
- 20. Hardie D. G. (2007) AMP-activated/SNF1 protein kinases: Conserved guardians of cellular energy. Nat. Rev. Mol. Cell Biol. 8, 774–785 [DOI] [PubMed] [Google Scholar]
- 21. Dzamko N. L., Steinberg G. R. (2009) AMPK-dependent hormonal regulation of whole-body energy metabolism. Acta Physiologica 196, 115–127 [DOI] [PubMed] [Google Scholar]
- 22. Carling D., Mayer F. V., Sanders M. J., Gamblin S. J. (2011) AMP-activated protein kinase: Nature's energy sensor. Nature chemical biology 7, 512–518 [DOI] [PubMed] [Google Scholar]
- 23. Russell R. R., 3rd, Li J., Coven D. L., Pypaert M., Zechner C., Palmeri M., Giordano F. J., Mu J., Birnbaum M. J., Young L. H. (2004) AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J. Clin. Invest. 114, 495–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jones R. G., Plas D. R., Kubek S., Buzzai M., Mu J., Xu Y., Birnbaum M. J., Thompson C. B. (2005) AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 18, 283–293 [DOI] [PubMed] [Google Scholar]
- 25. Wang S., Dale G. L., Song P., Viollet B., Zou M. H. (2010) AMPKα1 deletion shortens erythrocyte life span in mice: Role of oxidative stress. J. Biol. Chem. 285, 19976–19985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu C., Liang B., Wang Q., Wu J., Zou M. H. (2010) Activation of AMP-activated protein kinase α1 alleviates endothelial cell apoptosis by increasing the expression of anti-apoptotic proteins Bcl-2 and survivin. J. Biol. Chem. 285, 15346–15355 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 27. Viollet B., Andreelli F., Jørgensen S. B., Perrin C., Geloen A., Flamez D., Mu J., Lenzner C., Baud O., Bennoun M., Gomas E., Nicolas G., Wojtaszewski J. F., Kahn A., Carling D., Schuit F. C., Birnbaum M. J., Richter E. A., Burcelin R., Vaulont S. (2003) The AMP-activated protein kinase α2 catalytic subunit controls whole-body insulin sensitivity. J. Clin. Invest. 111, 91–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jørgensen S. B., Viollet B., Andreelli F., Frøsig C., Birk J. B., Schjerling P., Vaulont S., Richter E. A., Wojtaszewski J. F. (2004) Knock-out of the α2 but not α1 5′-AMP-activated protein kinase isoform abolishes 5-aminoimidazole-4-carboxamide-1-β-4-ribofuranoside but not contraction-induced glucose uptake in skeletal muscle. J. Biol. Chem. 279, 1070–1079 [DOI] [PubMed] [Google Scholar]
- 29. Todaro G. J., Green H. (1963) Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J. Cell Biol. 17, 299–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xie Z., Dong Y., Zhang M., Cui M. Z., Cohen R. A., Riek U., Neumann D., Schlattner U., Zou M. H. (2006) Activation of protein kinase C ζ by peroxynitrite regulates LKB1-dependent AMP-activated protein kinase in cultured endothelial cells. J. Biol. Chem. 281, 6366–6375 [DOI] [PubMed] [Google Scholar]
- 31. Steinberg G. R., Kemp B. E. (2009) AMPK in Health and Disease. Physiol. Rev. 89, 1025–1078 [DOI] [PubMed] [Google Scholar]
- 32. Garner E., Raj K. (2008) Protective mechanisms of p53-p21-pRb proteins against DNA damage-induced cell death. Cell Cycle 7, 277–282 [DOI] [PubMed] [Google Scholar]
- 33. Maltzman W., Czyzyk L. (1984) UV irradiation stimulates levels of p53 cellular tumor antigen in nontransformed mouse cells. Mol. Cell Biol. 4, 1689–1694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lavin M. F., Gueven N. (2006) The complexity of p53 stabilization and activation. Cell Death Differ. 13, 941–950 [DOI] [PubMed] [Google Scholar]
- 35. Shieh S. Y., Ikeda M., Taya Y., Prives C. (1997) DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 91, 325–334 [DOI] [PubMed] [Google Scholar]
- 36. Lee J. H., Paull T. T. (2007) Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene 26, 7741–7748 [DOI] [PubMed] [Google Scholar]
- 37. Brooks C. L., Gu W. (2003) Ubiquitination, phosphorylation, and acetylation: The molecular basis for p53 regulation. Curr. Opin. Cell Biol. 15, 164–171 [DOI] [PubMed] [Google Scholar]
- 38. Cheng H. L., Mostoslavsky R., Saito S., Manis J. P., Gu Y., Patel P., Bronson R., Appella E., Alt F. W., Chua K. F. (2003) Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 100, 10794–10799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vaziri H., Dessain S. K., Ng Eaton E., Imai S. I., Frye R. A., Pandita T. K., Guarente L., Weinberg R. A. (2001) hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 107, 149–159 [DOI] [PubMed] [Google Scholar]
- 40. Yi J., Luo J. (2010) SIRT1 and p53, effect on cancer, senescence, and beyond. Biochim. Biophys. Acta 1804, 1684–1689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cantó C., Gerhart-Hines Z., Feige J. N., Lagouge M., Noriega L., Milne J. C., Elliott P. J., Puigserver P., Auwerx J. (2009) AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458, 1056–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tokarska-Schlattner M., Zaugg M., da Silva R., Lucchinetti E., Schaub M. C., Wallimann T., Schlattner U. (2005) Acute toxicity of doxorubicin on isolated perfused heart: response of kinases regulating energy supply. Am. J. Physiol. Heart Circ. Physiol. 289, H37–47 [DOI] [PubMed] [Google Scholar]
- 43. Ji C., Yang B., Yang Y. L., He S. H., Miao D. S., He L., Bi Z. G. (2010) Exogenous cell-permeable C6 ceramide sensitizes multiple cancer cell lines to doxorubicin-induced apoptosis by promoting AMPK activation and mTORC1 inhibition. Oncogene 29, 6557–6568 [DOI] [PubMed] [Google Scholar]
- 44. Chen M. B., Wu X. Y., Gu J. H., Guo Q. T., Shen W. X., Lu P. H. (2011) Activation of AMP-activated protein kinase contributes to doxorubicin-induced cell death and apoptosis in cultured myocardial H9c2 cells. Cell Biochem. Biophys. 60, 311–322 [DOI] [PubMed] [Google Scholar]
- 45. Zhang X. P., Liu F., Wang W. (2011) Two-phase dynamics of p53 in the DNA damage response. Proc. Natl. Acad. Sci. U.S.A. 108, 8990–8995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Murray-Zmijewski F., Slee E. A., Lu X. (2008) A complex barcode underlies the heterogeneous response of p53 to stress. Nat. Rev. Mol. Cell Biol. 9, 702–712 [DOI] [PubMed] [Google Scholar]
- 47. Borrás C., Gómez-Cabrera M. C., Viña J. (2011) The dual role of p53: DNA protection and antioxidant. Free Radic Res. 45, 643–652 [DOI] [PubMed] [Google Scholar]
- 48. Speidel D. (2010) Transcription-independent p53 apoptosis: An alternative route to death. Trends Cell Biol. 20, 14–24 [DOI] [PubMed] [Google Scholar]
- 49. Shizukuda Y., Matoba S., Mian O. Y., Nguyen T., Hwang P. M. (2005) Targeted disruption of p53 attenuates doxorubicin-induced cardiac toxicity in mice. Mol. Cell Biochem. 273, 25–32 [DOI] [PubMed] [Google Scholar]
- 50. Kurz E. U., Douglas P., Lees-Miller S. P. (2004) Doxorubicin activates ATM-dependent phosphorylation of multiple downstream targets in part through the generation of reactive oxygen species. J. Biol. Chem. 279, 53272–53281 [DOI] [PubMed] [Google Scholar]
- 51. Okoshi R., Ozaki T., Yamamoto H., Ando K., Koida N., Ono S., Koda T., Kamijo T., Nakagawara A., Kizaki H. (2008) Activation of AMP-activated protein kinase induces p53-dependent apoptotic cell death in response to energetic stress. J. Biol. Chem. 283, 3979–3987 [DOI] [PubMed] [Google Scholar]
- 52. Gu W., Roeder R. G. (1997) Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 90, 595–606 [DOI] [PubMed] [Google Scholar]
- 53. Luo J., Nikolaev A. Y., Imai S., Chen D., Su F., Shiloh A., Guarente L., Gu W. (2001) Negative control of p53 by Sir2α promotes cell survival under stress. Cell 107, 137–148 [DOI] [PubMed] [Google Scholar]
- 54. Luo J., Su F., Chen D., Shiloh A., Gu W. (2000) Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature 408, 377–381 [DOI] [PubMed] [Google Scholar]
- 55. Cantó C., Jiang L. Q., Deshmukh A. S., Mataki C., Coste A., Lagouge M., Zierath J. R., Auwerx J. (2010) Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metab. 11, 213–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Carvalho C., Santos R. X., Cardoso S., Correia S., Oliveira P. J., Santos M. S., Moreira P. I. (2009) Doxorubicin: The good, the bad, and the ugly effect. Curr. Med. Chem. 16, 3267–3285 [DOI] [PubMed] [Google Scholar]
- 57. Chatterjee K., Zhang J., Honbo N., Karliner J. S. (2010) Doxorubicin cardiomyopathy. Cardiology 115, 155–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Singal P. K., Iliskovic N. (1998) Doxorubicin-induced cardiomyopathy. N. Engl. J. Med. 339, 900–905 [DOI] [PubMed] [Google Scholar]
- 59. Asensio-López M. C., Lax A., Pascual-Figal D. A., Valdés M., Sánchez-Más J. (2011) Metformin protects against doxorubicin-induced cardiotoxicity: Involvement of the adiponectin cardiac system. Free Radic Biol. Med. 51, 1861–1871 [DOI] [PubMed] [Google Scholar]
- 60. Chen K., Xu X., Kobayashi S., Timm D., Jepperson T., Liang Q. (2011) Caloric restriction mimetic 2-deoxyglucose antagonizes doxorubicin-induced cardiomyocyte death by multiple mechanisms. J. Biol. Chem. 286, 21993–22006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Konishi M., Haraguchi G., Ohigashi H., Ishihara T., Saito K., Nakano Y., Isobe M. (2011) Adiponectin protects against doxorubicin-induced cardiomyopathy by anti-apoptotic effects through AMPK up-regulation. Cardiovasc. Res. 89, 309–319 [DOI] [PubMed] [Google Scholar]