

Background: Mutations in menin cause multiple endocrine neoplasia type 1 (MEN1).
Results: We identified an MEN1 in-frame deletion mutant of menin.
Conclusion: The mutant was stable, had selective loss of TGF-β signaling and growth inhibition, and identified the menin/Smad3 interacting region.
Significance: The studies provide insights into the pathophysiology of MEN1 and suggest the menin/Smad3 interface as a potential therapeutic target.
Keywords: Endocrinology, Proliferation, SMAD Transcription Factor, Transforming Growth Factor beta (TGFbeta), Tumor Suppressor Gene
Abstract
Multiple endocrine neoplasia type 1 (MEN1) is characterized by tumors of the parathyroid, enteropancreas, and anterior pituitary. The MEN1 gene encodes the tumor suppressor menin of 610 amino acids that has multiple protein partners and activities. The particular pathways that, when lost, lead to tumorigenesis are not known. We demonstrated that members of a three-generation MEN1 kindred are heterozygous for a donor splice site mutation at the beginning of intron 3 (IVS3 + 1G→A). Lymphoblastoid cells of a mutant gene carrier had, in addition to the wild-type menin transcript, an aberrant transcript resulting from use of a cryptic splice site within exon III that splices to the start of exon IV. The predicted menin Δ(184–218) mutant has an in-frame deletion of 35 amino acids but is otherwise of wild-type sequence. The transfected menin Δ(184–218) mutant was well expressed and fully able to mediate the normal inhibition of the activity of the transcriptional regulators JunD and NF-κB. However, it was defective in mediating TGF-β-stimulated Smad3 action in promoter-reporter assays in insulinoma cells. Importantly, lymphoblastoid cells from an individual heterozygous for the mutation had reduced TGF-β-induced (Smad3) transcriptional activity but normal JunD and NF-κB function. In addition, the mutant gene carrier lymphoblastoid cells proliferated faster and were less responsive to the cytostatic effects of TGF-β than cells from an unaffected family member. In conclusion, the menin mutant exhibits selective loss of the TGF-β signaling pathway and loss of cell proliferation control contributing to the development of MEN1.
Introduction
Menin is the product of the multiple endocrine neoplasia type 1 (MEN1) gene that, when inactivated, is responsible for an autosomal dominant disorder characterized by the combined occurrence of tumors of the parathyroids, endocrine pancreas, and anterior pituitary (1, 2). The 610-amino acid protein has carboxyl-terminal nuclear localization sequences and is predominantly nuclear (3, 4). Over 500 independent germ line and somatic mutations scattered throughout the protein-coding region have been identified (5). Most of the mutations (∼80%) are nonsense or result in a frameshift predicting an inactive truncated product. In addition, missense mutations (∼20%) have been identified, and the mutants are targeted to the ubiquitin-proteasome pathway and degraded (6, 7). This would be consistent with menin acting as a tumor suppressor, and a complete lack of menin, caused by loss of both MEN1 alleles, resulting in eventual tumor development.
In the nucleus, menin acts as a scaffold protein to regulate gene transcription by coordinating chromatin remodeling (8–10). Menin is implicated in both histone deacetylase and histone methyltransferase activity (HMT),3 and via the latter, it regulates the expression of cyclin-dependent kinase inhibitor (CDKI) and homeobox domain genes (11–15). Menin interacts with several transcription factors, including JunD (16), NF-κB (17), and Smads (18–20), and modulates their activities.
JunD is a member of the activator protein-1 (AP-1) transcription factor family and in contrast to other Jun and Fos proteins has antimitogenic activity (21). Of all the AP-1 family members, menin interacts only with JunD and represses its transcriptional activity by association with an mSin3A-histone deacetylase complex (16, 22–24). Studies in fibroblasts have suggested that the nature of JunD can change depending upon whether it is bound by menin when it functions as a growth suppressor or is not bound by menin in which case it acts as a growth promoter like other AP-1 family members (25). However, the results of JunD-menin interaction may be cell type-specific as JunD has a differentiating effect in osteoblasts, an action that is inhibited by menin (26).
The NF-κB family of transcriptional regulators activates the genes for growth factors and their cellular receptors. NF-κB is deregulated and overexpressed in several human neoplasms. Menin interacts directly with the p65, p50, and p52 NF-κB proteins and acts as a repressor of NF-κB-mediated transcriptional activation (17).
The cytokine transforming growth factor (TGF)-β and family members provide cytostatic signals that limit G1 progression and cell proliferation (27). TGF-β activates a membrane complex of serine/threonine kinase receptors that phosphorylates Smad2 and Smad3 that associate with Smad4, and the complex translocates to the nucleus where it regulates transcription in combination with coactivators and corepressors. A subset of the regulated genes is critical for arresting cells in G1 phase, and in epithelial cells this involves induction of some CDKIs. Smad2, -3, and -4 are considered to be tumor suppressors, and mutations in several components of the TGF-β signaling pathway contribute to a wide variety of cancers.
We have demonstrated that menin is a Smad3-interacting protein, and inactivation of menin blocks TGF-β and activin signaling antagonizing their growth inhibitory properties in anterior pituitary cells (18, 28). We have shown that in cultured parathyroid cells, menin depletion achieved by antisense oligonucleotides led to loss of TGF-β inhibition of parathyroid cell proliferation and parathyroid hormone (PTH) secretion (29). Moreover, the ability of TGF-β to inhibit proliferation and PTH production was lost in parathyroid cells from MEN1 patients (29, 30).
In this study we identified a novel MEN1 splice site mutation with functional properties that shed new light on the mechanism of menin action. Lymphoblastoid lines were established from family members heterozygous for the mutation or homozygous for the wild-type MEN1 gene, respectively. The protein derived from the mutant transcript has an internal deletion of 35 amino acids, and we designated the mutant protein as menin Δ(184–218). The JunD, NF-κB, and Smad3 transcriptional activities, and basal and TGF-β-modulated proliferation rates, of the mutant versus the wild-type lymphoblastoid cell lines were determined. This allowed an unbiased evaluation of wild-type functions retained by the mutant and those that were lost. The mutant menin was also examined for its HMT activity, ability to interact with JunD, NF-κB, and Smad3, and ability to mediate the up-regulation of the CDKIs, p15 and p21, in response to TGF-β.
EXPERIMENTAL PROCEDURES
Subjects
All subjects gave informed consent for the study that was approved by the Ethics Committee of the Royal Victoria Hospital.
Sequence Analysis of the MEN1 Gene
Leukocyte DNA was isolated by standard techniques. Exons 2–10 of the MEN1 gene were amplified as described previously (31). Gel-purified PCR products were directly sequenced.
Reagents, Constructs, Including MEN1 Minigene, GST-menin, and Smad3, and EGFP-menin, Minigene, and Cellular Localization Assays
All are described in the supplemental material.
3TP-Lux Transcription Assay
Lymphoblastoid cells, either homozygous for the wild-type or heterozygous for the splice site MEN1 gene mutation, were seeded at a density of 2 × 105 cells per 6-well plate. Cells were transfected 24 h later with 0.5 μg of the reporter (p3TP-Lux) or control (pGL3) constructs either without or with the wild-type menin cDNA (50 ng) and the pCH110 plasmid expressing β-galactosidase (0.1 μg) using Lipofectamine. Fifteen hours later, the medium was changed to DMEM containing 1% FBS, and the cells were incubated for an additional 9 h. Thereafter, cells were cultured for 24 h in the absence or presence of TGF-β in DMEM containing 0.2% FBS. Cells were lysed, and the luciferase activity was measured and normalized to the β-galactosidase activity.
Rat insulinoma Rin-5F or somatolactotrope GH4C1 cells were seeded at a density of 2 × 105 cells per 6-well plate. Cells were transfected 24 h after with 0.5 μg of the reporter (p3TP-Lux) or control (pGL3) constructs either without or with empty vector or with the wild-type menin vector alone or with the Δ(184–218) mutant vector alone or with both. Fifteen hours later, the medium was changed to DMEM containing 1% FBS, and the cells were incubated for an additional 9 h. Thereafter, the cells were cultured for 24 h in the absence or presence of TGF-β in DMEM containing 0.2% FBS. Cells were lysed, and the luciferase activity was measured.
JunD and NF-κB Transcription Assays
Lymphoblastoid or Rin-5F cells were seeded as above and 24 h later transfected with 0.5 μg of the reporter (AP-1(7×)) or control (pGL3) constructs without or with JunD cDNA and either empty vector alone or wild-type menin or menin mutants, Δ(184–218), H139D, or A242V. Twenty four hours later, the cells were lysed, and the luciferase activity was measured.
Lymphoblastoid or Rin-5F cells were seeded as above and 24 h later transfected with 0.5 μg of the reporter (NF-κB(6×)) or control (pGL3) constructs either without or with empty vector or wild-type menin or menin mutants, Δ(184–218) or Δ(278–477). Fifteen hours later, the medium was changed to DMEM containing 1% FBS, and the cells were incubated for an additional 9 h. Thereafter, the cells were cultured for 24 h in the absence or presence of IL-1β in DMEM containing 0.2% FBS. Cells were lysed, and the luciferase activity was measured.
GST-tagged Protein Production in E. coli
E. coli BL21(DE3) (Invitrogen) expressing GST or the GST-menin plasmids (WT and mutants) in LB containing 100 μg/ml ampicillin were cultured with shaking overnight at 37 °C. The cultures were diluted 1:1000 into 500 ml of 2× YT medium (100 μg/ml ampicillin) and grown with vigorous agitation at 37 °C to an A600 of 0.5. Isopropyl 1-thio-β-d-galactopyranoside (Invitrogen) was added to a final concentration of 1 mm and incubation continued at 30 °C for 6 h. Bacteria were pelleted by centrifugation at 4 °C and resuspended and lysed in 1× PBS buffer with protease inhibitors (Sigma catalog no. P8465) and 1% Triton X-100, followed by sonication. Cell debris was pelleted by centrifugation, and supernatants were stored at −80 °C.
GST fusion proteins were batch-purified with glutathione-Sepharose 4B beads (GE Healthcare) by rocking at 4 °C overnight. The beads were pelleted by centrifugation at 1000 × g at 4 °C and washed four times with cold PBS.
GST Pulldown Assays with Cell Extracts
GH4C1 cells transiently transfected with GFP or GFP-tagged menin WT and deletion constructs were lysed in RIPA buffer containing protease inhibitors (Complete Mini, Roche Applied Science) and incubated with GST or GST-Smad3 fusion proteins coupled to glutathione beads overnight at 4 °C on a rotating shaker. The beads were pelleted by centrifugation at 1000 × g (4 °C) and washed four times with cold RIPA buffer. The beads were boiled in Laemmli buffer, and the released proteins were separated by SDS-PAGE. Western blot analysis was performed, and the membranes were probed with anti-GFP antibody (Clontech).
GST Pulldown Assay with in Vitro Transcribed/Translated Proteins
Human JunD, rat Smad3, and human p65 were in vitro transcribed/translated (TnT-coupled reticulocytes, Promega) in the presence of [35S]methionine, and the labeled proteins were incubated with GST or GST-menin fusion proteins coupled to glutathione beads overnight at 4 °C on a rotating shaker in binding buffer (150 mm KCl, 20 mm HEPES, 0.1% Nonidet P-40, 5 mm MgCl2, 10% glycerol, 1 mm DTT, 0.5 mm EDTA). The beads were pelleted by centrifugation at 1000 × g (4 °C) and washed four times with cold binding buffer. The beads were boiled in Laemmli buffer, and the released proteins were separated by SDS-PAGE. Gels were dried and autoradiographed. A similar protocol was used to generate 35S-labeled menin WT and mutant proteins that were incubated with GST-Smad3.
Western Blot Analysis
FLAG-tagged menin WT and mutated cDNAs were transfected into 100-mm diameter dishes of Rin-5F or GH4C1 cells. Total cell lysates were prepared, and equal amounts of protein were used for Western blot analysis of menin, p15, p21, and tubulin (as a loading control) with anti-FLAG, anti-p15, anti-p21, and anti-β-tubulin antibodies, respectively, as described previously (7). Endogenous menin was detected in lymphoblastoid cells using an anti-human menin antibody (Abcam, Cambridge, MA).
Cell Proliferation
Cell proliferation assays were conducted as described previously (18).
Histone Methyltransferase Assay
Cells (HEK293 and Rin5-F) were transfected with FLAG-tagged menin wild-type or Δ(184–218) mutant (or other mutants), harvested 48 h later, and lysed in RIPA buffer. Cell lysates were incubated with M2(FLAG)-agarose (Sigma) at 4 °C for 16 h. After washing three times with PBS containing 0.1% Nonidet P-40, immunoprecipitates were assayed for HMT activity using a kit (Upstate, Temecula, CA) according to the manufacturer's protocol.
The substrate for the HMT activity included core histones, including histone H1, purified from chicken erythrocytes. For the HMT assay, each reaction contained 0.55 μCi of S-adenosyl-l-[methyl-3H]methionine (55 Ci/mmol; GE Healthcare) in a total volume of 10 μl (25 mm Tris-HCl, pH 9.0, 250 μm dithiothreitol, 1 mm PMSF) with 2 μg of chicken core histones without or with immunoprecipitate from 1 mg of total cell lysate (see above) of cells that had been transfected with wild-type or mutant menin constructs. Reactions were incubated at 30 °C for 30 min. Five-μl aliquots were captured on cellulose phosphate paper, washed with 10% trichloroacetic acid and 95% ethanol, dried, and counted in a scintillation counter. Protein arginine N-methyltransferase 1 (PRMT1) (catalog no. 14-474, Upstate) was the positive control.
Homology Modeling of Human Menin
An alignment of the sequences of human menin (accession no. AAC51228) and the Nematostella vectensis menin (accession number A7RZU9) was made as described previously (32). A homology model of human menin based on the Nematostella menin crystal structure (32), Protein Data Bank accession code 3RE2, was built using Modeller 9, Version 8 (33). Energy minimization was performed on the model using the YASARA energy minimization server (34). In a second approach, the human menin sequence was submitted to MODBASE and a single model based upon the Nematostella structure was derived by MODPIPE using the Modeller program (35). Very similar structures were obtained using the two approaches. Figures were prepared with PyMOL Version 1.0. Analysis of conservation of the human menin sequence was made with ConSurf (36).
Statistical Analysis
The data are means ± S.E. Statistical comparisons employing GraphPad Prism Version 4.00 analysis software (GraphPad Software, Inc., San Diego, CA, USA) were made using the unpaired two-tailed Student's t test, and a p value of <0.05 was taken to indicate a significant difference.
RESULTS
The Family
The proband (II-2) (Fig. 1A), a 51-year-old male (father), presented with hyperparathyroidism and a gastrinoma. The grandfather (I-1) (82 years old) has a history of hyperparathyroidism. The three children (III-1, III-2, and III-3) (19, 17, and 14 years old) are presently healthy.
FIGURE 1.

Pedigree of family with MEN1 and MEN1 gene mutation. A, clinical status is indicated by open symbols (unaffected) and solid symbols (affected). Proband is indicated by the arrow. The presence (+) or absence (−) of a mutation in tested family members is shown. Lymphoblastoid lines were established from individuals indicated by an asterisk. B, detection of a mutation in the MEN1 gene. Direct sequence analysis of the exon 3 genomic DNA amplicon of proband II-2 (right) revealed a heterozygous donor splice site mutation compared with an unrelated normal individual (left). The sense strand is shown. C, wild-type and mutant sequences of the exon 3/intron 3 boundary. The restriction enzyme FauI recognition site present in wild-type but destroyed by the mutation (IVS3 + 1G→A) is bracketed (and the cleavage site arrowed). The FauI site retained in wild-type is shown on the restriction map of the exon 3 amplicon. Gel electrophoretic separation is shown of undigested or FauI restriction digests of exon 3 PCR product from a normal individual or proband II-2. Undigested and FauI-digested exon 3 amplicon sizes are shown to the right.
Identification of MEN1 Mutation
Direct sequence analysis of MEN1 PCR amplicons of proband II-2 (father) identified a heterozygous mutation of a G to A transition at position +1 of intron 3 (Fig. 1B). The grandfather (I-1), the son (III-1), and one of the daughters (III-3) were heterozygous for the mutation that led to the loss of an FauI restriction enzyme site present in the wild-type sequence (Fig. 1C). The son and daughter continue to be closely monitored in view of the fact that the onset of hyperparathyroidism and other clinical manifestations of MEN1 may not occur until after age 20 (37). The DNA of the other daughter (III-2) did not carry the mutation. This nucleotide change was not found in 100 MEN1 gene alleles from 50 unrelated normal individuals.
Transcription of the MEN1 Gene in Lymphoblastoid Cells
The presence of a donor splice site mutation at the exon 3-intron 3 boundary of the MEN1 gene indicated that the phenotype might result from abnormal MEN1 pre-mRNA splicing. To analyze this further, lymphoblastoid cell lines were established from lymphocytes of individuals III-2 (normal MEN1 gene, WT) and III-3 (heterozygous for the splice site mutation, MUT) by Epstein-Barr virus transformation. After RT-PCR of RNA from the WT cell line (individual III-2), predominant cDNA products of either 792 or 920 bp (depending upon which of two primer sets was used) were identified (Fig. 2, A and B), and in the MUT cell line (individual III-3) the amounts of wild-type products were reduced by ∼50%. In addition, a single smaller product was observed with each of the primer sets used (Fig. 2, A and B). Cloning and sequencing of all products revealed the following: 1) the 792- and 920-bp products from WT and MUT cell lines corresponded to correct splicing of exon 3 to exon 4 (Fig. 2, C and D), and 2) the 687- and 815-bp products from the MUT cell line represented abnormal splicing in which a cryptic donor splice site within exon 3 was used to splice to exon 4 (Fig. 2, C and D).
FIGURE 2.

Effect of the MEN1 gene mutation at the mRNA and protein level. A, schematic of the MEN1 gene and menin mRNA. The 10 gene exons with introns (not drawn to scale) are shown with the initiation codon (ATG) in exon 2, the donor splice site mutation at the exon 3/intron 3 boundary (▾), the two nuclear localization signals (NLSs), and the stop codon (TGA) in exon 10. The menin mRNA includes exons 1–10 and the positions of primers used in exons 2 and 4 (RT-PCR #1, product = 792 bp) and exons 2 and 6 (RT-PCR #2, product = 920 bp) are shown. B, RT-PCR amplification of menin mRNA in lymphoblastoid cells from individuals III-2 (WT) or III-3 (MUT) with primer set 1 or 2. Product sizes are as follows: a = 792 bp, b = 920 bp, c = 687 bp, and d = 815 bp. Note that small amounts of products c and d are present in wild-type lymphoblasts (*). M = DNA markers, and sizes are shown on the left. C, direct sequence analysis of wild-type and mutant menin mRNA RT-PCR products. The WT (792-bp species, see B) corresponds to a product in which exon 3 is correctly spliced to exon 4 (left panel). In contrast, the MUT (687-bp species, see B) corresponds to an exon 3/exon 4 product in which the 3′ 150 nucleotides of exon 3 have been deleted (right panel). D, schematic of the splicing patterns of the pre-mRNA transcript of the wild-type (WT) and mutant (MUT) MEN1 genes. The asterisk indicates the position of the cryptic donor splice site in exon 3 used in the mutant transcript. Part of the nucleotide sequence of exon 3, intron 3, and exon 4 of the MEN1 gene is shown. Cryptic splice site (*), donor, and acceptor splice sites (▾) are shown. E, DNA mutation leads to an mRNA lacking 105 nucleotides at the 3′ end of exon 3 (nucleotide numbers of human menin mRNA, GenBankTM accession number U93236.1). The resulting mutant Protein has an internal in-frame deletion of 35 amino acids (menin Δ(184–218)). F, Western blot analysis of Rin-5F cells transfected with either cDNA for wild-type menin (WT), menin Δ(184–218), menin W423R, menin S443Y, or empty vector (pcDNA3.1). G, FLAG-tagged menin constructs (same as for F) were transfected into Rin-5F cells that were treated with cycloheximide (CHX, 20 μg/ml) to block further protein synthesis. Cell lysates prepared at the indicated times were immunoblotted with anti-FLAG and anti-β-tubulin antibodies. A representative β-tubulin immunoblot is shown. H, relative levels of each menin species were quantitated by densitometry. Values for each construct at each time point differed by ≤10%.
Minigene Constructs, Some Aberrant Splicing of the Menin Pre-mRNA Occurs Normally
To further study the consequences of the donor splice site mutation, minigene constructs were made by cloning a 2554-bp genomic PCR product, including part of exon 2, intron 2, exon 3, intron 3, and exon 4 of the MEN1 gene into a mammalian expression vector (supplemental Fig. S1A). Both wild-type and mutant constructs were prepared and transfected into the insulinoma Rin-5F cell line. RT-PCR analysis of RNA from transfected cells and DNA sequence analysis of relevant products showed that with the mutant minigene the major transcript was 687 bp rather than the predominant product of 792 bp of the wild-type minigene-transfected cells (supplemental Fig. S1, B and C). An equivalent small amount of wild-type product was observed in both the empty vector and the minigene transfected cells representing amplification of the endogenous menin pre-mRNA. Therefore, no properly spliced menin mRNA was contributed by the mutant minigene. It can be noted that splicing of the wild-type minigene pre-mRNA was not 100% efficient; the same mutant transcript was produced ∼10% of the time (supplemental Fig. S1C).
Menin Δ(184–218) Protein, Like Wild-type Menin, Is Stable
The mutation in the gene results in an in-frame deletion of the last 105 nucleotides from exon 3 in the mature mRNA (Fig. 2E). The consequence at the protein level is an internal deletion of 35 amino acids encoded by exon 3, with the rest of the protein being of wild-type sequence (Fig. 2E).
Some menin missense mutants are unstable and are rapidly degraded (6, 7). To test whether this was the case for the menin Δ(184–218) protein, FLAG-tagged wild-type menin, menin mutant, Δ(184–218), W423R, and S443Y cDNAs or empty vector were transfected into Rin-5F cells. Western blot analysis showed that the 65-kDa menin Δ(184–218) was well expressed like the 69-kDa wild-type menin, whereas the menin missense mutants W423R and S443Y were very poorly expressed (Fig. 2F). During a 4-h addition of cycloheximide to inhibit de novo protein synthesis, the Δ(184–218) mutant was almost as stable as wild-type menin in contrast to the W423R and S443Y mutants that were cleared rapidly (Fig. 2, G and H). Hence, loss of menin activity cannot be ascribed to poor expression of the Δ(184–218) mutant.
Menin Δ(184–218) Mutant, Like Menin Wild-type, Localizes to the Nucleus
Nuclear localization signals located at the COOH terminus are critical for directing menin to the nucleus (3); potentially, however, mutations in other parts of the molecule could affect this function. To test this possibility, enhanced green fluorescent protein (EGFP)-tagged menin constructs were transfected into HEK293 cells, and the coincidence of the fluorescence of the expressed proteins and nuclear staining with DAPI was assessed. EGFP-tagged menin Δ(184–218) was localized to the nucleus like EGFP-menin wild-type, whereas an EGFP-menin Δ(478–610) lacking the critical nuclear localization signals did not (supplemental Fig. S2). Hence, the menin Δ(184–218) mutant is fully competent in achieving nuclear localization.
Menin Δ(184–218) Mutant, Like Wild-type Menin, Inhibits AP-1- and NF-κB-mediated Transcriptional Responses
Transcriptional regulators that interact with menin, resulting in altered gene transcription, include JunD (16), a member of the AP-1 family, and members of the NF-κB family (17). Therefore, we evaluated the ability of the Δ(184–218) mutant in comparison with wild-type menin to modulate AP-1- and NF-κB-mediated transcriptional responses in rat insulinoma cells.
The Rin-5F cells were transfected with the AP-1 (7×) construct having a synthetic promoter containing seven AP-1 sites in tandem driving a luciferase reporter gene. Cotransfection of a JunD expression vector resulted in a 6.5-fold increase in transcriptional activity relative to empty vector (Fig. 3A). Cotransfection of wild-type menin or the Δ(184–218) mutant with JunD reduced transcriptional activity to basal levels. For comparison, menin H139D or A242V missense mutants when transfected with JunD failed to reduce the JunD-stimulated transcriptional activity at all (Fig. 3, A and B). The H139D and A242V mutants were chosen because they have previously been shown to lack JunD binding and the ability to repress JunD-activated transcription in mammalian cells (16). Therefore, the menin Δ(184–218) mutant is fully functional with respect to modulating JunD transcriptional activity. Similar findings were obtained in GH4C1 cells (data not shown).
FIGURE 3.

JunD and NF-κB transcriptional activities are modulated by exogenous menin wild-type or the Δ(184–218) mutant. A, promoterless pGL3 or AP-1(7×) constructs were transfected without (−) or with (+) JunD, with empty vector (Vector), wild-type menin (WT), and menin mutants Δ(184–218), H139D, or A242V into Rin-5F cells. Relative luciferase activity was measured, and the values shown represent the mean ± S.E. ***, p < 0.001, JunD (+) versus (−). B, Western blot analysis of the cell extracts probed with antibodies against FLAG and β-tubulin. V, vector. C, promoterless pGL3 or NF-κB(6×) constructs were transfected either with empty vector (Vector), wild-type menin (WT), menin mutants Δ(184–218), or Δ(278–477) into Rin-5F cells that were treated (+) or not (−) with IL-1β (5 ng/ml). Relative luciferase activity was measured, and the values shown represent the mean ± S.E. ***, p < 0.001; *, p < 0.05; IL-1β (+) versus (−). D, Western blot analysis of the cell extracts probed with antibodies against FLAG and β-tubulin.
The Rin-5F cells were transfected with the NF-κB (6×) construct having six consensus κB elements in tandem upstream of a luciferase reporter gene. Cells were stimulated or not with interleukin-1β (IL-1β) that binds its cell-surface receptor that signals NF-κB proteins to be translocated to the nucleus and activate genes via κB elements in their promoters. IL-1β treatment provoked a 22-fold increase in luciferase activity (Fig. 3C). Cotransfection with either wild-type menin or the Δ(184–218) mutant almost completely abrogated the response to IL-1β. However, cotransfection of a menin Δ(278–477) mutant had no effect on the ∼20-fold stimulation in luciferase activity brought about by IL-1β (Fig. 3, C and D). Similar findings were obtained in GH4C1 cells (data not shown). It can be surmised from previous studies that the Δ(184–218) mutant would, similar to wild-type menin, bind NF-κB proteins like p65, whereas the Δ(278–477) mutant would not (17). Hence, the Δ(278–477) mutant would be unable to modulate NF-κB transcriptional responses. The present data are consistent with this supposition. Therefore, the Δ(184–218) mutant is as effective as wild-type menin in inhibiting NF-κB transcriptional activity. In addition, the Δ(184–218) mutant was as effective as wild-type menin in modulating AP-1 (supplemental Fig. S3, A and B) and NF-κB (supplemental Fig. S3, C and D) transcriptional activities in the lymphoblastoid cells of MEN1 family members.
Patient Lymphoblastoid Cells and TGF-β-mediated Transcriptional Responses
Menin inactivation by antisense RNA antagonizes inhibition of cell proliferation by TGF-β, and this occurs because of impaired gene transcriptional responses to TGF-β. Menin interacts specifically with Smad3, and antisense menin suppresses TGF-β-induced transcriptional activity by inhibiting DNA binding of the Smad3/4 complex at specific transcriptional regulatory sites (38).
To assess TGF-β-transcriptional responses in MEN1 family member lymphoblastoid cell lines, they were transfected with the 3TP-Lux construct containing a TGF-β-responsive promoter driving the luciferase reporter gene. Treatment of 3TP-Lux transfected wild-type cells with TGF-β brought about a 6-fold increase in luciferase activity; however, a stimulation of only 3-fold was achieved in the mutant cells under identical conditions (Fig. 4, A and B). Cotransfection with a construct expressing wild-type menin improved the responsiveness of the cells of the individual who was heterozygous for the mutation, but it also further increased that of the cells from the family member not carrying the menin mutation such that the differential between wild-type and mutant cells was maintained (Fig. 4, A and B). Therefore, the cells heterozygous for the splice site mutation were impaired with respect to TGF-β-mediated transcription, and the defect could be rescued by expression of exogenous wild-type menin.
FIGURE 4.

TGF-β transcriptional responses mediated by endogenous and exogenous wild-type menin or the menin Δ(184–218) mutant. A, promoterless pGL3 or 3TP-Lux constructs were transfected without (−) or with (+) wild-type (WT) menin cDNA into lymphoblastoid cells, either homozygous for the wild-type (WT), or heterozygous for the splice site mutant (Mutant) MEN1 genes. Cells were stimulated (+) or not (−) with 200 pm TGF-β. Relative luciferase activity was measured, and the values shown represent the mean ± S.E. ***, p < 0.001; **, p < 0.01, TGF-β (+) versus (−). Significant differences between responses to TGF-β in WT and mutant cells are indicated by the brackets. B, Western blot analysis of the cell extracts probed with antibodies against menin, FLAG, and β-tubulin. C, Rin-5F; E, GH4C1 cells. The pGL3 promoterless construct was transfected with the empty vector (V), and the 3TP-Lux construct was transfected with empty vector (V) or wild-type menin (WT) or menin Δ(184–218) (Δ) alone or with WT and (Δ) together. Amount of each expression vector transfected is indicated by 1 = 50 ng and 0.5 = 25 ng, respectively. Cells were stimulated (+) or not (−) with 200 pm TGF-β. Relative luciferase activity was measured, and the values shown represent the mean ± S.E. ***, p < 0.001, TGF-β (+) versus (−). D, Rin-5F; F, GH4C1 cells. Western blot analysis of the cell extracts probed with antibodies against FLAG and β-tubulin.
Menin Δ(184–218) Mutant Lacks the Ability of Wild-type Menin to Mediate a TGF-β-stimulated Transcriptional Response in Endocrine Cells
To assess the impact of the menin Δ(184–218) mutant on TGF-β-mediated transcription in endocrine cells, the Rin-5F or GH4C1 cells were transfected with the 3TP-Lux promoter-reporter construct with vector alone, with wild-type menin alone, with menin Δ(184–218) alone, or with both wild-type menin and mutant Δ(184–218). Cotransfection with increasing amounts (25–500 ng) of the wild-type menin construct resulted in increased responsiveness to TGF-β (200 pm) in Rin5F cells (supplemental Fig. S4A) and GH4C1 cells (supplemental Fig. S4B). Treatment of 3TP-Lux and vector-only transfected Rin-5F and GH4C1 cells with TGF-β (200 pm) resulted in a 10-fold increase in luciferase activity. Cotransfection with the wild-type menin construct (50 ng) led to a doubling of the response to TGF-β (Fig. 4, C–F). However, cotransfection with the menin Δ(184–218) construct (50 ng) caused no further increase in the response to TGF-β over vector-only transfected cells. Cotransfection with both wild-type menin (25 ng) and menin Δ(184–218) (25 ng) to mimic the conditions in cells of the patient carrying the germ line MEN1 mutation resulted in a response lower than with the 50-ng wild-type menin construct alone (Fig. 4, C–F). Hence, the menin Δ(184–218) mutant lacks the ability of wild-type menin to mediate the TGF-β response. Under these conditions, it does not affect the response to wild-type menin.
Cellular Menin and Smad3 Interactions
To assess the interaction of menin and Smad3 in cells, constructs expressing menin full-length and deletion mutants (tagged with GFP) were transfected into GH4C1 cells. Total cell extracts were incubated with GST-Smad3 and subjected to SDS-PAGE, and immunoblots were revealed with antibodies against GFP. All forms of menin tested interacted with Smad3 with the exception of menin Δ(41–277) (Fig. 5A). Hence, the Smad3-interacting region lies within the amino-terminal part of menin.
FIGURE 5.
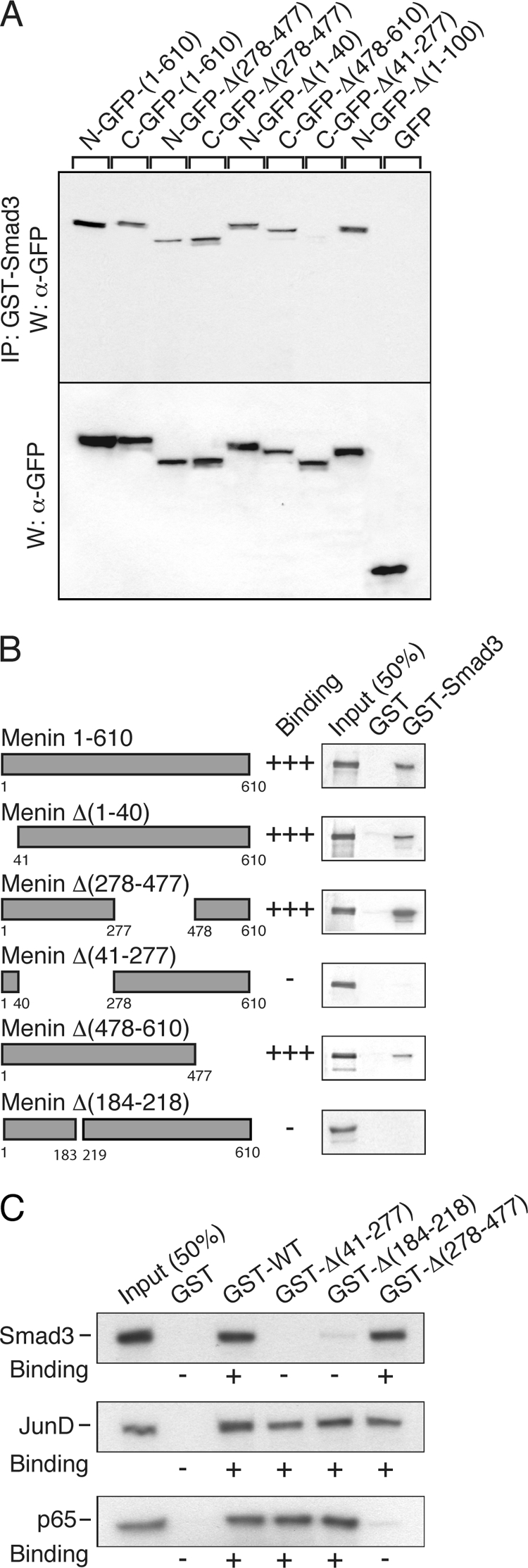
Menin coimmunoprecipitates with Smad3 and the interaction is direct. A, menin full-length and deletion mutants (tagged with GFP) were transfected into rat pituitary GH4C1 cells. Total cell extracts were incubated with GST-Smad3 expressed in and purified from bacteria and coupled to agarose beads. Upper panel, after washing, complexes were analyzed by SDS-PAGE, and immunoblots were revealed with antibodies against GFP. Lower panel, total cell extracts were examined in a similar fashion. All forms of menin interacted with Smad3 with the exception of menin Δ(41–277). IP, immunoprecipitate; W, Western blot. B, GST pulldown assays were conducted with glutathione-agarose beads coupled with GST alone or GST-Smad3 and in vitro transcribed/translated [35S]methionine-labeled menin and deletion mutants. GST-Smad3 interacted with all forms of menin, except the deletion mutants, menin Δ(41–277) and menin Δ(184–218). C, GST-menin construct was generated by inserting full-length menin cDNA into pGEX-5X1. GST pulldown assays were conducted with glutathionine-agarose beads coupled to GST alone or GST-menin constructs, GST-WT, GST-Δ(41–277), GST-Δ(184–218), or GST-Δ(278–477) and in vitro transcribed/translated [35S]methionine-labeled Smad3 or JunD or p65.
Menin Interacts Directly with Smad3
GST pulldown assays were conducted with glutathione-agarose beads coupled with GST alone or GST-Smad3 and in vitro transcribed/translated [35S]methionine-labeled menin and deletion mutants. GST-Smad3 interacted with all forms of menin, except the deletion mutants, menin Δ(41–277) and menin Δ(184–218) (Fig. 5B).
Interactions of Menin with Smad3, JunD, and NF-κB
GST pulldown assays were conducted with glutathione-agarose beads coupled with GST alone or GST-menin constructs, GST-WT, GST-Δ(41–277), GST-Δ(184–218), or GST-Δ(278–477), and in vitro transcribed/translated [35S]methionine-labeled Smad3 or JunD or p65. Menin wild-type and all mutants, including Δ(184–218), bound JunD consistent with previous findings (16). Menin wild-type and mutants Δ(41–277) and Δ(184–218) bound p65, but the Δ(278–477) mutant failed to interact consistent with previous findings (17). Menin wild-type and mutant Δ(278–477) bound Smad3, but the Δ(41–277) mutant failed to interact at all, and interaction with the Δ(184–218) mutant was minimal (Fig. 5C). Therefore, whereas JunD and p65 interacted with the Δ(184–218) mutant as well as with wild-type menin, Smad3 was unable to interact with the Δ(184–218) mutant as it did with wild-type menin.
Menin Δ(184–218) Mutant Demonstrates HMT Activity
An important activity ascribed to menin acting as a component of the mixed lineage leukemia (MLL) complex is that of trimethylation of the fourth lysine residue of histone H3 (H3K4) that is strongly associated with transcription activation. About 50% of the few MEN1-associated missense mutations tested lack or have markedly reduced HMT activity, but it remains unclear as to how essential this function is for the tumor suppressor activity of menin. FLAG-tagged menin wild-type (WT) or mutants (Δ184–218, A242V, H139D, or L22R) were transfected into HEK293 cells, and menin proteins were immunoprecipitated and equal amounts assayed for HMT activity. MEN1 mutants A242V and H139D had very low or markedly reduced activity, respectively, whereas mutant L22R was almost as fully active as menin wild-type, all consistent with previous findings (11). The Δ(184–218) mutant demonstrated good HMT activity, about 80% that of wild-type menin (Fig. 6, A–C).
FIGURE 6.

A, menin Δ(184–218) mutant demonstrates HMT activity. HEK293 cells were transfected with empty vector or FLAG-tagged menin wild-type (WT) or mutants (Δ184–218, A242V, H139D, or L22R) and harvested 48 h later. Cell lysates were immunoprecipitated with M2(FLAG)-agarose, and immunoprecipitates were assayed for HMT activity. Assay results are expressed as counts/min incorporated into chicken core histones after incubation of immunoprecipitates with S-adenosyl-l-[methyl-3H]methionine. Protein arginine N-methyltransferase 1 (PRMT1) was the positive control. ***, p < 0.001; **, p < 0.01; *, p < 0.05 versus vector (pCMVTag2B). B, Western blot of the extracts of the transfected cells probed with FLAG and β-tubulin antibodies. C, Western blot of cell extract immunoprecipitates adjusted to have equal amounts of each form of menin. D, the 184–218-residue Smad3 interacting region of menin is on the surface of the molecule distinct from the central cavity that binds the MLL protein critical for the HMT activity. A homology model of human menin was generated from the crystal structure of the menin homolog of N. vectensis (32). Menin is predominantly α-helical comprising three tetratricopeptide motifs that form a central cavity and are flanked by two α-helical bundles and covered by a β-sheet motif. Positions of residues Leu-22, His-139, and Ala-242, and the sequence 184–218 are indicated. The Smad3-interacting sequence 184–218 is on an exposed surface neighboring the β-sheet motif. E, structure of human menin rotated by 90° to show the central cavity that is lined by His-139 and Ala-242.
184–218 Smad3 Interacting Region of Menin Is on the Surface of the Molecule Distinct from the Central Cavity That Binds the MLL Protein Critical for the HMT Activity
A homology model of human menin was generated from the crystal structure of the menin homolog of N. vectensis (32). The H139D and A242V mutants are within a part of the molecule (Fig. 6, D and E) that forms the large central cavity that binds MLL (32), and their lack of HMT activity is fully consistent with this location. The L22R is within the first amino-terminal α-helix distinct from the central cavity (Fig. 6, D and E) and consistent with it having HMT activity similar to that of wild-type menin. Likewise, the 184–218 region is located on the surface of the molecule (Fig. 6, D and E) and is largely uninvolved with the binding pocket for MLL consistent with the Δ(184–218) mutant having ∼80% the HMT activity of wild-type menin.
Lymphoblastoid Cells Heterozygous for the Menin Δ(184–218) Mutant Have an Altered Basal Proliferation Rate and Impaired Proliferative Response to TGF-β Relative to Normal Cells
The basal proliferation of the lymphoblastoid cells, either homozygous for the wild-type or heterozygous for the Δ(184–218) mutant menin, was monitored over 72 h. The doubling rate of the mutant cells was approximately twice that of the wild-type cells (Fig. 7A). When serum-starved lymphoblastoid cells were cultured in TGF-β and cell proliferation was monitored over 72 h, clear differences were apparent by 48 h with the inhibition of the number of the mutant cells being only 50% that of the wild-type cells (Fig. 7B). Hence, the mutant lymphoblastoid cells had an altered basal proliferative rate and impaired responsiveness to TGF-β with respect to inhibition of proliferation.
FIGURE 7.

Proliferative and CDKI responses mediated by endogenous and exogenous wild-type menin or the menin Δ(184–218) mutant. A, basal proliferation of lymphoblastoid cells (cultured in 2% FBS, RPMI), either homozygous for the wild-type (WT) or heterozygous for the splice site mutant (Mutant) menin genes, was monitored over 72 h. Cell counts, mean ± S.E. ***, p < 0.001, mutant versus wild-type. B, proliferation of serum-starved (0.1% FBS) lymphoblastoid cells, either homozygous for the wild-type (WT) or heterozygous for the splice site mutant (Mutant) menin genes, cultured in TGF-β (10 ng/ml) was monitored over 72 h. Cell counts, mean ± S.E. *, p < 0.05; ***, p < 0.001; mutant versus wild-type. C and D, rat insulinoma Rin-5F cells were transfected with either empty vector (pcDNA3.1), wild-type (WT) menin, or the Δ(184–218) menin mutant. Serum-starved (0.1% FBS) cells were stimulated (+) or not (−) with 10 ng/ml TGF-β for 48 h; cell lysates were made and subjected to Western blot analysis with C (panel i), p15; D (panel i), p21, and FLAG and β-tubulin antibodies. Representative examples are shown. Densitometric quantitation is as follows: C (panel ii), p15; D (panel ii), p21 Western blot analyses (n = 3). Values are mean ± S.E. ***, p < 0.001; TGF-β (+) versus TGF-β (−) for each construct.
Transfected Menin Δ(184–218) Mutant Lacks the Ability of Wild-type Menin to Increase TGF-β-stimulated CDKI Expression in Endocrine Cells
Rin5F cells stimulated with TGF-β for 48 h demonstrated a doubling in their expression of p15 and p21 (Fig. 7, C and D). Cells transiently transfected with wild-type menin demonstrated an increase in basal expression (without added TGF-β) as well as increased TGF-β responsiveness of the level of p15 and p21 (Fig. 7, C and D). Cells transiently transfected with the menin Δ(184–218) mutant exhibited a similar basal and TGF-β-stimulated level of expression of p15 and p21 to cells transfected with vector-only plasmid (Fig. 7, C and D). Similar findings were observed in the GH4C1 cells (data not shown). Therefore, the menin Δ(184–218) mutant is unable to mediate the TGF-β up-regulation of the CDKIs, p15 and p21.
DISCUSSION
In this study, we identified a novel splice site mutation in the MEN1 gene. The resulting protein product comprises a menin mutant that has an internal deletion of 35 amino acids, menin Δ(184–218). Although the menin mutant modulated JunD and NF-κB transcriptional activities like the wild-type menin, it was deficient in mediating TGF-β-induced (Smad3) transcriptional activity. Lymphoblastoid cells from an individual heterozygous for the mutation proliferated at a faster rate and exhibited reduced cell proliferation inhibition in response to TGF-β than cells from an unaffected individual.
The majority (∼80%) of disease-causing mutations in the MEN1 gene give rise to truncated proteins that are likely to be unstable and rapidly turned over. Indeed, the recent report on the crystallization of a menin homolog from N. vectensis notes that “the three-dimensional structure revealed that menin is a single domain protein, and therefore the truncation of its amino acid sequence will invariably result in disruption of the menin fold leading to loss of menin function” (32). Most of the other 20% of the mutations are missense, and the majority of these mutants are targeted to the proteasome and are degraded (6, 7). Therefore, the identification of a mutant with a small in-frame deletion that is stable is of extreme interest and contributes to the novelty of the study.
Germ line mutations at the first nucleotide of IVS3 (but to a different nucleotide to that identified here) have been reported in other MEN1 cases: c.654 + 1G→C (39), c.654 + 1G→T (40, 41). Also, Kawamura et al. (42) identified a c.654 + 2T→A mutation in a sporadic gastrinoma case. However, no functional studies were conducted for any of these mutations.
Hai et al. (43) identified a substitution of guanine for adenosine at the third base of intron 3 (c.654 + 3A→G) in a sporadic MEN1 case. Osthus et al. (44) found a guanine to thymidine change at the last base of exon III (c.654 G→T) in family members with isolated hyperparathyroidism. RT-PCR of RNA from patient cells in both cases revealed the same aberrant splicing as found in the present case with a cryptic splice site in exon III being used and 105 bases being spliced out. Debelenko et al. (45) identified the intron 3, c.654 + 3A→G, change as a somatic mutation (carcinoid) and predicted that either two additional nucleotides (GT) would be added to the exon III coding sequence or that exon III might be entirely spliced out. Patient RNA was not available for functional studies to evaluate these predictions. Karges et al. (46), by cDNA sequencing from parathyroid adenomas in two unrelated primary hyperparathyroid patients, identified a transcript deleted for the 105 bp of the 3′ part of exon III. Analysis of genomic tumor DNA identified an intron 3 splice site mutation (c.654 + 3A→G) suggesting this as the cause of the aberrant splicing.
Roijers et al. (47) identified affected members of an MEN1 kindred heterozygous for a germ line 26-bp deletion in the MEN1 gene spanning the last 15 bp of exon III and the first 11 bp of intron 3. This mutation resulted in the same 105-base deletion in the menin mRNA and predicted 35-amino acid internal deletion in the menin protein as described here. The authors suggested that as the deleted region of menin had been implicated in binding to JunD, this might explain the tumorigenic effect of the mutation; however, functional studies were not done.
Therefore, although other MEN1 cases have been described with the involvement of nucleotide changes at the MEN1 gene exonIII/IVS3 splice junction (and in three cases the altered mutant transcript was identified), no functional analysis of the mutant protein has been carried out prior to this study.
Since its discovery as a novel protein over a decade ago, many cellular functions of menin have been identified. However, which ones of these relate specifically to the role of menin as a tumor suppressor and when lost contribute to the development of MEN1 neoplasia is an ongoing debate. In this case, we detected a menin mutant with a small internal deletion that formed a stable protein that achieved nuclear localization. Furthermore, lymphoblastoid cell lines from one family member carrying one copy of the mutant allele and from one normal family member were available. This afforded the unique opportunity to examine the effect of the mutant on the different signaling pathways impacted upon by menin.
Menin interacts with JunD (which is unusual in being an AP-1 family member that has antimitogenic activity) and represses its transcriptional activity (16). Although it might seem paradoxical that one antimitogenic factor would repress the activity of another, one study has suggested that when not bound to menin, JunD acts as a growth promoter like other AP-1 family members (25). In this study, we found that the menin Δ(184–218) mutant bound to JunD and was capable of modulating JunD transcriptional activity as effectively as wild-type menin.
Menin interacts with the NF-κB, p50, p52, and p65 transcription factors and represses p65 transcriptional activity (17). The role of NF-κB transcription factors in neoplasia is difficult to assess with deregulation and overexpression in some neoplasms and lower levels of activity and expression in others. One study reported reduced (rather than increased) NF-κB activity in parathyroid neoplasia (primary, secondary, and MEN1-related) (48). However, in non-MEN1-related parathyroid tumors, lower menin levels were associated with higher phosphorylated p65 levels suggesting that menin might act as a suppressor of the proliferative actions of the NF-κB pathway (48). In this study, we found that the menin Δ(184–218) mutant bound to NF-κB p65 and was capable of modulating p65 transcriptional activity as effectively as wild-type menin.
TGF-β and family members provide cytostatic signals that limit G1 progression and cell proliferation (27). TGF-β activates a membrane complex of serine/threonine kinase receptors that phosphorylates Smad2 and Smad3 that associate with Smad4, and the complex translocates to the nucleus where it regulates transcription in combination with coactivators and corepressors. Menin is a Smad3-interacting protein, and inactivation of menin blocks TGF-β and activin signaling antagonizing their growth inhibitory properties in anterior pituitary cells (18, 28). In cultured parathyroid cells, menin inactivation leads to loss of TGF-β inhibition of parathyroid cell proliferation and PTH secretion (29). Moreover, TGF-β does not affect (decrease) the proliferation and PTH production of parathyroid cells from MEN1 patients (29, 30). In this study, we found that the menin Δ(184–218) mutant did not bind Smad3 and had lost the ability to facilitate transcriptional activity of Smad3 like wild-type menin.
In the nucleus menin acts as a scaffold protein to regulate gene transcription by coordinating chromatin remodeling (8–10). It is implicated in HMT activity and via this activity regulates the basal expression of CDKI genes such as those for p18 and p27 (11, 14). In a mouse Men1+/− model in which pancreatic islet tumors developed over time, the loss of the wild-type Men1 gene preceded or accompanied the reduction in HMT activity (14). Of the few MEN1-associated missense mutants examined thus far, some exhibit absent or reduced and others normal HMT activity, respectively (11). In this study, the menin Δ(184–218) mutant exhibited HMT activity close to that of wild-type menin.
Recently, the first crystal structure of a menin homolog, from the sea anenome N. vectensis, was reported (32). The major focus of that study was on how menin, in contrast to its role as a tumor suppressor, is able to act as an oncogenic cofactor of MLL fusion proteins in acute leukemias. Menin is predominantly an α-helical protein with the core consisting of three tetratricopeptide motifs that are flanked by two α-helical bundles and covered by a β-sheet motif. There is a large central cavity that binds MLL, and the MEN1 missense mutant A242V that lines the cavity was shown to lack the ability of wild-type menin to bind MLL (32). This is fully consistent with the finding of this study that the A242V mutant lacks HMT activity. The knowledge of the crystal structure of the Nematostella menin gave us the opportunity to place the Smad3 interacting 184–218 sequence on both the Nematostella structure and a homology model of the highly conserved human menin. The 184–218 sequence is not involved with the central cavity but rather is located on the exposed surface neighboring the β-sheet motif. This is in agreement with the Δ(184–218) mutant having good HMT activity like the MEN1 L22R missense mutant that is within the first α-helix in the molecule. However, like the A242V mutant, as noted above, we showed that the MEN1 H139D mutant that is involved in the central cavity-binding site of MLL was markedly deficient in HMT activity. Therefore, these findings suggest that the mechanism whereby menin facilitates the transcriptional activity of Smad3 does not involve the MLL complex, and further studies will be required to identify the mechanism(s).
The cytostatic effects of TGF-β are mediated in part by its ability to up-regulate the expression of a subset of the CDKIs, including p15 and p21. In this study, patient lymphoblastoid cells heterozygous for the menin Δ(184–218) mutant had an increased basal proliferative rate and an impaired cytostatic response to TGF-β relative to the lymphoblastoid cells from the normal relative. The menin Δ(184–218) mutant was impaired with respect to mediating the TGF-β up-regulation of the CDKIs, p15 and p21, in transfected rat insulinoma and somatolactotroph cells. The defective proliferative and CDKI modulatory properties of the menin mutant provide further evidence of the critical importance of the TGF-β signaling pathway in this particular case.
Menin is widely expressed from an early developmental stage and found in both nonendocrine and endocrine tissues (49). Mice heterozygous for ablation of the Men1 gene develop endocrine tumors during their lifetime similar to MEN1 patients (50, 51). Reduced TGF-β signaling has been noted in the tumors that develop in these models (52, 53). Interestingly, the homozygous deletion of Men1 is embryonic lethal with fetuses dying at mid-gestation with defects in multiple organs (54). Evidence is accumulating of the importance of menin for the proper functioning of the signaling pathways activated by TGF-β ligand family members that are critical for development and maintenance of many organ systems (19, 20, 55–58). Therefore, the importance of menin in this particular pathway extends beyond control of endocrine and nonendocrine cells that when dysregulated lead to tumorigenesis.
In summary, we have identified a novel MEN1 splice site mutation that leads to an altered RNA transcript. The resulting menin protein mutant that carries an in-frame internal deletion is selectively defective for the TGF-β-signaling pathway. This would identify the loss of this particular pathway as being critical as the first hit (according to the Knudsen hypothesis). The later somatic mutation (second hit) in the wild-type MEN1 gene responsible for the MEN1 tumorigenesis may involve either the same or any one of several other growth-regulatory pathways in which menin is implicated.
Supplementary Material
Acknowledgments
We thank all family members for their participation and Drs. Natasha Garfield and J. Brent Richards for clinical data. We thank Xiang Zhou, Alice Aylott, Michael Grant, and Hayeon Kwak for technical assistance and Drs. Hugh P. J. Bennett and Bernard Turcotte for critical review of the manuscript.
This work was supported in part by Canadian Institutes of Health Research Grants MOP-9315 (to G. N. H.) and MOP-86703 (to D. G.).

This article contains supplemental “Methods,” Figs. S1–S4, and additional references.
- HMT
- histone methyl transferase
- MLL
- mixed lineage leukemia
- CDKI
- cyclin-dependent kinase inhibitor
- PTH
- parathyroid hormone
- MUT
- mutant
- EGFP
- enhanced GFP.
REFERENCES
- 1. Chandrasekharappa S. C., Guru S. C., Manickam P., Olufemi S. E., Collins F. S., Emmert-Buck M. R., Debelenko L. V., Zhuang Z., Lubensky I. A., Liotta L. A., Crabtree J. S., Wang Y., Roe B. A., Weisemann J., Boguski M. S., Agarwal S. K., Kester M. B., Kim Y. S., Heppner C., Dong Q., Spiegel A. M., Burns A. L., Marx S. J. (1997) Positional cloning of the gene for multiple endocrine neoplasia type 1. Science 276, 404–407 [DOI] [PubMed] [Google Scholar]
- 2. Lemmens I., Van de Ven W. J., Kas K., Zhang C. X., Giraud S., Wautot V., Buisson N., De Witte K., Salandre J., Lenoir G., Pugeat M., Calender A., Parente F., Quincey D., Gaudray P., De Wit M. J., Lips C. J., Höppener J. W., Khodaei S., Grant A. L., Weber G., Kytölä S., Teh B. T., Farnebo F., Thakker R. V. (1997) Identification of the multiple endocrine neoplasia type 1 (MEN1) gene. The European Consortium on MEN1. Hum. Mol. Genet. 6, 1177–1183 [DOI] [PubMed] [Google Scholar]
- 3. Guru S. C., Goldsmith P. K., Burns A. L., Marx S. J., Spiegel A. M., Collins F. S., Chandrasekharappa S. C. (1998) Menin, the product of the MEN1 gene, is a nuclear protein. Proc. Natl. Acad. Sci. U.S.A. 95, 1630–1634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kaji H., Canaff L., Goltzman D., Hendy G. N. (1999) Cell cycle regulation of menin expression. Cancer Res. 59, 5097–5101 [PubMed] [Google Scholar]
- 5. Lemos M. C., Thakker R. V. (2008) Multiple endocrine neoplasia type 1 (MEN1): analysis of 1336 mutations reported in the 1st decade following identification of the gene. Hum. Mutat. 29, 22–32 [DOI] [PubMed] [Google Scholar]
- 6. Yaguchi H., Ohkura N., Takahashi M., Nagamura Y., Kitabayashi I., Tsukada T. (2004) Menin missense mutants associated with multiple endocrine neoplasia type 1 are rapidly degraded via the ubiquitin-proteasome pathway. Mol. Cell. Biol. 24, 6569–6580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Canaff L., Vanbellinghen J. F., Kanazawa I., Kwak H., Garfield N., Vautour L., Hendy G. N. (2011) Menin missense mutants encoded by the MEN1 gene that are targeted to the proteasome: restoration of expression and activity by CHIP siRNA. J. Clin. Endocrinol. Metab. 97, E282–E291 [DOI] [PubMed] [Google Scholar]
- 8. Dreijerink K. M., Höppener J. W., Timmers H. M., Lips C. J. (2006) Mechanisms of disease. Multiple endocrine neoplasia type 1-relation to chromatin modifications and transcription regulation. Nat. Clin. Pract. Endocrinol. Metab. 2, 562–570 [DOI] [PubMed] [Google Scholar]
- 9. Wu X., Hua X. (2008) Menin, histone h3 methyltransferases, and regulation of cell proliferation. Current knowledge and perspective. Curr. Mol. Med. 8, 805–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hendy G. N., Kaji H., Canaff L. (2009) Cellular functions of menin. Adv. Exp. Med. Biol. 668, 37–50 [DOI] [PubMed] [Google Scholar]
- 11. Hughes C. M., Rozenblatt-Rosen O., Milne T. A., Copeland T. D., Levine S. S., Lee J. C., Hayes D. N., Shanmugam K. S., Bhattacharjee A., Biondi C. A., Kay G. F., Hayward N. K., Hess J. L., Meyerson M. (2004) Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol. Cell 13, 587–697 [DOI] [PubMed] [Google Scholar]
- 12. Yokoyama A., Wang Z., Wysocka J., Sanyal M., Aufiero D. J., Kitabayashi I., Herr W., Cleary M. L. (2004) Leukemia proto-oncoprotein MLL forms a SET1-like histone methyltransferase complex with menin to regulate Hox gene expression. Mol. Cell. Biol. 24, 5639–5649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Milne T. A., Hughes C. M., Lloyd R., Yang Z., Rozenblatt-Rosen O., Dou Y., Schnepp R. W., Krankel C., Livolsi V. A., Gibbs D., Hua X., Roeder R. G., Meyerson M., Hess J. L. (2005) Menin and MLL cooperatively regulate expression of cyclin-dependent kinase inhibitors. Proc. Natl. Acad. Sci. U.S.A. 102, 749–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Karnik S. K., Hughes C. M., Gu X., Rozenblatt-Rosen O., McLean G. W., Xiong Y., Meyerson M., Kim S. K. (2005) Menin regulates pancreatic islet growth by promoting histone methylation and expression of genes encoding p27Kip1 and p18INK4c. Proc. Natl. Acad. Sci. U.S.A. 102, 14659–14664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shen H. C., Rosen J. E., Yang L. M., Savage S. A., Burns A. L., Mateo C. M., Agarwal S. K., Chandrasekharappa S. C., Spiegel A. M., Collins F. S., Marx S. J., Libutti S. K. (2008) Parathyroid tumor development involves deregulation of homeobox genes. Endocr. Relat. Cancer 15, 267–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Agarwal S. K., Guru S. C., Heppner C., Erdos M. R., Collins R. M., Park S. Y., Saggar S., Chandrasekharappa S. C., Collins F. S., Spiegel A. M., Marx S. J., Burns A. L. (1999) Menin interacts with the AP1 transcription factor JunD and represses JunD-activated transcription. Cell 96, 143–152 [DOI] [PubMed] [Google Scholar]
- 17. Heppner C., Bilimoria K. Y., Agarwal S. K., Kester M., Whitty L. J., Guru S. C., Chandrasekharappa S. C., Collins F. S., Spiegel A. M., Marx S. J., Burns A. L. (2001) The tumor suppressor protein menin interacts with NF-κB proteins and inhibits NF-κB-mediated transactivation. Oncogene 20, 4917–4925 [DOI] [PubMed] [Google Scholar]
- 18. Kaji H., Canaff L., Lebrun J. J., Goltzman D., Hendy G. N. (2001) Inactivation of menin, a Smad3-interacting protein, blocks transforming growth factor type β signaling. Proc. Natl. Acad. Sci. U.S.A. 98, 3837–3842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sowa H., Kaji H., Hendy G. N., Canaff L., Komori T., Sugimoto T., Chihara K. (2004) Menin is required for bone morphogenetic protein 2- and transforming growth factor β-regulated osteoblastic differentiation through interaction with Smads and Runx2. J. Biol. Chem. 279, 40267–40275 [DOI] [PubMed] [Google Scholar]
- 20. Kaji H., Canaff L., Hendy G. N. (2009) Role of menin in bone development. Adv. Exp. Med. Biol. 668, 59–67 [DOI] [PubMed] [Google Scholar]
- 21. Pfarr C. M., Mechta F., Spyrou G., Lallemand D., Carillo S., Yaniv M. (1994) Mouse JunD negatively regulates fibroblast growth and antagonizes transformation by ras. Cell 76, 747–760 [DOI] [PubMed] [Google Scholar]
- 22. Gobl A. E., Berg M., Lopez-Egido J. R., Oberg K., Skogseid B., Westin G. (1999) Menin represses JunD-activated transcription by a histone deacetylase-dependent mechanism. Biochim. Biophys. Acta 1447, 51–56 [DOI] [PubMed] [Google Scholar]
- 23. Kim H., Lee J. E., Cho E. J., Liu J. O., Youn H. D. (2003) Menin, a tumor suppressor, represses JunD-mediated transcriptional activity by association with an mSin3A-histone deacetylase complex. Cancer Res. 63, 6135–6139 [PubMed] [Google Scholar]
- 24. Yazgan O., Pfarr C. M. (2001) Differential binding of the Menin tumor suppressor protein to JunD isoforms. Cancer Res. 61, 916–920 [PubMed] [Google Scholar]
- 25. Agarwal S. K., Novotny E. A., Crabtree J. S., Weitzman J. B., Yaniv M., Burns A. L., Chandrasekharappa S. C., Collins F. S., Spiegel A. M., Marx S. J. (2003) Transcription factor JunD, deprived of menin, switches from growth suppressor to growth promoter. Proc. Natl. Acad. Sci. U.S.A. 100, 10770–10775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Naito J., Kaji H., Sowa H., Hendy G. N., Sugimoto T., Chihara K. (2005) Menin suppresses osteoblast differentiation by antagonizing the AP-1 factor, JunD. J. Biol. Chem. 280, 4785–4791 [DOI] [PubMed] [Google Scholar]
- 27. Massagué J., Gomis R. R. (2006) The logic of TGFβ signaling. FEBS Lett. 580, 2811–2820 [DOI] [PubMed] [Google Scholar]
- 28. Lacerte A., Lee E. H., Reynaud R., Canaff L., De Guise C., Devost D., Ali S., Hendy G. N., Lebrun J. J. (2004) Activin inhibits pituitary prolactin expression and cell growth through Smads, Pit-1 and menin. Mol. Endocrinol. 18, 1558–1569 [DOI] [PubMed] [Google Scholar]
- 29. Sowa H., Kaji H., Kitazawa R., Kitazawa S., Tsukamoto T., Yano S., Tsukada T., Canaff L., Hendy G. N., Sugimoto T., Chihara K. (2004) Menin inactivation leads to loss of transforming growth factor β inhibition of parathyroid cell proliferation and parathyroid hormone secretion. Cancer Res. 64, 2222–2228 [DOI] [PubMed] [Google Scholar]
- 30. Naito J., Kaji H., Sowa H., Kitazawa R., Kitazawa S., Tsukada T., Hendy G. N., Sugimoto T., Chihara K. (2006) Expression and functional analysis of menin in a multiple endocrine neoplasia type 1 (MEN1) patient with somatic loss of heterozygosity in chromosome 11q13 and unidentified germline mutation of the MEN1 gene. Endocrine 29, 485–490 [DOI] [PubMed] [Google Scholar]
- 31. Poncin J., Abs R., Velkeniers B., Bonduelle M., Abramowicz M., Legros J. J., Verloes A., Meurisse M., Van Gaal L., Verellen C., Koulischer L., Beckers A. (1999) Mutation analysis of the MEN1 gene in Belgian patients with multiple endocrine neoplasia type 1 and related diseases. Hum. Mutat. 13, 54–60 [DOI] [PubMed] [Google Scholar]
- 32. Murai M. J., Chruszcz M., Reddy G., Grembecka J., Cierpicki T. (2011) Crystal structure of menin reveals binding site for mixed lineage leukemia (MLL) protein. J. Biol. Chem. 286, 31742–31748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Eswar N., Webb B., Marti-Renom M. A., Madhusudhan M. S., Eramian D., Shen M. Y., Pieper U., Sali A. (2006) Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinformatics, Chapter 5, Unit 5.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Krieger E., Joo K., Lee J., Lee J., Raman S., Thompson J., Tyka M., Baker D., Karplus K. (2009) Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling. Four approaches that performed well in CASP8. Proteins 77, Suppl. 9, 114–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pieper U., Webb B. M., Barkan D. T., Schneidman-Duhovny D., Schlessinger A., Braberg H., Yang Z., Meng E. C., Pettersen E. F., Huang C. C., Datta R. S., Sampathkumar P., Madhusudhan M. S., Sjölander K., Ferrin T. E., Burley S. K., Sali A. (2011) ModBase, a database of annotated comparative protein structure models, and associated resources. Nucleic Acids Res. 39, D465–D474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ashkenazy H., Erez E., Martz E., Pupko T., Ben-Tal N. (2010) ConSurf 2010. Calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 38, W529–W533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Marx S. J., Simonds W. F. (2005) Hereditary hormone excess: genes, molecular pathways, and syndromes. Endocr. Rev. 26, 615–661 [DOI] [PubMed] [Google Scholar]
- 38. Hendy G. N., Kaji H., Sowa H., Lebrun J. J., Canaff L. (2005) Menin and TGF-β superfamily member signaling via the Smad pathway in pituitary, parathyroid, and osteoblast. Horm. Metab. Res. 37, 375–379 [DOI] [PubMed] [Google Scholar]
- 39. Klein R. D., Salih S., Bessoni J., Bale A. E. (2005) Clinical testing for multiple endocrine neoplasia type 1 in a DNA diagnostic laboratory. Genet. Med. 7, 131–138 [DOI] [PubMed] [Google Scholar]
- 40. Teh B. T., Kytölä S., Farnebo F., Bergman L., Wong F. K., Weber G., Hayward N., Larsson C., Skogseid B., Beckers A., Phelan C., Edwards M., Epstein M., Alford F., Hurley D., Grimmond S., Silins G., Walters M., Stewart C., Cardinal J., Khodaei S., Parente F., Tranebjaerg L., Jorde R., Salmela P. (1998) Mutation analysis of the MEN1 gene in multiple endocrine neoplasia type 1, familial acromegaly, and familial isolated hyperparathyroidism. J. Clin. Endocrinol. Metab. 83, 2621–2626 [DOI] [PubMed] [Google Scholar]
- 41. Teh B. T., Zedenius J., Kytölä S., Skogseid B., Trotter J., Choplin H., Twigg S., Farnebo F., Giraud S., Cameron D., Robinson B., Calender A., Larsson C., Salmela P. (1998) Thymic carcinoids in multiple endocrine neoplasia type 1. Ann. Surg. 228, 99–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kawamura J., Shimada Y., Komoto I., Okamoto H., Itami A., Doi R., Fujimoto K., Kosugi S., Imamura M. (2005) Multiple endocrine neoplasia type 1 gene mutations in sporadic gastrinomas in Japan. Oncol. Rep. 14, 47–52 [PubMed] [Google Scholar]
- 43. Hai N., Aoki N., Shimatsu A., Mori T., Kosugi S. (2000) Clinical features of multiple endocrine neoplasia type 1 (MEN1) gene mutations: analysis of 20 Japanese sporadic cases with MEN1. Clin. Endocrinol. 52, 509–518 [DOI] [PubMed] [Google Scholar]
- 44. Osthus R. C., Reyes Y., Glynn M. W., Bale A. E. (1998) Mutations in typical and variant MEN1 kindreds: Splice site mutation in familial isolated hyperparathyroidism. Am. J. Hum. Genet. 63, (suppl.) p. A80, (Abstr. 436) [Google Scholar]
- 45. Debelenko L. V., Brambilla E., Agarwal S. K., Swalwell J. I., Kester M. B., Lubensky I. A., Zhuang Z., Guru S. C., Manickam P., Olufemi S. E., Chandrasekharappa S. C., Crabtree J. S., Kim Y. S., Heppner C., Burns A. L., Spiegel A. M., Marx S. J., Liotta L. A., Collins F. S., Travis W. D., Emmert-Buck M. R. (1997) Identification of MEN1 gene mutations in sporadic carcinoid tumors of the lung. Hum. Mol. Genet. 6, 2285–2290 [DOI] [PubMed] [Google Scholar]
- 46. Karges W., Jostarndt K., Maier S., Flemming A., Weitz M., Wissmann A., Feldmann B., Dralle H., Wagner P., Boehm B. O. (2000) Multiple endocrine neoplasia type 1 (MEN1) gene mutations in a subset of patients with sporadic and familial primary hyperparathyroidism target the coding sequence but spare the promoter region. J. Endocrinol. 166, 1–9 [DOI] [PubMed] [Google Scholar]
- 47. Roijers J. F., Apel T., Neumann H. P., Arnim U. V., Lips C. J., Hoppener J. W. (2000) Internally shortened menin protein as a consequence of alternative RNA splicing due to a germ line deletion in the multiple endocrine neoplasia type 1 gene. Int. J. Mol. Med. 5, 611–614 [DOI] [PubMed] [Google Scholar]
- 48. Corbetta S., Vicentini L., Ferrero S., Lania A., Mantovani G., Cordella D., Beck-Peccoz P., Spada A. (2005) Activity and function of the nuclear factor κB pathway in human parathyroid tumors. Endocr. Relat. Cancer 12, 929–937 [DOI] [PubMed] [Google Scholar]
- 49. Stewart C., Parente F., Piehl F., Farnebo F., Quincey D., Silins G., Bergman L., Carle G. F., Lemmens I., Grimmond S., Xian C. Z., Khodei S., Teh B. T., Lagercrantz J., Siggers P., Calender A., Van de Vem V., Kas K., Weber G., Hayward N., Gaudray P., Larsson C. (1998) Characterization of the mouse Men1 gene and its expression during development. Oncogene 17, 2485–2493 [DOI] [PubMed] [Google Scholar]
- 50. Crabtree J. S., Scacheri P. C., Ward J. M., Garrett-Beal L., Emmert-Buck M. R., Edgemon K. A., Lorang D., Libutti S. K., Chandrasekharappa S. C., Marx S. J., Spiegel A. M., Collins F. S. (2001) A mouse model of multiple endocrine neoplasia, type 1, develops multiple endocrine tumors. Proc. Natl. Acad. Sci. U.S.A. 98, 1118–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bertolino P., Tong W. M., Galendo D., Wang Z. Q., Zhang C. X. (2003) Heterozygous Men1 mutant mice develop a range of endocrine tumors mimicking multiple endocrine neoplasia type 1. Mol. Endocrinol. 17, 1880–1892 [DOI] [PubMed] [Google Scholar]
- 52. Hussein N., Lu J., Casse H., Fontanière S., Morera A. M., Guittot S. M., Calender A., Di Clemente N., Zhang C. X. (2008) Deregulation of anti-Mullerian hormone/BMP and transforming growth factor-β pathways in Leydig cell lesions developed in male heterozygous multiple endocrine neoplasia type 1 mutant mice. Endocr. Relat. Cancer 15, 217–227 [DOI] [PubMed] [Google Scholar]
- 53. Mould A. W., Duncan R., Serewko-Auret M., Loffler K. A., Biondi C., Gartside M., Kay G. F., Hayward N. K. (2009) Global expression profiling of sex cord stromal tumors from Men1 heterozygous mice identifies altered TGF-β signaling, decreased Gata6 and increased Csf1r expression. Int. J. Cancer 124, 1122–1132 [DOI] [PubMed] [Google Scholar]
- 54. Bertolino P., Radovanovic I., Casse H., Aguzzi A., Wang Z. Q., Zhang C. X. (2003) Genetic ablation of the tumor suppressor menin causes lethality at mid-gestation with defects in multiple organs. Mech. Dev. 120, 549–560 [DOI] [PubMed] [Google Scholar]
- 55. Engleka K. A., Wu M., Zhang M., Antonucci N. B., Epstein J. A. (2007) Menin is required in cranial neural crest for palatogenesis and perinatal viability. Dev. Biol. 311, 524–537 [DOI] [PubMed] [Google Scholar]
- 56. Ji Y., Prasad N. B., Novotny E. A., Kaur S., Elkahloun A., Chen Y., Zhang R. Z., Chu M. L., Agarwal S. K., Marx S. J., Collins F. S., Chandrasekharappa S. C. (2007) Mouse embryo fibroblasts lacking the tumor suppressor menin show altered expression of extracellular matrix protein genes. Mol. Cancer Res. 5, 1041–1051 [DOI] [PubMed] [Google Scholar]
- 57. Aziz A., Miyake T., Engleka K. A., Epstein J. A., McDermott J. C. (2009) Menin expression modulates mesenchymal cell commitment to the myogenic and osteogenic lineages. Dev. Biol. 332, 116–130 [DOI] [PubMed] [Google Scholar]
- 58. Kanazawa I., Nayak G., Canaff L., Murshed M., Hendy G. N. (2011) Reduced accumulation of bone in mice lacking the Men1 gene in osteoblasts. J. Bone Miner. Res. 26, (suppl.1) p.S309, (Abstr. SU0233) [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.