

Background: The coupling mechanism of proton and electron transfer in the redox-linked proton pump cytochrome c oxidase (COX) is still not understood.
Results: Both H+ uptake and release steps during single-electron injection into oxidized COX precede net H+ uptake.
Conclusion: The first H+ uptake coincides with electron input into CuA at the opposite membrane side.
Significance: This suggests efficient H+ uptake mechanisms, such as proton-collecting antennae.
Keywords: Bioenergetics, Cytochrome Oxidase, Kinetics, Proton Transport, Spectroscopy, Cytochrome c Oxidase, pH Indicator Dye, Proton Transfer Kinetics, Redox-linked Proton Pump, Time-resolved Absorption Spectroscopy
Abstract
Cytochrome c oxidase (COX), the last enzyme of the respiratory chain of aerobic organisms, catalyzes the reduction of molecular oxygen to water. It is a redox-linked proton pump, whose mechanism of proton pumping has been controversially discussed, and the coupling of proton and electron transfer is still not understood. Here, we investigated the kinetics of proton transfer reactions following the injection of a single electron into the fully oxidized enzyme and its transfer to the hemes using time-resolved absorption spectroscopy and pH indicator dyes. By comparison of proton uptake and release kinetics observed for solubilized COX and COX-containing liposomes, we conclude that the 1-μs electron injection into CuA, close to the positive membrane side (P-side) of the enzyme, already results in proton uptake from both the P-side and the N (negative)-side (1.5 H+/COX and 1 H+/COX, respectively). The subsequent 10-μs transfer of the electron to heme a is accompanied by the release of 1 proton from the P-side to the aqueous bulk phase, leaving ∼0.5 H+/COX at this side to electrostatically compensate the charge of the electron. With ∼200 μs, all but 0.4 H+ at the N-side are released to the bulk phase, and the remaining proton is transferred toward the hemes to a so-called “pump site.” Thus, this proton may already be taken up by the enzyme as early as during the first electron transfer to CuA. These results support the idea of a proton-collecting antenna, switched on by electron injection.
Introduction
Cytochrome c oxidase (COX),2 the terminal enzyme of the respiratory chains of mitochondria and many aerobic prokaryotes, catalyzes electron transfer from cytochrome c to molecular oxygen, reducing the latter to water. Cytochrome c, which binds to COX on the positively charged side (P-side) of the membrane (the extramitochondrial or periplasmic side of bacteria), injects electrons into the bimetallic CuA center, which in turn donates electrons (1 at the time) to the low-spin heme a (Fig. 1). From there, electrons are passed on to the high-spin heme a3-CuB binuclear center, the binding site for oxygen. The protons required for water formation originate from the opposite, negatively charged side (N-side; the matrix side in the case of mitochondria or the cytoplasmic side in the case of bacteria) of the membrane. This redox reaction is coupled to translocation of additional protons across the membrane (“proton pumping”) to further increase the electrochemical proton gradient, which is the driving force for the ATP synthesis by the ATPase. To reduce 1 molecule of oxygen, 4 electrons are taken up from cytochrome c. Extensive studies have been performed to elucidate the mechanisms by which the enzyme translocates protons and couples this process with the chemical reaction (1–3). It is generally accepted that there is an overall involvement of 8 protons during the catalytic cycle: 4 “substrate” protons to complete the reaction (water formation) and 4 to be translocated (“pumped”) across the membrane.
FIGURE 1.

Overall architecture of COX embedded in lipid bilayer. Subunit I is shown in green, and subunit II is shown in cyan (Protein Data Bank code 1QLE). CuA, CuB, heme a, heme a3, and residues Asp124 and Lys354 are indicated. The figure was prepared with VMD (47).
Based on the crystal structure (4, 5) and mutagenesis studies (6–8), two proton pathways have been suggested for the bacterial COXs from Paracoccus denitrificans and Rhodobacter sphaeroides, leading from the N-side toward the heme-copper site: the K-pathway and the D-pathway. The K-pathway includes the conserved amino acid Lys354 and may be involved in the delivery of the first 1 or 2 protons during the reduction of the oxidized enzyme. The D-pathway, including Asp124, is likely to be involved in the uptake of both “chemical” and pumped protons in the F → O state transition. It appears to be the only pathway required when the fully reduced COX reacts with molecular oxygen (9–12).
The assignment of proton uptake and proton pumping to the individual steps of the catalytic cycle of COX is a matter of controversy (10, 13–22). Injection of 1 electron into the fully oxidized O state leads to formation of the 1 electron-reduced E state. During this step of the catalytic cycle, proton uptake was proposed to take place from the N-side of the membrane via the K-pathway and to be linked to the reduction of heme a (23). This idea is based on the fact that the slower electrogenic phase (τ ∼ 180 μs) observed in voltage measurements showed a clear kinetic deuterium isotope effect, indicative of the transfer of a proton from the N-side toward the P-side. However, the existence of this protonic phase was questioned (24) and is still a matter of debate (22, 25).
Resolving this debate is of crucial importance not only for proton uptake linkage in the O → E step but also for the determination of the role of proton transfer pathways through COX in the different reactions of the catalytic cycle. Our experiments are designed to directly determine the proton uptake and release in COX by nanosecond time-resolved absorption spectroscopy in combination with pH indicator dyes. To address the role of the D- and K-pathways, COX variants with mutations in the respective pathway were investigated.
Our results presented here clearly show that the first proton uptake from the aqueous environment already takes place in the 1-μs time range and coincides with electron input into the CuA center (26, 27). At later times, no proton uptake was observed. Instead and most surprisingly, electron transfer to heme a was accompanied by proton release. These data challenge and extend the current models of coupling proton and electron transfer (5, 17, 22–24, 28). We present a model that contains important aspects of each of the previous models and is, in a sense, a unification of these hypotheses.
EXPERIMENTAL PROCEDURES
Materials
Phenol red (phenolsulfonphthalein), 3-carboxy-2,2,5,5-tetramethyl-1-pyrrolidinyloxy (3CP), and asolectin (phosphatidylcholine type IIS) were obtained from Sigma. n-Dodecyl-β-d-maltoside (LM) was from GLYCON Biochemicals GmbH (Luckenwalde, Germany). (Ruthenium (2,2′-bipyridine)2)2 quarterpyridine (Ru2D) was a kind gift from K. Fendler and C. Bamann. All other chemicals were of the highest grade available.
Sample Preparation
Enzyme preparation of COX and variants from P. denitrificans strain AO1 was performed as described (29).
Proteoliposomes were prepared by the cholate dialysis method as described (30) using asolectin, which was further purified as described (31), at a concentration of 40 mg/ml. Lipids were dried under vacuum and resuspended in 100 mm HEPES/KOH (pH 7.3), 10 mm KCl, and 2% (w/v) cholate. The suspension was stirred on ice under argon for 1–2 h and sonicated to clarity with a Branson sonifier. COX was added to a concentration of 4 μm. Subsequently, asolectin vesicles were dialyzed against buffer without cholate and subsequent reduction of HEPES/KOH (10-kDa cutoff). In the last dialysis step, no buffer was present.
The respiratory control ratio was determined as the ratio of the rates of cytochrome c oxidation in the coupled and uncoupled states, respectively (32). Reduced cytochrome c was used at a concentration of 40 μm in 10 mm HEPES/KOH, 50 mm KCl, and 50 mm sucrose (pH 7.3); the rate of its oxidation was measured by following the change in absorbance at 550 nm after the addition of 1.2 nm reconstituted COX. Uncoupling was achieved by the addition of 5 μm valinomycin and 10 μm carbonyl cyanide m-chlorophenylhydrazone. The turnover number of COX was determined to be ∼500 electrons/s, and the respiratory control ratio was 8.5–9, i.e. the enzyme has sustained no damage during preparation. The COX concentration was determined from the reduced-minus-oxidized optical difference spectrum with ϵ605–630 nm = 11.7 mm−1 cm−1 (33).
Flash Spectroscopy
Flash spectroscopy was performed with a homemade flash photolysis spectrometer (34). Prior to the experiments, samples of WT COX and functional variants were incubated overnight with potassium ferricyanide to ensure a fully oxidized enzyme. Ferricyanide was then quickly removed by gel filtration (GE Healthcare PD-10 columns). In general, preparation and measurements were carried out in the dark to prevent the enzyme from prereducing (24).
Samples containing 10 μm COX in 0.05% LM, 25 μm Ru2D as an electron donor, 10 mm aniline as a sacrificial donor for ruthenium, 1 mm 3CP to prevent acidification due to proton release from aniline (35), and 50 mm KCl (pH 7.5) were excited with 10-ns pulses of 10–15 mJ of energy at 492 nm. Under these conditions, up to ∼10% of the COX becomes photoreduced. Electron uptake was monitored at 605 nm, as a rise in absorbance at this wavelength indicates the reduction of heme a. To reduce the amount of scattered light from the exciting laser flash, a cutoff filter (OG515) was placed in front of the entrance slit of the monochromator in the monitoring path. The signal-to-noise ratio obtained in a single-flash experiment was sufficient for data analysis.
Proton Uptake and Release Measurements
Proton concentration changes were recorded via the absorbance change in the soluble pH indicator dye phenol red (50 μm) at 558 nm with and without 50 mm Tris-HCl. Typically 5–10 time traces from single-flash experiments were averaged for the protonation kinetics accompanying the O → E transition.
The pKa of phenol red observed in the presence of COX and 50 mm salt was 7.95 compared with the value of 7.8 in the absence of COX. This small change in pKa indicates that the phenol red molecules may interact, at least partially, with the COX/detergent micelle, leading to a shift in pKa due to the protein surface potential (36, 37). The time constant of proton release from bacteriorhodopsin/LM micelles measured with phenol red (τ ∼ 70 μs) agrees with the time constant for proton release from bacteriorhodopsin (36) measured with a covalently bound pH indicator dye facing the detergent shell or residing at the cytoplasmic surface (opposite the proton release side), thus supporting our assumption. When phenol red interacts with the COX micelle, i.e. resides in the membrane-water interfacial layer, fast proton release and uptake events are detectable, in contrast to measurements with pH indicator dyes residing entirely in the aqueous bulk phase (36). Proton concentration changes in proteoliposomes were observed by the addition of 50 μm phenol red in the medium outside of the liposomes (P-side of COX).
Proton uptake stoichiometry was calculated according to Equation 1,
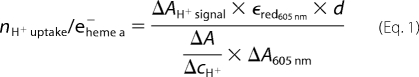 |
where ΔAH+ signal denotes the absorbance change at 558 nm (phenol red), ϵred605 nm is the extinction coefficient of the reduced heme a (21,600 m−1 cm−1 (38)), d is the diameter of the cuvette used, ΔA/ΔcH+ is the proton calibration factor (determined as the absorbance change for a defined proton concentration change), and ΔA605 nm is the absorbance change resulting from the reduction of heme a.
RESULTS
Spectral Characterization of WT COX
WT COX shows clear spectral differences between the oxidized and reduced forms (Fig. 2A). The shift of the strong absorption band from 425 to 440 nm is attributed to the reduced form of both heme groups (hemes a and a3), whereas the two absorption bands at 598 nm (oxidized form) and 605 nm (reduced form) are derived from heme a to almost 90% (marked by black and red arrows in Fig. 2A).
FIGURE 2.

Salt dependence of electron transfer signal. A, absorption spectra of oxidized COX (ox.; black line), reduced COX (red.; red line), and oxidized COX with phenol red (PR; dashed line). The absorption maxima of oxidized heme a (black arrow), reduced heme a (red arrow), and phenol red (arrowhead) are indicated. B, amplitude of the slow phase of the transient absorbance change at 605 nm (10-μs component) as a function of salt concentration. Inset, absorbance changes at 605 nm induced by flash photolysis of 10 μm COX in the presence of 0.05% LM, 25 μm Ru2D, and 10 mm aniline (pH 8) at various salt concentrations. The transients were obtained from a sample containing no NaCl and a sample containing 500 mm NaCl, respectively. mOD, milli-optical density units.
To investigate proton uptake or release, we used the soluble pH indicator dye phenol red, which has a pH-dependent absorption band at 558 nm (marked by an arrowhead in Fig. 2A). At this wavelength, the absorption spectrum of COX displays only marginal changes during reduction or oxidation; thus, the detected pH-dependent absorption change is almost solely due to the pH indicator dye.
Time-resolved Measurements of Electron Transfer
To observe the kinetics of electrons transferred to heme a in a single-electron photochemical reduction of the enzyme from the light-reactive electron donor Ru2D, the latter was excited with a single laser flash, and the absorbance change at 605 nm was recorded (Fig. 2B, inset). The lower the ionic strength, the better the binding of the ruthenium complex to COX and the subsequent electron transfer (Fig. 2B). Optimal conditions for maximum electron transfer were found at pH 7.5. However, we observed aggregation below 50 mm salt. Thus, we set the salt concentration to 50 mm. A representative time trace is presented in Fig. 3A. For a better comparison with results in the literature, the trace is presented with a linear time scale, although our data were recorded with 50-ns time resolution and sampled on a logarithmic time scale with 100 data points per decade. The time-dependent absorbance changes at 605 nm were fitted with two time constants of τ1 = 1.5 ± 0.1 μs and τ2 = 13.2 ± 0.7 μs. The first rise time was assigned to the relaxation of Ru2D, which correlates with the kinetics of electron uptake by CuA (26). The second time constant describes the kinetics of the electron transfer from CuA to heme a. The average time constant of electron transfer under these optimal conditions is τ = 13.7 ± 2.4 μs (mean value of five single-flash experiments at 492 nm excitation, 22 °C, and 50 mm KCl (pH 7.5)). No further absorbance changes were observed, in agreement with the formation of the 1 electron-reduced E state.
FIGURE 3.

Electron transfer and proton signal in WT COX solubilized and incorporated in liposomes. A, electron transfer signal at 50 mm KCl recorded at 605 nm. The second component of the time trace with a time constant of 13 μs describes the kinetics of the reduction of heme a. B, absorbance changes of the pH indicator phenol red at 558 nm after injection of 1 electron into solubilized COX with and without 50 mm buffer monitored in a sample containing 10 μm COX in 0.05% LM, 50 μm phenol red, 25 μm Ru2D, 10 mm aniline, 1 mm 3CP, and 50 mm salt. The time traces are the average of multiple single-flash experiments from three independent sample preparations. C, difference absorbance changes obtained from traces in B describing the kinetics of transient proton concentration changes in solubilized COX. For comparison, a fit with two (black line) and three (red line) exponentials is given, showing that the three-exponential fit describes the data best. A scheme of the experiment (solubilized COX and phenol red (PR)) is given in the inset. D, difference absorbance changes of 50 μm phenol red at 558 nm after injection of 1 electron into COX incorporated into liposomes with and without 50 mm buffer monitored in a sample containing 25 μm Ru2D, 10 mm aniline, 1 mm 3CP, and 50 mm salt. The proton signal was normalized to the maximum amplitude of the proton signal obtained from solubilized COX to allow for a direct comparison. For comparison, a fit with two (black line) and three (red line) exponentials is given. A scheme of the experiment (COX incorporated into liposomes and phenol red residing outside of the liposomes) is given in the inset. As the electron donor is added to the outside of the liposomes, electrons are injected only into those COX molecules with their P-side facing to the outside of the liposome. E, control proton signal. Difference absorbance changes of 50 μm phenol red at 558 nm after flash photolysis of 25 μm ruthenium-cytochrome c complex containing 10 mm aniline, 1 mm 3CP, and 50 mm salt. All measurements were performed at 20 °C and pH 7.5. Note that the time axis is linear in A and logarithmic in B–E. The data points were collected on a linear time scale and averaged such that the data points are equidistantly distributed on the logarithmic time scale to cover several decades. mOD, milli-optical density units.
Proton Uptake and Release Kinetics of WT COX
Proton uptake from the aqueous bulk phase was detected with the pH indicator dye phenol red in the O → E step of the WT COX catalytic cycle. In Fig. 3 (A and C), the kinetics of electron transfer (ΔA at 605 nm, “electron transfer signal”) are compared with the kinetics of proton concentration changes as detected with the pH indicator dye (ΔΔA at 558 nm, “proton signal”). Both measurements were performed under the same conditions (20 °C, pH 7.5, and 10 μm COX in 0.05% LM, 25 μm Ru2D, 10 mm aniline, 1 mm 3CP, and 50 mm salt/buffer). The proton signal was calculated as the difference between the two phenol red time traces obtained with and without buffer (Fig. 3B). To discriminate between true proton uptake by COX and possible transient protonation changes caused by the electron donor system, we measured a control proton signal using a covalent ruthenium-cytochrome complex. Under our experimental conditions, no contribution of the electron donor system to the transient proton signal was observed (Fig. 3E).
The proton signal (Fig. 3C) contains both proton uptake and release phases as seen by the positive and negative absorption changes. A fit of the proton signal (Fig. 3C) required three exponentials, marked by the respective arrows. The proton uptake time of τ1 = 1.2 ± 0.1 μs correlates with the first phase of the electron transfer signal (τ1 = 1.5 ± 0.1 μs), i.e. with the time constant for the electron transfer to CuA. Surprisingly, the proton uptake is followed by proton release. The decay of the proton signal contains two components, τ2 = 11.5 ± 2.8 μs and τ3 = 249 ± 18 μs. The 11.5-μs proton release component correlates with the electron transfer from CuA to heme a (τ2 = 13.7 ± 2.4 μs). However, the sum of the amplitudes of the decay components (proton release) is smaller than the amplitude of the rise component (proton uptake), therefore resulting in a positive net amplitude, i.e. in a net proton uptake.
Proton Kinetics of WT COX Incorporated in Liposomes
To ascertain the sidedness of the above observed proton uptake and release steps, proton measurements were also performed using COX-containing liposomes. The overall shape of the time trace (Fig. 3D) resembles that of the solubilized enzyme (Fig. 3C), with fast proton uptake in the beginning (τ1 = 0.9 ± 0.1 μs), followed by two consecutive proton release steps with τ2 = 6.7 ± 1.4 μs and τ3 = 192 ± 28 μs. In contrast to the kinetics of proton uptake and release observed in solubilized COX, no net proton uptake was detected in COX-containing liposomes with phenol red residing at the P-side of the enzyme (Fig. 3D). Thus, net proton uptake occurs from the N-side.
Stoichiometry of Proton Uptake by COX and Its Variants K345M and D124N
The analysis of the two proton signals measured for the solubilized enzyme and COX incorporated in liposomes (Fig. 3, C and D) in terms of the numbers of protons taken up per injected electron using Equation 1 clearly shows that, for each injected electron, more than 1 proton is taken up by the enzyme: ∼1 proton from the N-side and, on average, ∼1.5 H+ from the P-side (Table 1). Simultaneously with electron transfer from CuA to heme a, ∼1 proton is released from the P-side. The last proton release step with a time constant of ∼200 μs takes place on both sides of the enzyme. The number of remaining protons in the enzyme at the N-side is 0.4 H+/COX on average. Time constants and stoichiometries are summarized in Table 1.
TABLE 1.
Time constants of proton uptake and release and proton stoichiometries
Time constants of proton uptake and release as well as of electron transfer in the O → E step and proton stoichiometries for the individual proton transfer reactions triggered by the injection of 1 electron into the oxidized enzyme are given below.
| H+ uptake τ1 | H+ release |
Net proton uptake | e− transfer |
|||
|---|---|---|---|---|---|---|
| τ2 | τ3 | τ1 | τ2 | |||
| μs | μs | μs | ||||
| Solubilized COX | 1.2 ± 0.1 | 11.5 ± 2.8 | 249 ± 18 | 1.5 ± 0.1 | 13.2 ± 0.7 | |
| COX-containing liposomes | 0.9 ± 0.1 | 6.7 ± 1.4 | 192 ± 28 | 2.7 ± 0.2 | 13.6 ± 2.9 | |
| H+/COX | ||||||
| Solubilized COX | 2.6 ± 0.4 | 0.8 ± 0.2 | 1.4 ± 0.2 | 0.38 ± 0.05 | ||
| COX-containing liposomes (detection at P-side) | 1.6 ± 0.4 | 1.1 ± 0.2 | 0.5 ± 0.2 | 0 | ||
| Solubilized D124N | 1.0 ± 0.2 | 0.5 ± 0.08 | 0.4 ± 0.06 | 0.1 ± 0.02 | ||
| Solubilized K354M | 1.3 ± 0.2 | 0.7 ± 0.1 | 0.2 ± 0.03 | 0.2 ± 0.03 | ||
Measurements of COX variants blocked in either the K-pathway (K354M) or the D-pathway (D124N) showed that the blockage results in reduced initial proton uptake but does not abolish net proton uptake (Table 1). In both variants, ∼1 proton is taken up in the first step, which kinetically correlates with the initial electron injection into CuA as observed in the wild type. Net proton uptake was determined to 0.1 H+/COX and 0.2 H+/COX for K354M and D124N, respectively.
DISCUSSION
The coupling of electron injection and proton uptake is still elusive. Although several studies investigating the proton uptake by COX have been published, the results are not consistent (10, 13–22). In particular, for the O → E transition, where 1 electron is injected into the oxidized enzyme, the published results are controversial (22–25). Electron transfer from CuA to heme a followed by proton transfer through the K-pathway in the O → E transition with kinetics of ∼200 μs has been found in voltage measurements (23). In disagreement with that, a signal consistent with the absence of proton uptake was observed upon reduction of heme a, also using voltage measurements (24), although a more recent study indicated that proton uptake occurs with a time constant of 150 μs (25). To explain the differences in the various studies, it was suggested (22) that the proton uptake observed previously (23) did not correlate to the O → E transition but rather to electron transfer from heme a to the heme a3-CuB binuclear center due to a not fully oxidized sample or differences in preparation of the fully oxidized COX sample leading to partial transfer of the first electron from heme a to the binuclear center. In our experiments, care was taken to use a fully oxidized COX sample (see “Experimental Procedures”), and the kinetics of the electron transfer steps were monitored.
In this study, using time-resolved absorption spectroscopy and pH indicator dyes, we directly observed proton concentration changes equivalent to proton uptake and release by COX during the O → E step. A careful optimization of the parameters affecting the proton signal allowed us to gain fundamental insights into the proton transfer steps associated with the injection of 1 electron into the oxidized enzyme.
A striking feature is the observation that proton uptake by COX occurs already in the 1-μs time range, when electron injection into CuA occurs. This fast proton uptake is followed by a gradual proton release. As the amplitude of the proton release signal is smaller than the amplitude of the proton uptake component, a final net proton uptake from the N-side is determined for the O → E step and becomes apparent at times slower than 300 μs (Fig. 3C).
The proton uptake stoichiometry was calculated according to Equation 1 and amounts to an initial value of 2.6 H+/COX per electron input into the WT enzyme. Comparison of proton uptake by the solubilized enzyme and that by COX incorporated into liposomes unambiguously shows that protons are taken up from both sides of the enzyme, ∼1.5 H+ from the P-side and 1 H+ from the N-side (Fig. 3 and Table 1). This large number of protons taken up is clearly surprising. It is very unlikely, however, that the electron donor system is responsible for the observed proton excess. Rapid proton release by aniline upon re-reduction of Ru2D, an effect that even opposes the observed decrease in proton concentration in the aqueous bulk phase as a result of proton uptake by COX, was eliminated by the addition of 3CP and further tested in our experiments by monitoring the pH indicator dye absorption changes upon photoreduction of cytochrome c by using a covalent ruthenium complex.
However, even more surprising is the finding that electron transfer to CuA already leads to proton uptake at the opposite membrane surface. One might have expected the proton uptake accompanying electron injection into CuA on the P-side by titratable residues nearby to electrostatically balance the negative charge of the extra electron at CuA, but obviously, the organization of the protein in the low membrane dielectric allows pK changes leading to proton uptake at the opposite side of the membrane via long-range interactions. These findings are in accordance with the electroneutrality principle (39), which states that each electron transfer into the hydrophobic interior of COX is charge-compensated by the uptake of a proton. In addition, long-range effects may result in protonation dynamics at the opposite membrane surface.
For proton uptake from the N-side, the idea of a proton-collecting antenna has been widely discussed (see Ref. 40 for a detailed review). This idea is supported by our observations: as initial excess proton uptake occurs not only from the P-side but also from the N-side and net proton uptake accompanying formation of the E state is abolished in neither the D124N nor K354M variant, some of the residues in the vicinity of the proton transfer pathways probably play a role as the primary proton acceptor(s). Our experimental result differs from an earlier theoretical prediction (41), where all protonation changes upon reduction of CuA are accompanied by proton uptake mostly by residues located on the cytosolic side (N-side) of the membrane. On the basis of our data, we propose that important transmembrane charge compensation occurs already during electron transfer to the CuA center and not primarily during reduction of heme a (Fig. 4). Reduction of CuA may, via long-range interactions, influence and prepare proton transfer via the D-pathway.
FIGURE 4.

Model for coupling electron injection and proton transfer steps in O → E step. A nanosecond laser flash excites Ru2D and initiates electron transfer to CuA within 1 μs. This event causes electron uptake from both sides of the enzyme. Red arrows indicate proton movement in the direction of proton pumping through COX, and black arrows indicate proton transfer in the opposite direction. After 10 μs, the electron is being transferred to heme a, and partial proton release occurs on the P-side of the membrane. After 200 μs, all but 0.38 protons/COX are released. According to Ref. 23, the residual proton is transferred toward the catalytic center.
From the primary proton acceptor sites at the N-side as well as from the P-side, partial proton release occurs within ∼200 μs, leaving ∼0.4 H+ on the N-side of the enzyme (Figs. 3 and 4). This time constant correlates with the time constant observed for the deuterium-sensitive phase (∼180 μs) in voltage measurements (23). Combining these results, the following picture emerges: at the same time as the last proton release step occurs from the enzyme, the remaining proton is transferred toward the hemes, observed as a shift of a positive charge from the N-side toward the membrane interior (Fig. 4). The number of 0.4 H+/COX also agrees with the results for redox-linked proton uptake of ∼0.2–0.4 H+/heme a-CuA pair in carbon monoxide-treated COX (42).
In summary, we have shown that multistep proton transfer reactions take place during the single-electron transfer in the O → E step of the catalytic cycle of COX. Excess proton uptake from both sides of the membrane is coupled to electron input into CuA and precedes proton transfer from the N-side to the hemes. The former suggests the existence of efficient proton uptake mechanisms, such as proton-collecting antennae at the protein surface (40), which were also discussed for proton uptake by bacteriorhodopsin (43–45) and green fluorescent protein (46). The observed crosstalk of the two enzyme surfaces, probably mediated by long-range electrostatic interactions, and the consecutive protonation-deprotonation reactions may constitute a common mechanism linking the proton and electron transfer reactions in the different stages of the catalytic cycle of COX.
Acknowledgment
We thank Dr. Elena Olkhova for discussion.
This work was supported by Deutsche Forschungsgemeinschaft Grants SFB 472 (to H. M.) and SFB 449 (to U. A.), the Max-Planck-Gesellschaft, Cluster of Excellence Frankfurt “Macromolecular Complexes” and the Fonds der Chemischen Industrie.
- COX
- cytochrome c oxidase
- 3CP
- 3-carboxy-2,2,5,5-tetramethyl-1-pyrrolidinyloxy
- LM
- n-dodecyl-β-d-maltoside
- Ru2D
- (ruthenium (2,2′-bipyridine)2)2 quarterpyridine.
REFERENCES
- 1. Michel H. (1999) Cytochrome c oxidase: catalytic cycle and mechanisms of proton pumping–a discussion. Biochemistry 38, 15129–15140 [DOI] [PubMed] [Google Scholar]
- 2. Wikström M. (2004) Cytochrome c oxidase: 25 years of the elusive proton pump. Biochim. Biophys. Acta 1655, 241–247 [DOI] [PubMed] [Google Scholar]
- 3. Yoshikawa S., Muramoto K., Shinzawa-Itoh K. (2011) Proton-pumping mechanism of cytochrome c oxidase. Annu. Rev. Biophys. 40, 205–223 [DOI] [PubMed] [Google Scholar]
- 4. Iwata S., Ostermeier C., Ludwig B., Michel H. (1995) Structure at 2.8 Å resolution of cytochrome c oxidase from Paracoccus denitrificans. Nature 376, 660–669 [DOI] [PubMed] [Google Scholar]
- 5. Koepke J., Olkhova E., Angerer H., Müller H., Peng G., Michel H. (2009) High resolution crystal structure of Paracoccus denitrificans cytochrome c oxidase: new insights into the active site and the proton transfer pathways. Biochim. Biophys. Acta 1787, 635–645 [DOI] [PubMed] [Google Scholar]
- 6. Fetter J. R., Qian J., Shapleigh J., Thomas J. W., García-Horsman A., Schmidt E., Hosler J., Babcock G. T., Gennis R. B., Ferguson-Miller S. (1995) Possible proton relay pathways in cytochrome c oxidase. Proc. Natl. Acad. Sci. U.S.A. 92, 1604–1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Garcia-Horsman J. A., Puustinen A., Gennis R. B., Wikström M. (1995) Proton transfer in cytochrome bo3 ubiquinol oxidase of Escherichia coli: second-site mutations in subunit I that restore proton pumping in the mutant Asp135 → Asn. Biochemistry 34, 4428–4433 [DOI] [PubMed] [Google Scholar]
- 8. Thomas J. W., Puustinen A., Alben J. O., Gennis R. B., Wikström M. (1993) Substitution of asparagine for aspartate 135 in subunit I of the cytochrome bo ubiquinol oxidase of Escherichia coli eliminates proton-pumping activity. Biochemistry 32, 10923–10928 [DOI] [PubMed] [Google Scholar]
- 9. Konstantinov A. A., Siletsky S., Mitchell D., Kaulen A., Gennis R. B. (1997) The roles of the two proton input channels in cytochrome c oxidase from Rhodobacter sphaeroides probed by the effects of site-directed mutations on time-resolved electrogenic intraprotein proton transfer. Proc. Natl. Acad. Sci. U.S.A. 94, 9085–9090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brzezinski P., Adelroth P. (1998) Pathways of proton transfer in cytochrome c oxidase. J. Bioenerg. Biomembr. 30, 99–107 [DOI] [PubMed] [Google Scholar]
- 11. Michel H., Behr J., Harrenga A., Kannt A. (1998) Cytochrome c oxidase: structure and spectroscopy. Annu. Rev. Biophys. Biomol. Struct. 27, 329–356 [DOI] [PubMed] [Google Scholar]
- 12. Forte E., Scandurra F. M., Richter O. M., D'Itri E., Sarti P., Brunori M., Ludwig B., Giuffrè A. (2004) Proton uptake upon anaerobic reduction of the Paracoccus denitrificans cytochrome c oxidase: a kinetic investigation of the K354M and D124N mutants. Biochemistry 43, 2957–2963 [DOI] [PubMed] [Google Scholar]
- 13. Brzezinski P., Adelroth P. (1998) Proton-controlled electron transfer in cytochrome c oxidase: functional role of the pathways through Glu286 and Lys362. Acta Physiol. Scand. Suppl. 643, 7–16 [PubMed] [Google Scholar]
- 14. Karpefors M., Adelroth P., Aagaard A., Sigurdson H., Svensson Ek M., Brzezinski P. (1998) Electron-proton interactions in terminal oxidases. Biochim. Biophys. Acta 1365, 159–169 [DOI] [PubMed] [Google Scholar]
- 15. Mills D. A., Ferguson-Miller S. (1998) Proton uptake and release in cytochrome c oxidase: separate pathways in time and space? Biochim. Biophys. Acta 1365, 46–52 [DOI] [PubMed] [Google Scholar]
- 16. Papa S., Capitanio N., Villani G., Capitanio G., Bizzoca A., Palese L. L., Carlino V., De Nitto E. (1998) Cooperative coupling and role of heme a in the proton pump of heme-copper oxidases. Biochimie 80, 821–836 [DOI] [PubMed] [Google Scholar]
- 17. Michel H. (1999) Proton pumping by cytochrome c oxidase. Nature 402, 602–603 [DOI] [PubMed] [Google Scholar]
- 18. Hill B. C. (2004) Intermediate forms of cytochrome oxidase observed in transient kinetic experiments and those visited in the catalytic cycle. Biochim. Biophys. Acta 1655, 256–262 [DOI] [PubMed] [Google Scholar]
- 19. Hosler J. P., Ferguson-Miller S., Mills D. A. (2006) Energy transduction: proton transfer through the respiratory complexes. Annu. Rev. Biochem. 75, 165–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Papa S., Capitanio G., Luca Martino P. (2006) Concerted involvement of cooperative proton-electron linkage and water production in the proton pump of cytochrome c oxidase. Biochim. Biophys. Acta 1757, 1133–1143 [DOI] [PubMed] [Google Scholar]
- 21. Verkhovsky M. I., Belevich I., Bloch D. A., Wikström M. (2006) Elementary steps of proton translocation in the catalytic cycle of cytochrome oxidase. Biochim. Biophys. Acta 1757, 401–407 [DOI] [PubMed] [Google Scholar]
- 22. Siletsky S. A., Konstantinov A. A. (August 30, 2011) Biochim. Biophys. Acta 10.1016/j.bbabio. 2011.08.003 [DOI] [PubMed] [Google Scholar]
- 23. Ruitenberg M., Kannt A., Bamberg E., Ludwig B., Michel H., Fendler K. (2000) Single-electron reduction of the oxidized state is coupled to proton uptake via the K-pathway in Paracoccus denitrificans cytochrome c oxidase. Proc. Natl. Acad. Sci. U.S.A. 97, 4632–4636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Verkhovsky M. I., Tuukkanen A., Backgren C., Puustinen A., Wikström M. (2001) Charge translocation coupled to electron injection into oxidized cytochrome c oxidase from Paracoccus denitrificans. Biochemistry 40, 7077–7083 [DOI] [PubMed] [Google Scholar]
- 25. Belevich I., Bloch D. A., Belevich N., Wikström M., Verkhovsky M. I. (2007) Exploring the proton pump mechanism of cytochrome c oxidase in real time. Proc. Natl. Acad. Sci. U.S.A. 104, 2685–2690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nilsson T. (1992) Photoinduced electron transfer from tris(2,2′-bipyridyl)ruthenium to cytochrome c oxidase. Proc. Natl. Acad. Sci. U.S.A. 89, 6497–6501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zaslavsky D., Sadoski R. C., Wang K., Durham B., Gennis R. B., Millett F. (1998) Single-electron reduction of cytochrome c oxidase compound F: resolution of partial steps by transient spectroscopy. Biochemistry 37, 14910–14916 [DOI] [PubMed] [Google Scholar]
- 28. Wikström M., Jasaitis A., Backgren C., Puustinen A., Verkhovsky M. I. (2000) The role of the D- and K-pathways of proton transfer in the function of the heme-copper oxidases. Biochim. Biophys. Acta 1459, 514–520 [DOI] [PubMed] [Google Scholar]
- 29. Kleymann G., Ostermeier C., Ludwig B., Skerra A., Michel H. (1995) Engineered Fv fragments as a tool for the one-step purification of integral multisubunit membrane protein complexes. Biotechnology 13, 155–160 [DOI] [PubMed] [Google Scholar]
- 30. Kannt A., Soulimane T., Buse G., Becker A., Bamberg E., Michel H. (1998) Electrical current generation and proton pumping catalyzed by the ba3-type cytochrome c oxidase from Thermus thermophilus. FEBS Lett. 434, 17–22 [DOI] [PubMed] [Google Scholar]
- 31. Darley-Usmar V., Capaldi R., Takamiya S., Millett F., Wilson M., Malatesta F., Sarti P. (1987) Mitochondria: A Practical Approach, IRL Press, Oxford [Google Scholar]
- 32. Müller M., Thelen M., O'Shea P., Azzi A. (1986) Functional reconstitution of proton-pumping cytochrome c oxidase in phospholipid vesicles. Methods Enzymol. 126, 78–87 [DOI] [PubMed] [Google Scholar]
- 33. Ludwig B., Schatz G. (1980) A two-subunit cytochrome c oxidase (cytochrome aa3) from Paracoccus denitrificans. Proc. Natl. Acad. Sci. U.S.A. 77, 196–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Alexiev U., Mollaaghababa R., Khorana H. G., Heyn M. P. (2000) Evidence for long-range allosteric interactions between the extracellular and cytoplasmic parts of bacteriorhodopsin from the mutant R82A and its second site revertant R82A/G231C. J. Biol. Chem. 275, 13431–13440 [DOI] [PubMed] [Google Scholar]
- 35. Zaslavsky D., Sadoski R. C., Rajagukguk S., Geren L., Millett F., Durham B., Gennis R. B. (2004) Direct measurement of proton release by cytochrome c oxidase in solution during the F → O transition. Proc. Natl. Acad. Sci. U.S.A. 101, 10544–10547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Alexiev U., Marti T., Heyn M. P., Khorana H. G., Scherrer P. (1994) Surface charge of bacteriorhodopsin detected with covalently bound pH indicators at selected extracellular and cytoplasmic sites. Biochemistry 33, 298–306 [DOI] [PubMed] [Google Scholar]
- 37. Alexiev U., Scherrer P., Marti T., Khorana H. G., Heyn M. P. (1995) Time-resolved surface charge change on the cytoplasmic side of bacteriorhodopsin. FEBS Lett. 373, 81–84 [DOI] [PubMed] [Google Scholar]
- 38. Morrison M., Horie S. (1965) Determination of heme a concentration in cytochrome preparations by hemochromogen method. Anal. Biochem. 12, 77–82 [DOI] [PubMed] [Google Scholar]
- 39. Rich P. (1995) Towards an understanding of the chemistry of oxygen reduction. Funct. Plant Biol. 22, 479–486 [Google Scholar]
- 40. Adelroth P., Brzezinski P. (2004) Surface-mediated proton transfer reactions in membrane-bound proteins. Biochim. Biophys. Acta 1655, 102–115 [DOI] [PubMed] [Google Scholar]
- 41. Popović D. M., Stuchebrukhov A. A. (2004) Electrostatic study of the proton-pumping mechanism in bovine heart cytochrome c oxidase. J. Am. Chem. Soc. 126, 1858–1871 [DOI] [PubMed] [Google Scholar]
- 42. Verkhovsky M. I., Belevich N., Morgan J. E., Wikstrom M. (1999) Proton linkage of cytochrome a oxidoreduction in carbon monoxide-treated cytochrome c oxidase Biochim. Biophys. Acta 1412, 184–189 [DOI] [PubMed] [Google Scholar]
- 43. Sass H. J., Büldt G., Gessenich R., Hehn D., Neff D., Schlesinger R., Berendzen J., Ormos P. (2000) Structural alterations for proton translocation in the M state of wild-type bacteriorhodopsin. Nature 406, 649–653 [DOI] [PubMed] [Google Scholar]
- 44. Riesle J., Oesterhelt D., Dencher N. A., Heberle J. (1996) D38 is an essential part of the proton translocation pathway in bacteriorhodopsin. Biochemistry 35, 6635–6643 [DOI] [PubMed] [Google Scholar]
- 45. Alexiev U., Mollaaghababa R., Scherrer P., Khorana H. G., Heyn M. P. (1995) Rapid long-range proton diffusion along the surface of the purple membrane and delayed proton transfer into the bulk. Proc. Natl. Acad. Sci. U.S.A. 92, 372–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Agmon N. (2005) Proton pathways in green fluorescence protein. Biophys. J. 88, 2452–2461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Humphrey W., Dalke A., Schulten K. (1996) VMD: Visual Molecular Dynamics. J. Mol. Graph. 14, 33–38, 27–38 [DOI] [PubMed] [Google Scholar]