

Background: The divergent activities of the BlaR1 sensor protein and those of the resistance enzymes are assessed.
Results: Experiments shed light on the importance of a specific amino acid in imparting disparate functions for the family of related enzymes.
Conclusion: Same structural template can impart disparate functions to proteins.
Significance: This discovery reveals how selection of function for proteins in nature takes place.
Keywords: Antibiotic Resistance, Antibiotics Action, Enzyme Mechanisms, Enzyme Mutation, Enzyme Structure, Antibiotic Sensing
Abstract
The integral membrane protein BlaR1 of Staphylococcus aureus senses the presence of β-lactam antibiotics in the milieu and transduces the information to its cytoplasmic side, where its activity unleashes the expression of a set of genes, including that for BlaR1 itself, which manifest the antibiotic-resistant phenotype. The x-ray structure of the sensor domain of this protein exhibits an uncanny similarity to those of the class D β-lactamases. The former is a membrane-bound receptor/sensor for the β-lactam antibiotics, devoid of catalytic competence for substrate turnover, whereas the latter are soluble periplasmic enzymes in Gram-negative bacteria with avid ability for β-lactam turnover. The two are clearly related to each other from an evolutionary point of view. However, the high resolution x-ray structures for both by themselves do not reveal why one is a receptor and the other an enzyme. It is documented herein that a single amino acid change at position 439 of the BlaR1 protein is sufficient to endow the receptor/sensor protein with modest turnover ability for cephalosporins as substrates. The x-ray structure for this mutant protein and the dynamics simulations revealed how a hydrolytic water molecule may sequester itself in the antibiotic-binding site to enable hydrolysis of the acylated species. These studies document how the nature of the residue at position 439 is critical for the fate of the protein in imparting unique functions on the same molecular template, to result in one as a receptor and in another as a catalyst.
Introduction
A total of 2700 unique protein folds have been proposed to exist in nature (1). Considering that there are more open reading frames in most genomes sequenced to date than this figure, the implication is that nature exploits individual protein folds for multiple functions. A well documented structural scaffolding is that of the catalytic domains of penicillin-binding proteins (PBPs),4 which is shared with β-lactamases. Whereas β-lactamases are bacterial resistance enzymes to β-lactam antibiotics, PBPs are enzymes of biosynthesis of the cell wall (2). The issue of the evolutionary kinship between PBPs and β-lactamases from a structural perspective has been reviewed earlier (3, 4).
An interesting feature of these evolutionary events is that the three classes of active site serine β-lactamases known (classes A, C, and D) have evolved distinct catalytic mechanisms (5), which diverge from the reactions that take place in the active sites of PBPs, the vast majority of which are also serine-dependent enzymes. β-Lactam antibiotics are covalent inhibitors of PBPs, but they are substrates for β-lactamases. The antibiotic acylates the active site serine in PBPs, giving rise to an acyl-enzyme species that exhibits stability, resulting in enzyme inhibition. Serine acylation in β-lactamases is followed by a second and hydrolytic step that completes turnover of β-lactam antibiotics. Another point of interest is that virtually all PBPs are membrane-bound proteins, whereas β-lactamases are not. It has been argued that three separate evolutionary events led to the shedding of β-lactamases from their distinct membrane-bound progenitor PBPs in the course of emergence of antibiotic resistance genes (4). The structures of the catalytic domains of PBPs are highly similar to those of serine-dependent β-lactamases.
Investigations from our laboratories and from others (6–8) have documented also that the structure of the antibiotic sensor domain of the Staphylococcus aureus BlaR1 bears an uncanny resemblance to those of class D β-lactamases (Fig. 1A). This includes the general fold of the proteins, as well as the respective antibiotic-binding sites, yet the former protein is merely a sensor for the presence of β-lactam antibiotics in the milieu (Fig. 1B, steps I–III). It undergoes acylation at a serine on binding of the β-lactam antibiotic, the product of which exhibits significant longevity (9). However, the same protein fold with the same catalytic machinery in class D β-lactamases is endowed with multiple substrate turnover ability. The acyl-enzyme species formed on binding of the β-lactam antibiotic undergoes deacylation in a second step, liberating the protein to undergo additional cycles of antibiotic turnover (Fig. 1B, steps I–IV).
FIGURE 1.

A, stereo view of the ribbon structure of the sensor domain of S. aureus BlaR1 (Protein Data Bank entry 3Q7V, in blue) superimposed with that of the P. aeruginosa class D β-lactamase (Protein Data Bank entry 1E4D, in red). B, Lys-392 of BlaR1—and the corresponding lysine in class D β-lactamases—has to experience N-carboxylation for the protein to be functional. The N-carboxylation of Lys-392 promotes Ser-389 acylation by the antibiotic (step II). N-Decarboxylation Lys-392 at the acyl-enzyme stage is the final species for BlaR1 (step III), but the corresponding N-recarboxylated lysine in class D β-lactamases or in the BlaRS N439V mutant variant does carry out the deacylation step (step IV) to complete a catalytic cycle of turnover of β-lactam antibiotic. C, a close-up stereo view of the antibiotic-binding site of BlaR1 shown as a Connolly surface in green with important residues depicted as sticks and atoms colored by atom types (nitrogen in blue, oxygen in red, and carbon in gray; hydrogens not shown). D, a close-up stereo view of the antibiotic-binding site of the class D OXA10 β-lactamase.
Insight into this divergence of mechanisms has emerged recently, and it has much to do with the disposition of the critical active site N-carboxylated lysine in these two proteins (Fig. 1, B–D) (8, 10, 11). The two types of proteins position this important post-translationally modified amino acid differently within the antibiotic-binding site, as we discern from x-ray structures, predisposing it in the case of the BlaR1 protein to an N-decarboxylation event that entraps the acyl-protein species, hence the longevity of the species that is necessary for the sensor function of BlaR1 (Fig. 1B, step III). Whereas N-decarboxylation of the corresponding lysine in class D β-lactamases also takes place (12), it does so only after many antibiotic turnover events have taken place; hence there exists substrate turnover (12). It is of interest that BlaR1 cannot readily N-recarboxylate at the requisite lysine—the reason for which is discussed later—but class D β-lactamases do, which enhances the catalytic competence of class D β-lactamases (8, 12).
The two antibiotic-binding sites are decidedly highly similar but clearly not identical. A conspicuous difference exists at the amino acid at position 439. This residue in the BlaR1 protein is either an asparagine (as in the S. aureus variant) or a threonine (as in BlaR1 in other organisms). The side chain of the amino acid at this position is hydrogen-bonded to that of the N-carboxylated lysine (Lys-392) in the BlaR1 protein. Hence, the ability of residue 439 to hydrogen bond to the N-carboxylated Lys-392 would appear to be conserved in all BlaR1 proteins (Fig. 1C). We note that in all class D β-lactamases, this residue has been converted strictly to a hydrophobic amino acid (valine, isoleucine, or leucine; Fig. 1D). We also note that the hydrogen bonding ability of the residue at 439 (Asn-439 in S. aureus) allows the side chain of N-carboxylated Lys-392 to be accommodated in an extended conformation, away from Ser-389, the site of acylation by the antibiotic (Fig. 1C) (8). The absence of this hydrogen bonding in class D β-lactamases allows the alternative hydrogen bonding between the side chains of the active site serine and the N-carboxylated lysine. This differential set of interactions, as will be elaborated later, would appear to be important in evolution of the distinct functions for these clearly related proteins.
We have investigated in this report the importance of these differences with respect to position 439 in emergence of the catalytic turnover of β-lactam antibiotics engineered into the template of the sensor domain of the BlaR1 protein (BlaRS) of S. aureus. This effort documents unequivocally the role of Asn-439 in the BlaR1 protein as important for the sensing of the antibiotic, one that arrests the progress of the process at the acylated protein stage (Fig. 1B, step III), as will be detailed. The mutation at this position to a hydrophobic amino acid is a gateway for the emergence of catalytic ability that is seen in class D β-lactamases (Fig. 1B, steps I–IV).
EXPERIMENTAL PROCEDURES
Materials
The antibiotics and other reagents were purchased from Sigma, unless otherwise stated. The growth medium was purchased either from Difco Laboratories (Detroit, MI) or Fisher. The chromatography media were either from Bio-Rad or Amersham Biosciences. Escherichia coli DH5α was from Invitrogen, and E. coli BL21(DE3) and plasmid pET24a(+) were from Novagen. NaH13CO3 was from Cambridge Isotope Laboratories.
Site-directed Mutagenesis of BlaRS from S. aureus NRS128
The surface domain of BlaR1 was cloned and introduced into the pUC19 vector, as described previously (9). The N439V mutation was introduced using the QuikChange II XL site-directed mutagenesis kit (Stratagene, La Jolla, CA). The plasmid pUC19_blaRS was used as the DNA template (9). The vector was amplified using Pfu Turbo DNA polymerase and two overlapping mutagenic primers, BlaRSN439V-D (5′ GCAAAATTCAGTTGTTTGGTACTTCGAACG) and BlaRSN439V-R (5′ CGTTCGAAGTACCAAACAACTGAATTTTGC), to introduce the Asn-439 to Val substitution (mutation site italicized). The PCR products were digested with restriction endonuclease DpnI to remove the template plasmid, which was methylated. The DpnI-treated DNA mixture was then transformed into E. coli JM83. The blaRS gene was sequenced to confirm the introduction of the desired mutation. The mutated gene was then released by digestion with NdeI and HindIII endonucleases and ligated into pET-24a(+) vector, previously digested with the same restriction enzymes. The resulting vector was designated as pET-24a(+)_blaRSN439V.
To produce the mutation N439V of BlaRS for crystallization studies, the point mutation was introduced into the BlaRS triple-mutant variant K369A/K370A/K372A. Two PCRs were carried out to introduce the N439V mutation, using the plasmid pET24a(+)_blaRS_SM411 (8), which codes for BlaRS K369A/K370A/K372A, as template. Substitution of the AAT codon (Asn) for GTT (Val) introduced the desired mutation in the antibiotic-binding site. The region surrounding the codon AAT that codes for Asn-439 in BlaR1 was analyzed for possible silent mutations that would introduce a unique endonuclease restriction site that can be used for the mutagenesis strategy. Substitution of the CAA codon, located 12 bases before the Asn-439 codon, for CAG introduced an EcoRI restriction site by silent mutagenesis, which was appropriate for the mutagenesis strategy, because there is no such site in the wild-type blaRS sequence. In the first PCR, we amplified a fragment encompassing nucleotides 1302–1755 of blaR1 (or 315–768 of blaRS) using a forward oligonucleotide that introduces an EcoRI site by silent mutagenesis, and the N439V mutation (primer EcoRI_N110V_fw; 5′-gcagaattcagttgtttggtacttcg), and a reverse primer with a HindIII site (primer HindIII_blaRs_rv; 5′-ttcaaagcttattggccatttaaaacac). The purified PCR product was digested with EcoRI and HindIII endonucleases, and the resulting fragment was purified from a 1% agarose gel. In the second PCR, we amplified a fragment encompassing nucleotides 988–1313 of blaR1 (or 1–326 of blaRS) using a forward oligonucleotide that introduces an NdeI site (primer NdeI_blaRs_fw; 5′-gatatacatatggggcaatccataactg) and a reverse primer that introduces an EcoRI site by silent mutagenesis (primer EcoRI_blaRs_rv; 5′-actgaattctgcattgctgtatttaaatc). The purified PCR product was digested with NdeI and EcoRI endonucleases, and the resulting fragment was purified from a 1% agarose gel. The two PCR fragments, digested with endonucleases as described above, were cloned between the NdeI and HindIII sites of the pET-24a(+) expression vector, to give the plasmid named pET-24a(+)_blaRS_SM411_N439V. The correct cloning and DNA sequence of the gene was verified before protein expression, using primers T7-promoter and T7-terminator.
Expression and Purification of Wild-type and Mutant BlaRS Proteins (N439V and BlaRS K369A/K370A/K372A/N439V)
E. coli BL21 (DE3) cells were transformed with the corresponding plasmid (pET24a(+)_blaRS for WT protein (9), pET-24a(+)_blaRSN439V for BlaRS N439V and pET24a(+)_blaRS_SM411_N439V for the quadruple mutant). The expression and purification procedures were as described previously (9). BlaRS protein concentration in each case was determined from the absorbance, using the theoretical molar extinction coefficient of ϵ280 nm = 60,280 m−1 cm−1.
13C NMR Measurements with BlaRS N439V
All of the buffers were degassed, purged with argon, and kept under argon prior to use. To achieve N-decarboxylation of lysine 392 in the active site, BlaRS N439V (10 mg) was subjected to cycles of dilution in 25 mm sodium acetate buffer, pH 4.5, and concentration using a 10-kDa cutoff Amicon ultracentrifugal filter device (Millipore). The buffer was then exchanged to 10 mm sodium phosphate buffer, pH 7.5, 0.1 mm EDTA using an Amicon ultracentrifugal filter device. Finally, the sample was reconstituted with 10 mm sodium phosphate buffer, containing 0.1 mm EDTA, 20 mm NaH13CO3, and 10% D2O, pH 7.5. The protein concentration used in the NMR experiment was 600 μm.
Determination of Partition Ratio with BlaRS N439V
The partition ratio determination was done as previously described (13). Briefly, BlaRS (wild-type or N439V) (final concentration, 1.0 μm) was mixed with various concentrations of antibiotics in 100 mm sodium phosphate, pH 7.0, 50 mm sodium bicarbonate in a total volume of 20 μl. The reaction mixtures were incubated at room temperature for 30 min, at which time BOCILLIN FL, a fluorogenic penicillin, was added to each sample to a final concentration of 20 μm. The mixtures were incubated at 37 °C for 5 min, at which point the reactions were quenched by the addition of 15 μl of 2× protein loading buffer (125 mm Tris, 4% SDS, 20% glycerol, 2% 2-mercaptoethanol, pH 6.8), and the samples were boiled for 4 min. The samples were run in 15% SDS-PAGE gels at 125 V, and the fluorescently labeled proteins were visualized using a Storm 840® Scanner (GE Healthcare) in the fluorescence mode. The fluorescent bands of BOCILLIN FL-labeled BlaRS wild type and N439V were analyzed using Image Quant 5.2 software. Each data point was determined at least in duplicate. The protein activity (%) was plotted against [I]/[E], where [I] is the antibiotic concentration, and [E] is the concentration of protein. Protein activity (%) was determined from the intensity of the bands labeled with BOCILLIN FL normalized by the activity of the enzyme in the absence of antibiotic. The partition ratio was estimated by extrapolating the [I]/[E] at 0% enzyme activity using a linear fit (14).
Determination of Pre-steady-state and Steady-state Kinetic Parameters of BlaRS N439V
The hydrolysis of different β-lactam antibiotics catalyzed by BlaRS N439V was evaluated using a Cary 50 UV-visible spectrophotometer (Varian Inc.) equipped with an SFA-20 stopped flow apparatus (Hi-Tech Scientific, Salisbury, UK) at room temperature, in 100 mm sodium phosphate, pH 7.0, 50 mm NaHCO3. The parameters for the reaction between the BlaRS N439V protein and nitrocefin were determined by monitoring product formation at 500 nm (Δϵ500 = 15,900 m−1 cm−1). For all other β-lactam antibiotics, we monitored substrate consumption: ceftazidime, Δϵ260 = −8,660 m−1 cm−1; cephalosporin C, Δϵ260 = −5,200 m−1 cm−1; cephalothin, Δϵ262 = −7,960 m−1 cm−1; ampicillin, Δϵ235 = −670 m−1 cm−1; penicillin G, Δϵ235 = −775 m−1 cm−1; imipenem, Δϵ300 = −9,000 m−1 cm−1; and meropenem, Δϵ280 = −7,200 m−1 cm−1. For the antibiotics nitrocefin, ceftazidime, and cephalothin, the velocity v at time t changed exponentially from an initial value vi to a steady-state value vs. Hence, the progress curves were fit to Equation 1 (15), using SigmaPlot (Systat Software Inc.), where p is the concentration of product at time t, and k is the rate constant characterizing the change (burst rate constant).
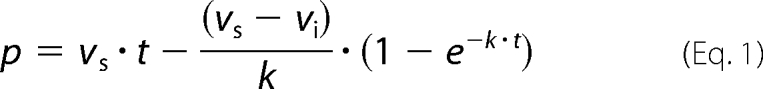 |
The Michaelis-Menten parameters for the initial and steady-state phases were then determined by fitting the changes of vi or vs with substrate concentration, respectively, to the Michaelis-Menten equation. For hydrolysis of cephalosporin C, no burst was observed; the steady-state parameters were determined by fitting the changes in the steady-state velocity with substrate concentration to the Michaelis-Menten equation.
Crystallization Conditions and Determination of the X-ray Structure of BlaRS K369A/K370A/K372A/N439V
The N439V variant of the BlaRs protein was crystallized at 24 °C from 30% PEG 8000, 0.2 m ammonium sulfate, pH 6.5, buffered with sodium cacodylate. As performed previously (8), three surface-exposed lysines (K369A/K370A/K372A) were mutated to alanine to facilitate crystallization. Prior to data collection, the crystals were briefly transferred to crystallization buffer supplemented with 20% glycerol and flash frozen in liquid nitrogen. X-ray data collection, processing, and refinement were performed as previously described (8). Briefly, the data were collected at the 19BM beamline at the Structural Biology Center of the Advanced Photon Source and processed with HKL2000 (16). The structure was solved via molecular replacement using MOLREP (17), using the structure of the previously determined native apo protein as a search model. Rigid body refinement, TLS refinement, and restrained refinement were performed with Refmac5 (18). Graphical evaluation of the model and fitting to maps was performed with Coot (19) and XtalView (20). The quality of the structure was evaluated with Coot, Procheck (21), and MolProbity (22). Structure factors and coordinates have been submitted to the Protein Data Bank (entry 3UY6). Data collection and refinement statistics are presented in Table 1.
TABLE 1.
X-ray data and refinement statistics
| Protein | BlaR1 N439V |
| Protein Data Bank Entry | 3UY6 |
| Radiation source | APS 19BM |
| Space group | P21 |
| a (Å) | 47 |
| b (Å) | 108.0 |
| c (Å) | 56.0 |
| b (°) | 108.4 |
| Molecules/a.u. | 2 |
| Resolution (Å) | 20-2.1 |
| Total number of reflections | 28919 |
| Mosaicity (°) | 0.36 |
| Completeness (%)a | 99.3 (92.4) |
| I/σ | 16.5 (2.0) |
| Rmerge (%) | 8.8 (59) |
| Average redundancy | 3.7 (3.6) |
| Rwork (%) (no. reflections) | 18.3 (27386) |
| Rfree (%) (no. reflections) | 22.9 (1533) |
| Average B factor (all atoms) (Å2) | 21.5 |
| Ramachandran plot | |
| Most favored (%) | 94.9 |
| Allowed (%) | 5.1 |
| Generously allowed (%) | 0.0 |
| Root mean square deviations from ideality | |
| Bonds (Å) | 0.014 |
| Angles (°) | 1.534 |
| Coordinate error (Å)b | 0.14 |
a The numbers in parentheses refer to highest resolution shell.
b Mean estimate based on maximum likelihood methods.
Computational Methods
The x-ray crystal structure of the native BlaRS acylated with imipenem (Protein Data Bank entry 3Q81) provided the initial atomic coordinates. The ceftazidime complex was generated by importing the antibiotic coordinates extracted from a previous BlaRS crystal structure (Protein Data Bank entry 1XKZ) to replace imipenem (7). Sybyl-X (23) was used for all coordinate manipulations, to generate the N439V mutation and N-carboxylated Lys-392. Amber ff99 and gaff forcefields provided all forcefield parameters and partial atomic charges for protein atoms. The RESP methodology was used for the N-carboxylated Lys-392 side chain, imipenem, and ceftazidime (24). Xleap module of Amber 11 (25) software suite was used to add counter ions to neutralize the simulated systems, followed by the addition of TIP3P (26) water molecules to solvate the systems in truncated octahedral periodic boxes. Four simulation systems were prepared for each ligand: wild type acylated intermediate with N-carboxylated Lys-392, wild type acylated intermediate with neutral Lys-392, N439V mutant acylated intermediate with N-carboxylated Lys-392, and N439V mutant acylated intermediate with neutral Lys-392.
The pmemd module of Amber 11 was used to carry out molecular dynamics (MD) simulations. The equilibration stage consisted of restrained minimization for 10,000 steps, followed by slowly heating up the restrained system to 300 K. The restraints were gradually released over 150 ps, and a 500-ps additional equilibration was performed before data collection. Production MD was carried out with a time step of 2 fs and Langevin temperature control. Coordinates were extracted every 3 ps over a period of 45 ns. The simulations were run in triplicate with different random number seeds to generate different initial velocity distributions. Finally, the trajectories were analyzed using Amber 11 ptraj module.
RESULTS
Evaluation of Hydrolytic Activity of BlaRS N439V
As described earlier, the surface domain of BlaR1 from S. aureus and class D β-lactamases present a remarkable similarity in structure and in their antibiotic-binding sites. A key difference is the presence of a highly conserved hydrophilic residue at position 439 of BlaR1, instead of a highly conserved hydrophobic residue at the equivalent position in class D β-lactamases (11, 27). To evaluate the role of this change in amino acid on the reaction of the sensor domain of BlaR1 with β-lactam antibiotics, we introduced the substitution N439V into the sensor domain of BlaR1. The protein was expressed and was purified to homogeneity. We evaluated the reaction of the BlaRS N439V mutant with cephalosporins, penicillins, and carbapenems, by means of steady-state and stopped flow kinetics. The substitution N439V imparted β-lactamase activity to the sensor domain of BlaR1 against cephalosporins (Table 2), albeit a modest one.
TABLE 2.
Parameters for turnover of cephalosporins by the BlaRS N439V mutant protein
Carbapenems and penicillins were investigated and were shown not to be substrates.
| Substrate | Initial phase parameters |
Steady-state parameters |
||||
|---|---|---|---|---|---|---|
| kcat | Km | kcat/Km × 10−3 | kcat × 103 | Km | kcat/Km × 10−3 | |
| s−1 | μm | m−1s−1 | s−1 | μm | m−1s−1 | |
| Nitrocefin | 2.8 ± 0.1 | 57.4 ± 0.6 | 48 ± 2 | 24 ± 2.0 | 5.0 ± 0.5 | 4.9 ± 0.4 |
| Cephalosporin C | 3.7 ± 0.2 | 80.0 ± 0.3 | 0.046 ± 0.003 | |||
| Cephalothin | 0.5 ± 0.1 | 86.1 ± 0.5 | 5.3 ± 0.3 | 4.7 ± 0.3 | 20.0 ± 0.1 | 24 ± 2 |
| Ceftazidime | > 800 | 0.085 ± 0.007 | 3.8 ± 0.3 | 120.0 ± 0.5 | 0.032 ± 0.003 | |
For investigation of hydrolysis of cephalosporins, we used nitrocefin, ceftazidime, cephalothin, and cephalosporin C. For nitrocefin, ceftazidime, and cephalothin, the velocity v at time t changed exponentially from an initial value vi to a steady-state value vs, as shown in Fig. 2A for hydrolysis of nitrocefin. The observed biphasic time course for hydrolysis of nitrocefin, cephalothin, and ceftazidime catalyzed by BlaRS N439V could be fit to the branched kinetic mechanisms displayed in Fig. 2 (B–D) (15). The first scheme (Fig. 2B) stipulates that on acylation of the protein (E-S1), there is the opportunity for the formation of a new acyl-enzyme species (E-S2), which returns to the initial acyl-enzyme species (E-S1) prior to the deacylation step that completes turnover. One plausibly could envision E-S2 to be one of two distinct entities. For example, it is known that some substrates acylate β-lactamases, but the acyl group of the acyl-enzyme species flips out of the oxyanion hole, where it was ensconced initially for the acylation event (28, 29). The second possibility, one that actually is precedented for class D β-lactamases and for BlaR1 (10, 12), is that N-decarboxylation of the lysine takes place on acylation, which actually arrests turnover at the acyl-protein stage (E-S2; Fig. 1B). The requisite N-recarboxylation has to take place, before catalysis can be completed in the hydrolytic step. Alternatively, the branch could be at the stage of the noncovalent enzyme-substrate complex (Fig. 2C). Conversely, the covalent enzyme complex (E-S1; Fig. 2D) could be converted to another intermediate essentially irreversibly, which would then be converted into free enzyme and product. It is difficult to distinguish these models (Fig. 2, B–D) by kinetics alone.
FIGURE 2.

A, time course of nitrocefin turnover by BlaRS N439V mutant variant. Product formation was monitored at 500 nm upon reaction of 0.24 μm BlaRS N439V with nitrocefin (2.5–100 μm). The burst in activity is followed by a steady-state phase, indicative of substrate-induced inhibition of the protein. B–D, schemes of the branched mechanisms of substrate-induced inhibition that could account for the biphasic turnover of cephalosporins by BlaRS N439V.
For these kinetic determinations, we set up the experiments for rapid kinetics in the stopped flow equipment to document the initial burst but allowed the monitoring to continue for 600 s in each case. This duration for monitoring includes the establishment of the steady-state processes as well. The time courses were fit to Equation 1, which accounts for the decrease in velocity seen for substrate-induced inhibition (15). The data of Table 2 revealed that the single amino acid substitution was sufficient to impart a modest, but quantifiable, ability for the N439V mutant protein to turn over cephalosporins. This kinetic ability translated to kcat/Km values of 102–104, as evaluated from the initial phase rate measurements. The exception was cephalosporin C, which did not show biphasic kinetics. The steady-state parameters for turnover of cephalosporin C were similar to those observed in the steady-state phase for ceftazidime. The kcat values for turnover of nitrocefin and cephalothin attenuated approximately 2 orders of magnitude upon the onset of steady state. In the case of ceftazidime, we could only determine the ratio kcat/Km for the initial phase, because of the high Km value. The kcat/Km values for nitrocefin, cephalothin, and ceftazidime consistently diminished in the steady-state process (3–20-fold). This could be attributed to the difficulty of N-recarboxylation of Lys-392 in the BlaRS protein, even in the presence of excess bicarbonate—as the source of carbon dioxide—during the reaction. This subject is discussed further later. For hydrolysis of nitrocefin, cephalothin, and ceftazidime by BlaRS N439V, there was an increase in the burst rate constant with increasing substrate concentration, consistent with the branching schemes shown in Fig. 2 (B–D). An alternative to these three schemes that could potentially account for the biphasic time courses is one where the free enzyme is converted into an alternative form not capable of reacting with β-lactams (e.g. the N-decarboxylated form or another conformational state for the protein). Because the assays were carried out in the presence of the bicarbonate ion as the precursor for carbon dioxide, we would expect that BlaRS N439V would be fully N-carboxylated at Lys-392. Notwithstanding, in this mechanistic possibility, the burst rate constant decreases with increasing substrate concentration. The fits of the progress curves for hydrolysis of nitrocefin, ceftazidime, and cephalothin by BlaRS N439V to Equation 1 showed an increase in the burst rate constant with substrate concentration in all cases, which is inconsistent with this scheme. Finally, BlaRS N439V showed negligible turnover ability for the penicillins and carbapenems that were assayed, and as is the case for the wild-type sensor domain, we observed essentially irreversible acylation.
To further characterize the capacity of the mutant protein to turnover β-lactam antibiotics, we evaluated the parameter partition ratio as described earlier (30). The protein was first incubated with different concentrations of a given β-lactam antibiotic to allow the equilibration of the system, followed by quantification of the uncomplexed BlaRS by fluorescence measurements (13). In this analysis, the antibiotic was treated as a substrate that partitioned into an “inhibited” species with BlaRS (30). The partition ratio (kcat/kinact) was evaluated with both the wild-type BlaRS and with the N439V mutant variant (Table 3). To a first approximation, this partition ratio would reveal the facility of the branching route in the mechanism that pertains to the process. The partition ratios increased significantly for nitrocefin, cephalosporin C, and ceftazidime upon introduction of the mutation N439V but only slightly for cephalothin. However, the partition ratios remained small for the penicillins and carbapenems, at values comparable with the ones observed for the wild-type sensor domain. The partition ratios revealed that BlaRS N439V could afford multiple turnover cycles for the cephalosporins, before sequestration into the inhibited species (the formation of which might be reversible).
TABLE 3.
Comparison of the partition ratios (kcat/kinact) for the reaction of the wild-type BlaRS and the N439V mutant variant with β-lactam antibiotics
| Substrate | Wild type | N439V |
|---|---|---|
| Nitrocefin | 1.3 ± 0.1 | 73 ± 2 |
| Cephalosporin C | 2.6 ± 0.9 | 1010 ± 70 |
| Cephalothin | 1.0 ± 0.1 | 6.3 ± 0.1 |
| Ceftazidime | 1.1 ± 0.1 | 150 ± 10 |
| Ampicillin | 1.0 ± 0.1 | 2.0 ± 0.4 |
| Penicillin G | 1.4 ± 0.2 | 1.9 ± 0.5 |
| Imipenem | 1.0 ± 0.1 | 1.6 ± 0.3 |
| Meropenem | 2.0 ± 0.5 | 1.2 ± 0.1 |
X-ray Structure of the BlaRS N439V Variant
To understand the effect of the N439V substitution at a molecular level, we obtained the crystal structure of the BlaRS N439V mutant protein, following the same strategy previously described to obtain crystals that diffracted to good resolution (8). The N439V mutant of the BlaRS protein crystallized in space group P21. Crystals diffracted to a resolution of 2.1 Å. The structure was solved by molecular replacement (Table 1), using the previously determined structure of the wild-type protein as a search model (8). Two molecules related by noncrystallographic symmetry were present in the asymmetric unit. They were nearly identical, superimposing with root mean square deviations of 0.6 Å for all atoms and 0.3 Å for backbone atoms. The overall structure of the protein was identical to that of the wild-type apo-protein, displaying the characteristic α/β fold and, for the first molecule in the asymmetric unit, superimposing onto the wild-type structure with root mean square deviations of 0.5 Å for all atoms and 0.2 Å for backbone atoms (Fig. 3A). Examining the antibiotic-binding site in detail, the N439V mutation did not alter the conformation of any of the critical active site residues (Fig. 3, B and C). In contrast to the wild-type protein (8), Lys-392 was not N-carboxylated in either molecule in the asymmetric unit. At the limit of the resolution of the data, the mutation did appear to alter solvation within the active site, which is important in the context of the acquired β-lactamase activity, as will be elaborated later. In the first molecule in the asymmetric unit (Fig. 3B), which is the most directly comparable with the case of N-decarboxylated wild-type protein (8), the positions of two water molecules were altered, and two others were eliminated (Fig. 3C). The observed electron density for the two altered water molecules is given in supplemental Fig. S1. The importance of these water molecules is discussed later.
FIGURE 3.

Crystal structure of BlaRS N439V mutant. A, stereo view of the protein structure, with the antibiotic-binding site in the center. B, the close-up stereo view of the active site of the first molecule of the asymmetric unit. The crystallographic waters and the sulfate ion are depicted as red and yellow spheres, respectively. The side chains of residues Lys-392, Ser-389, and Val-439 are shown as capped sticks, color-coded by atom type (nitrogen in blue, oxygen in red, and carbon in gray; hydrogens not shown). C, the stereo view of the same perspective as in B for the second molecule of the asymmetric unit. Four water molecules are seen here, with Wat-2 shared between the two molecules.
Documentation of N-carboxylation of Lys-392 in BlaRS N439V
Documentation of N-carboxylation of Lys-392 in the wild-type sensor domain was first made in solution by 13C NMR spectroscopy (9). It was only recently that we also showed by an x-ray structure that Lys-392 is N-carboxylated (8). This is intuitively explained, because N-carboxylation of lysine is reversible. Hence, 13C NMR offers a convenient and diagnostic method for the observation of the presence of Lys-392 N-carboxylation in solution, notwithstanding that the x-ray structure of the N439V mutant did not show it to be N-carboxylated in solid state.
We used the method that we have reported for reversing N-carboxylation of lysine by lowering the pH, followed by N-recarboxylation with the use of 13CO2 for the NMR experiment (9). As shown in Fig. 4, the diagnostic 13C NMR resonance for the lysine carbamate (N-carboxylation) at 164 ppm is observed for the N439V mutant protein. Therefore, this mutant protein is N-carboxylated at Lys-392 in solution, as is the case for the wild-type BlaR1 and for class D β-lactamases at the same position.
FIGURE 4.

The 13C NMR spectrum of the BlaRS N439V mutant protein (600 μm) in 10 mm sodium phosphate, 0.1 mm EDTA, supplemented with 20 mm NaH13CO3.
Molecular Dynamics Simulations
We performed several molecular dynamics simulations on the structure of the sensor domain in its acylated forms with ceftazidime and imipenem. All of the trajectories were inspected for stability and convergence with respect to macroscopic dynamical properties such as temperature and density, among others. Throughout MD sampling, the acylated ceftazidime complexes did not deviate significantly from the initial model. The triplicated MD trajectories did not show significant deviations among them either. Additionally, a cross-correlation analysis of the backbone atoms did not reveal significant differences among the trajectories of all investigated simulated systems, suggesting that neither the N439V mutation nor the acylation with different antibiotics impacted the overall motions.
With the focus on events immediately following acylation, we examined the trajectories for the existence of a hydrolytic water molecule, which would allow the deacylation half-reaction to be completed, which is obviously a requisite for substrate turnover. When Lys-392 was N-carboxylated in the acylated ceftazidime species, we observed a water molecule poised between the N-carboxyl of lysine and the acyl carbonyl carbon of the ester of the acylated species (Fig. 5A). Comparing this arrangement to the x-ray structure of the N439V mutant, we identified Wat-1 (Fig. 3B) as the hydrolytic water. In the crystal structure, this water is present only in the first molecule in the asymmetric unit, indicating a degree of lability consistent with the simulation results discussed below. This water molecule remained near the acyl carbonyl throughout the simulations, for both the wild-type protein and for its N439V mutant variant, both acylated with ceftazidime (Fig. 5, B and C). However, the extent of its interaction with an N-carboxyl oxygen of Lys-392 was significantly different between the wild-type ceftazidime complex and that for the N439V mutant. One N-carboxyl oxygen in the N439V mutant was in hydrogen bonding contact with water for more than twice the duration as in the case of the wild-type complex. With a 2.0 Å cut-off distance between water hydrogen and the N-carboxyl oxygen, the hydrogen bonding contact was conserved in ∼38% of the molecular dynamics snapshots in the mutant, whereas in the wild-type complex it was preserved in only ∼17% of the cases. Therefore, this critical arrangement for the hydrolytic water molecule was significantly more favored in the N439V mutant than was in the wild-type protein. Considering that Lys-392 actually experiences N-decarboxylation on acylation of serine (7, 10), the opportunity for promotion of Wat-1 for the deacylation reaction is actually nil in the wild-type protein, consistent with the experimental results.
FIGURE 5.

Key interactions during molecular-dynamics simulations. A, stereo image of the active site of a representative molecular dynamics snapshot showing the hydrolytic water molecule positioned for the deacylation reaction. The color code is identical to that in Fig. 1. B, the variation of the distance between side chain carboxyl oxygen of N-carboxylated Lys-392 and water hydrogen for the wild-type BlaRS-ceftazidime acyl complex as a function of time. C, variation of the same distance for the N439V mutant in complex with ceftazidime.
In contrast, the data extracted from MD trajectories of the imipenem complex with N-carboxylated Lys-392 showed that the water molecule only rarely adopted a similar positioning, regardless of the nature of the amino acid at position 439. Only 8–10% of snapshots showed the water hydrogen atoms within a 2.0 Å radius of a Lys-392 N-carboxyl oxygen, whereas in a meager ∼3% of snapshots, a water molecule approached at least 3.0 Å to the acyl carbonyl carbon atom (supplemental Fig. S2). Thus, the acylated imipenem (a carbapenem) species might only rarely adopt a conformation that could promote deacylation, consistent with the experimental findings (lack of turnover of carbapenems and Table 3). This subject is discussed further in the supplemental text.
Furthermore, the trajectories of simulations with the neutral (free-base) unmodified side chain of Lys-392, the product of N-decarboxylation, did not retain a water molecule within an interacting distance to the acyl carbonyl carbon. This finding is consistent with the indispensable need for N-carboxylation of Lys-392 for the protein function, which was previously documented experimentally (10).
DISCUSSION
The surface domain of BlaR1 from S. aureus has amino acid sequence homology to class D β-lactamases (BlaRS from strain S. aureus NRS128 has 26% identity and 49% similarity to OXA10 β-lactamase from Pseudomonas aeruginosa, for example), but they exhibit a remarkable similarity in the overall structures and in their antibiotic-binding sites (13). Despite the high similarity in their binding sites, a key difference is the presence of a highly conserved hydrophilic amino acid at BlaR1 position 439, instead of hydrophobic residues at the equivalent position in class D β-lactamases (27). In all of the BlaR1 sequences available—and those of the related MecR1—there is an Asn at position 439 (except for BlaR1 from Bacillus licheniformis and Brevibacillus brevis in which Asn-439 is substituted by threonine), whereas at the equivalent position in class D β-lactamases (position 120 using the DBL consensus numbering), one finds a Val, Ile, or Leu. We undertook the current work with the intuitive understanding that position 439 should play a functional role in the activities of these disparate, yet related, proteins. Would introduction of a hydrophobic residue at position 439 of BlaRS be sufficient to impart enhanced β-lactamase activity to the protein? It is important to note that the wild-type BlaRS normally exhibits the marginal ability to turn over two or three molecules of β-lactam antibiotics, prior to the formation of the stable acylated species (Table 3). Facile conversion to a stable acylated species is required for the signaling events of BlaR1 in response to the antibiotic challenge. Hence, a residual β-lactamase activity is already inherent to BlaR1. Can this activity be enhanced by a mere point mutation at position 439? Why is the presence of Asn at position 439 of BlaR1 important for the protein in its antibiotic sensing function?
The BlaRS N439V mutant protein indeed exhibits enhanced β-lactamase activity with cephalosporins as substrates but not with penicillins and carbapenems (Tables 2 and 3). We documented with the x-ray structure of the BlaRS N439V mutant that introduction of valine did not alter the three-dimensional structure of the protein. Lys-392 was found in this structure in its N-decarboxylated form, but with 13C NMR spectroscopy we showed that the protein is indeed N-carboxylated at this position in solution, a form that is critical for both the functions of BlaR1 and of class D β-lactamases (10, 12).
In the wild-type BlaRS structure, N-carboxylated Lys-392 is hydrogen bonded to Asn-439. This bond has to break, and the N-carboxylated Lys-392 would approach Ser-389 to promote it for the acylation event by the antibiotic. This indeed takes place for acylation of BlaR1 for its function as an antibiotic sensor/signal transducer protein (8). The identical process occurs for β-lactamases, except that the presence of the hydrophobic residue in place of Asn at the requisite site would mandate a different conformation for N-carboxylated Lys-392 (7, 12). Insertion of a water molecule into the active site of the β-lactamase between the N-carboxylated lysine and the ester carbonyl of the acyl species initiates the deacylation process (31). Hence, class D β-lactamases exhibit symmetry in catalysis as N-carboxylated Lys-392 performs the proton-transfer steps needed for both the acylation and deacylation steps (Fig. 1B, steps II and IV, respectively). Would this be possible for the N439V mutant of BlaRS? The answer is in the affirmative, as disclosed by our findings.
As revealed in the dynamics simulations for the wild-type BlaRS (Fig. 5), the contact between the side chain of N-carboxylated Lys-392 and a water molecule that can serve the hydrolytic role exists only for 17% of the snap shots, but this contact increases to 38% of snap shots in the simulations of the N439V mutant variant. The likelihood of promotion of water for the deacylation event is enhanced, because the side chain of Lys-392 in the N439V mutant variant is not sequestered in interactions with Asn-439, as is seen in the wild-type BlaR1. This is the likely reason why the mutant variant has—as do class D β-lactamases—the enhanced ability to turn over cephalosporins.
In both BlaR1 protein and class D β-lactamases, we have documented N-decarboxylation of the active site lysine. The N-decarboxylation in the case of BlaR1 arrests the process after protein acylation by the antibiotic, which is required for its sensing function. The now N-decarboxylated Lys-392 makes a strong hydrogen bond to Asn-439 (8), which we have shown to prevent its N-recarboxylation, despite the presence of excess carbon dioxide in the reaction vessel. This is advantageous for the function of BlaR1 as an antibiotic sensor/signal transducer. Not so for the class D β-lactamase activity, because N-decarboxylation at Lys-392 would arrest the process in midcatalysis. However, because of the presence of the hydrophobic residue at position 439, the N-decarboxylated Lys-392 is now predisposed for facile N-recarboxylation and resumption of catalysis. Golemi et al. (12) indeed have shown that the presence of sodium bicarbonate as the precursor for carbon dioxide accomplishes this very task in the case of the class D OXA10 β-lactamase. The results reported here are in agreement with a recent study on the OXA10 β-lactamase, where the equivalent valine residue was mutated to threonine (11). The lowered affinity for carbon dioxide displayed by this mutant β-lactamase, with the attendant decreased turnover ability, underscores the importance of this post-translational modification for catalytic ability.
The BlaRS N439V mutant turns over cephalosporins significantly better than the wild-type protein, but ultimately the branching mechanism for kinetics that we have documented herein subverts catalysis by sequestering the complex in an inhibited form. Whereas this kinetic nuance of the interaction of cephalosporins and the BlaRS N439V mutant protein could not be predicted a priori, the work evidently places the residue at position 439 for the two protein classes at the crossroads of divergence of functions: one outcome leading to a receptor protein and the other to a protein with catalytic capability. It is highly likely that such molecular pivoting points for evolutionary outcome of function are common in nature, because of the restrictions that the aforementioned limit of 2700 unique protein folds place on evolution of function otherwise.
Supplementary Material
Acknowledgment
S. aureus isolate NRS128 was obtained through the Network on Antimicrobial Resistance in Staphylococcus aureus program.
This work was supported, in whole or in part, by National Institutes of Health Grant AI33170. This work was also supported by the Network on Antimicrobial Resistance in Staphylococcus aureus program under NIAID, National Institutes of Health Contract HHSN272200700055C.

This article contains supplemental text and Figs. S1 and S2.
The atomic coordinates and structure factors (code 3UY6) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
- PBP
- penicillin-binding proteins
- BlaRS
- C-terminal sensor domain of BlaR1
- MD
- molecular dynamics.
REFERENCES
- 1. Liu X., Fan K., Wang W. (2004) The number of protein folds and their distribution over families in nature. Proteins 54, 491–499 [DOI] [PubMed] [Google Scholar]
- 2. Fisher J. F., Mobashery S. (2010) Enzymology of Antibacterial Resistance, Elsevier, Oxford [Google Scholar]
- 3. Knox J. R., Moews P. C., Frere J. M. (1996) Molecular evolution of bacterial β-lactam resistance. Chem. Biol. 3, 937–947 [DOI] [PubMed] [Google Scholar]
- 4. Massova I., Mobashery S. (1998) Kinship and diversification of bacterial penicillin-binding proteins and β-lactamases. Antimicrob. Agents Chemother. 42, 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fisher J. F., Meroueh S. O., Mobashery S. (2005) Bacterial resistance to β-lactam antibiotics. Compelling opportunism, compelling opportunity. Chem. Rev. 105, 395–424 [DOI] [PubMed] [Google Scholar]
- 6. Wilke M. S., Hills T. L., Zhang H. Z., Chambers H. F., Strynadka N. C. (2004) Crystal structures of the Apo and penicillin-acylated forms of the BlaR1 β-lactam sensor of Staphylococcus aureus. J. Biol. Chem. 279, 47278–47287 [DOI] [PubMed] [Google Scholar]
- 7. Birck C., Cha J. Y., Cross J., Schulze-Briese C., Meroueh S. O., Schlegel H. B., Mobashery S., Samama J. P. (2004) X-ray crystal structure of the acylated β-lactam sensor domain of BlaR1 from Staphylococcus aureus and the mechanism of receptor activation for signal transduction. J. Am. Chem. Soc. 126, 13945–13947 [DOI] [PubMed] [Google Scholar]
- 8. Borbulevych O., Kumarasiri M., Wilson B., Llarrull L. I., Lee M., Hesek D., Shi Q., Peng J., Baker B. M., Mobashery S. (2011) Lysine Nζ-decarboxylation switch and activation of the β-lactam sensor domain of BlaR1 protein of methicillin-resistant Staphylococcus aureus. J. Biol. Chem. 286, 31466–31472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Golemi-Kotra D., Cha J. Y., Meroueh S. O., Vakulenko S. B., Mobashery S. (2003) Resistance to β-lactam antibiotics and its mediation by the sensor domain of the transmembrane BlaR signaling pathway in Staphylococcus aureus. J. Biol. Chem. 278, 18419–18425 [DOI] [PubMed] [Google Scholar]
- 10. Thumanu K., Cha J., Fisher J. F., Perrins R., Mobashery S., Wharton C. (2006) Discrete steps in sensing of β-lactam antibiotics by the BlaR1 protein of the methicillin-resistant Staphylococcus aureus bacterium. Proc. Natl. Acad. Sci. U.S.A. 103, 10630–10635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vercheval L., Bauvois C., di Paolo A., Borel F., Ferrer J. L., Sauvage E., Matagne A., Frère J. M., Charlier P., Galleni M., Kerff F. (2010) Three factors that modulate the activity of class D β-lactamases and interfere with the post-translational carboxylation of Lys70. Biochem. J. 432, 495–504 [DOI] [PubMed] [Google Scholar]
- 12. Golemi D., Maveyraud L., Vakulenko S., Samama J. P., Mobashery S. (2001) Critical involvement of a carbamylated lysine in catalytic function of class D β-lactamases. Proc. Natl. Acad. Sci. U.S.A. 98, 14280–14285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cha J., Mobashery S. (2007) Lysine Nζ-decarboxylation in the BlaR1 protein from Staphylococcus aureus at the root of its function as an antibiotic sensor. J. Am. Chem. Soc. 129, 3834–3835 [DOI] [PubMed] [Google Scholar]
- 14. Silverman R. B. (1995) Mechanism-based enzyme inactivators. Methods Enzymol. 249, 240–283 [DOI] [PubMed] [Google Scholar]
- 15. Waley S. G. (1991) The kinetics of substrate-induced inactivation. Biochem. J. 279, 87–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Otwinowski Z., Minor W. (1997) The kinetics of substrate-induced inactivation. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 17. Vagin A., Teplyakov A. (2010) Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 66, 22–25 [DOI] [PubMed] [Google Scholar]
- 18. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 19. Emsley P., Cowtan K. (2004) Coot. Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 20. McRee D. E. (1999) XtalView/Xfit. A versatile program for manipulating atomic coordinates and electron density. J. Struct. Biol. 125, 156–165 [DOI] [PubMed] [Google Scholar]
- 21. Laskowski R. A., MacArthur M. W., Moss D. S., Thornton J. M. (1993) PROCHECK: a program to check the stereochemical quality of protein structure. J. Appl. Crystallogr. 26, 283–291 [Google Scholar]
- 22. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity. All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sybyl-X (2010) Tripos International, Saint Louis, MO [Google Scholar]
- 24. Bayly C. I., Cieplak P., Cornell W. D., Kollman P. A. (1993) A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J. Phys. Chem. 97, 10269–10280 [Google Scholar]
- 25. Case D. A., Darden T. A., Cheatham I., Simmerling T. E., Wang C. L., Duke R. E., Luo R., Merz K. M., Pearlman D. A., M., Walker R. C., Zhang W., Wang B., Hayik S., Roitberg A., Seabra G., Wong K. F., Paesani F., Wu X., Brozell S., Tsui V., Gohlke H., Yang L., Tan C., Mongan J., Hornak V., Cui G., Beroza P., Mathews D. H., Schafmeister C., Ross W. S., Kollman P. A. (2008) in Amber 11, University of California, San Francisco, CA [Google Scholar]
- 26. Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 [Google Scholar]
- 27. Bou G., Santillana E., Sheri A., Beceiro A., Sampson J. M., Kalp M., Bethel C. R., Distler A. M., Drawz S. M., Pagadala S. R., van den Akker F., Bonomo R. A., Romero A., Buynak J. D. (2010) Design, synthesis, and crystal structures of 6-alkylidene-2′-substituted penicillanic acid sulfones as potent inhibitors of Acinetobacter baumannii OXA-24 carbapenemase. J. Am. Chem. Soc. 132, 13320–13331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wilkinson A. S., Bryant P. K., Meroueh S. O., Page M. G., Mobashery S., Wharton C. W. (2003) A dynamic structure for the acyl-enzyme species of the antibiotic aztreonam with the Citrobacter freundii β-lactamase revealed by infrared spectroscopy and molecular dynamics simulations. Biochemistry 42, 1950–1957 [DOI] [PubMed] [Google Scholar]
- 29. Maveyraud L., Mourey L., Kotra L. P., Pedelacq J. D., Guillet V., Mobashery S., Samama J. P. (1998) Structural basis for clinical longevity of carbapenem antibiotics in the face of challenge by the common class-A β-lactamase from the antibiotic-resistant bacteria. J. Am. Chem. Soc. 120, 9748–9752 [Google Scholar]
- 30. Mobashery S., Ghosh S. S., Tamura S. Y., Kaiser E. T. (1990) Design of an effective mechanism-based inactivator for a zinc protease. Proc. Natl. Acad. Sci. U.S.A. 87, 578–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Maveyraud L., Golemi-Kotra D., Ishiwata A., Meroueh O., Mobashery S., Samama J. P. (2002) High-resolution X-ray structure of an acyl-enzyme species for the class D OXA-10 β-lactamase. J. Am. Chem. Soc. 124, 2461–2465 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.