

Background: It is not well understood how HIV-1 Tat performs complex functions despite its considerable sequence diversity.
Results: Individually nonconserved but functionally coevolving sites in Tat were identified.
Conclusion: In contrast to most coevolving sites, these sites are critical for two distinct functions regulating viral gene expression.
Significance: Balancing the two mechanisms is a strategy to maintain Tat function and may impact viral latency.
Keywords: Computational Biology, Gene Regulation, HIV-1, Protein Evolution, Protein-Protein Interactions, HIV-1 Tat, HIV-1 Subtypes, Mutual Information, Tat-P-TEFb Interaction, Protein Coevolution
Abstract
Viral genomes are continually subjected to mutations, and functionally deleterious ones can be rescued by reversion or additional mutations that restore fitness. The error prone nature of HIV-1 replication has resulted in highly diverse viral sequences, and it is not clear how viral proteins such as Tat, which plays a critical role in viral gene expression and replication, retain their complex functions. Although several important amino acid positions in Tat are conserved, we hypothesized that it may also harbor functionally important residues that may not be individually conserved yet appear as correlated pairs, whose analysis could yield new mechanistic insights into Tat function and evolution. To identify such sites, we combined mutual information analysis and experimentation to identify coevolving positions and found that residues 35 and 39 are strongly correlated. Mutation of either residue of this pair into amino acids that appear in numerous viral isolates yields a defective virus; however, simultaneous introduction of both mutations into the heterologous Tat sequence restores gene expression close to wild-type Tat. Furthermore, in contrast to most coevolving protein residues that contribute to the same function, structural modeling and biochemical studies showed that these two residues contribute to two mechanistically distinct steps in gene expression: binding P-TEFb and promoting P-TEFb phosphorylation of the C-terminal domain in RNAPII. Moreover, Tat variants that mimic HIV-1 subtypes B or C at sites 35 and 39 have evolved orthogonal strengths of P-TEFb binding versus RNAPII phosphorylation, suggesting that subtypes have evolved alternate transcriptional strategies to achieve similar gene expression levels.
Introduction
Genomes are continuously subjected to mutations that can in many cases undermine the structure and function of their encoded proteins. These processes may be especially important in rapidly evolving genomes, such as those of RNA viruses, which feature high rates of mutation and recombination during replication (1, 2). For example, the retrovirus human immunodeficiency virus-1 (HIV-1),4 exhibits enormous sequence diversity, both within individual patients and among numerous subtypes and recombinants circulating throughout the world, which in many cases reduces viral fitness but can also promote its ability to adapt to different selective pressures applied by the host immune system and anti-retroviral drugs (2–6). In many cases, purifying selection purges mutations that reduce overall fitness; however, deleterious mutations at one residue may also be compensated for by mutations at other sites to maintain protein structure and function (7). Such compensating positions within a protein may conceal significant evolutionary and functional information, yet are not readily apparent from an analysis of protein sequence. We use an approach whereby discovering coevolving sites within the HIV-1 protein Tat (transactivator of transcription) allows us to elucidate the underlying functional mechanism that constrains these sites to certain residue pairs for optimal function of this important viral protein.
Tat, which displays complex interactions with several cellular and viral factors that are critical to activating gene expression from the viral promoter, is an interesting substrate for analysis of the effects of protein evolution on complex, multifaceted protein functions. Briefly, once HIV-1 infects a host cell and integrates into its genome, transcription factor binding sites within the viral promoter recruit host cellular factors and mediate a low, basal level of gene expression in which transcriptional elongation is inefficient and yields primarily abortive transcripts (8). However, a small number of full-length viral transcripts are produced and spliced to yield a mRNA species that encodes Tat. The few Tat molecules that are formed from this basal transcription bind to the positive transcription-elongation factor b (P-TEFb), which consists of the cellular proteins cyclin T1 (CycT1) and cyclin-dependent kinase 9 (Cdk9) (9). The resulting Tat·P-TEFb complex then binds to a RNA hairpin, transactivation response element, present at the 5′ end of all viral transcripts and is subsequently transferred to the pre-initiation complex, wherein Cdk9 phosphorylates the C-terminal domain (CTD) of RNA polymerase II (RNAPII) and thereby greatly increases RNAPII processivity (10, 11). In a recently proposed, alternate mechanism, P-TEFb and Tat may be recruited to the viral promoter in an inactive form, and the newly synthesized transactivation response element may then bind to Tat and P-TEFb and displace the inhibitory 7SK snRNP to activate P-TEFb, which phosphorylates RNAPII (12). In either case, the increased RNAPII processivity greatly elevates viral gene expression and initiates a cascade of HIV-1 replication.
In addition to its interactions with RNAPII through P-TEFb, the multifunctional Tat interacts with numerous other host factors, and a balance among these various interactions and post-translational modifications is likely necessary for effective overall function (13–23). However, it is unclear how Tat, despite its considerable sequence diversity among HIV-1 isolates globally, is able to mediate these critical processes of co-opting a series of host cellular mechanisms to orchestrate viral replication. Sequence alignment of a protein can enable the identification of single amino acids that are conserved and hence potentially important for specific functions, and a number of such sites have been identified within Tat (e.g. supplemental Fig. S1) (13, 16). However, we hypothesized that correlated and coordinated amino acid changes may have played a role in diversifying the sequence of Tat while preserving its interactions with many host proteins and thus its overall function (13–23). Furthermore, sequence alignments can readily miss situations where neither of the two given residues is individually conserved, but instead where correlated pairs that make important contributions to protein structure and/or function appear together. Thus, bioinformatic and statistical approaches may help identify such sites and thereby gain greater molecular insights into a protein critical for HIV pathogenicity.
To address these hypotheses, we applied a statistical measure termed Mutual Information (MI) (24), one of several methods that can be used to identify correlated position pairs from multiple sequence alignments of proteins (25–27). Previously, such statistical measures have been applied to HIV-1 proteins, including the V3 loop of the env gene and the gag gene (28–30); however, these elegant computational analyses were not accompanied by experimental investigation. Similarly, such analysis has also been applied to other biological systems (31). Because the background noise in multiple sequence alignments makes it difficult for most statistical measures to predict correlated residues accurately, accompanying experimental validation of these sites is critical to identify structurally or functionally constrained residue pairs.
Here, we present a combined computational and experimental approach to identify and investigate coevolving residues in Tat. Sites 35 and 39 emerged in this analysis, and the functional importance of these coevolving residues was verified experimentally by introducing single point mutations in Tat. Whereas the single mutants proved nonfunctional, adding the second mutation restored viral gene expression. Surprisingly, despite their structural proximity, positions 35 and 39 appeared to be important for two distinct, underlying mechanisms, Tat binding to P-TEFb and Tat-mediated activation of P-TEFb to enable it to phosphorylate the CTD of RNAPII, and a combination of these two functions constrains the identities of these residues to certain pairs of amino acids. Extending this analysis indicates that the Tat proteins of HIV-1 subtypes B and C appear to have evolved compensatory strengths for different steps of Tat-mediated transactivation to achieve similar overall viral gene expression levels.
EXPERIMENTAL PROCEDURES
Cell Culture
Jurkat cells, used for infections, mRNA extraction and ChIP assays, were cultured in RPMI 1640 (Mediatech). HEK 293T cells, used for viral packaging, were cultured in Isocove's DMEM (Mediatech). HeLa cells, used for co-immunoprecipitation experiments, and the HL3T1 cell line, used for the Luciferase assay, were cultured in DMEM. All cell media were supplemented with 10% fetal bovine serum (FBS) and 100 units/ml of penicillin-streptomycin. All cells were grown at 37 °C and 5% CO2.
Viral Harvesting, Titering, and Infections
To package the lentiviral vectors, 100-mm plates with HEK 293T cells were cotransfected with 10 μg of the plasmid of interest and the following helper plasmids: 5 μg of pMDLg/pRRE, 3.5 μg of pVSV-G, and 1.5 μg of pRSV-Rev (32). 36 h post-transfection, virus was harvested by ultracentrifugation, and the viral pellets were resuspended in PBS and stored at −80 °C for future use. Viral titers were obtained by infecting 3 × 105 cells with different viral volumes and measuring GFP expression of cells 8 days post-infection. On day 8 post-infection, cells were stimulated with TNF-α (20 ng/ml) and trichostatin A (400 nm) for 18 h prior to analysis of GFP expression by flow cytometry. Based on the resulting titering curves, Jurkat cells were infected at a multiplicity of infection of 0.05–0.1 for experiments to ensure single integration events per cell.
Co-Immunoprecipitation and Western Blots
Protocol details are provided in supplemental “Experimental Procedures.” The following antibodies were used for this assay: anti-Cdk9, anti-ENL (Abcam, research sample), anti-CycT1 (Santa Cruz Biotechnology, catalog number sc-10750), anti-Sp1 (Millipore, catalog number 07-645), anti-ELL2 (Bethyl Laboratories, catalog number A302-505A), anti-AFF4 (Santa Cruz Biotechnology, catalog number sc-101062), and anti-AF9 (Bethyl Laboratories, catalog number A300-595A).
Chromatin Immunoprecipitation
Upstate EZ ChIP Kit reagents (Upstate) and protocol were used for the assay with variations. The following antibodies were used for ChIP: anti-RNAPII (Millipore, catalog number 05-623), anti-Ser-5-P CTD RNAPII (Covance, catalog number MMS-134R), and anti-Ser-2-P CTD RNAPII (Covance, catalog number MMS-129R). See supplemental “Experimental Procedures” for details of protocol and primers.
Mutual Information Analysis
Matlab codes for calculation of raw and background MI scores to estimate the corrected MI scores will be made available upon request.
Statistical Analyses
All statistical significances were computed using one-way analysis of variance followed by the Tukey-Kramer multiple comparison method to compare different pairs.
Details of plasmids, flow cytometry and cell sorting, transfections, mRNA extraction and RT-qPCR, co-immunoprecipitation and Western blots, chromatin immunoprecipitation, nuclease sensitivity assay, luciferase assay, and structure modeling can be found in supplemental “Experimental Procedures.”
RESULTS
Mutual Information Analysis Identifies Sites 35 and 39 in Tat as Coevolving
To identify correlated sites within Tat that are potentially important for maintaining Tat structure or function, we calculated MI between position pairs for 917 pre-aligned Tat sequences, from 9 viral subtypes and 14 recombinant forms, from the Los Alamos Sequence Database (hiv.lanl.gov). To encode a large amount of information within a relatively small genome, HIV-1 uses overlapping reading frames and alternative splicing. Because Tat shares overlapping reading frames with another viral protein, Rev, beyond amino acid 47, analysis was restricted to the first 46 amino acids of Tat within its activation domain (amino acids 1–48) to ensure that the sequence conservation and structural constraints in Rev did not introduce false positives in the analysis.
Within a multiple sequence alignment, MI predicts the likelihood of observing an amino acid at a particular site X in an alignment, given the identity of the amino acid at another site Y, and is computed by,
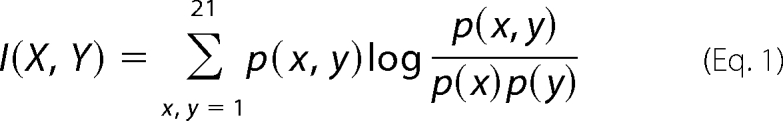 |
where X, Y, ∈(1,2 … 21) corresponds to one of the 20 amino acids or an alignment gap. p(x) and p(y) correspond to the probability of observing amino acid (or gap) x or y at sites X and Y, respectively, and p(x,y) is the corresponding joint probability of observing amino acids (or gaps) x and y at sites X and Y. Higher MI values indicate stronger correlation between two sites.
Using a method described by Dunn et al. (33) to minimize background noise, three position pairs, (35, 39), (31, 35), and (31, 39), were identified with MI scores higher than a threshold score, computed using a method described by Weigt et al. (Fig. 1A, B and supplemental Fig. S2). See supplemental “Results” for details (33, 34). For positions 35 and 39, frequencies of the different amino acids, based on the Tat sequence database, show that a Leu at position 35 constrains position 39 primarily to a Gln. Similarly, a Gln at position 35 results in a majority of Tat sequences having an Ile, Leu, or Thr but not a Gln at position 39 (Fig. 2A). Based on the MI analysis, positions 31, 35, and 39 were chosen for experimental testing. In addition, a relatively conserved site that is coevolving with another site may in general have a low MI score due to certain mathematical constraints (see supplemental “Results” for details) (24). We therefore also experimentally tested several sites whose MI did not reach threshold. Furthermore, we tested several nonconserved sites within background MI as controls to validate the low predicted functional relationship between such sites. Experimentally tested positions are shown by solid black dots (Fig. 1B).
FIGURE 1.

MI analysis applied to the Los Alamos Sequence Database reveals correlated position pairs in Tat. A, MI scores between all possible position pairs within the first 46 residues in Tat. Sites (35, 39), (31, 35), and (31, 39) have the highest scores. B, plot of entropy of a site (a measure of amino acid conservation at a site) versus maximum MI score for that site with any other site in Tat. The dotted line denotes the threshold MI score used to separate signal from background. Solid circles represent experimentally tested positions in Fig. 2.
FIGURE 2.

Experimental identification of coevolving sites in Tat using gene expression studies. A, truncated 2 × 4 matrix showing amino acid residues usually observed at sites 35 and 39 from 917 sequences in the Los Alamos Sequence Database, rather than a sparse 21 × 21 matrix representing all the amino acids (and gaps) at sites 35 and 39 in Tat. In the complete matrix, each row sums to 100%. Shaded cells represent residues pairs commonly observed at sites 35 and 39. Gray cells represent amino acid pairs not observed in the database. B, schematic of the lentiviral vector (LGIT) used to study gene expression for different Tat variants. Jurkat cells were infected with the LGIT vector at low multiplicity of infections (0.05–0.1) to ensure a single integration event per cell. C, gene expression levels based on GFP fluorescence for different Tat mutants normalized by WT Tat. D, Percentage Infected but Off, a measure of the fraction of cells that are silenced and not expressing GFP. The shading of bars in C and D correlate with the matrix in A for easy visualization. Error bars represent S.D. for 3 independent infections. ** denotes statistically significant differences (p < 0.01) from WT Tat.
Gene Expression Analysis of Coevolving Sites 35 and 39
If two sites functionally coevolve, then mutating either individually may impair biological activity, whereas mutating both together may rescue function. To test this hypothesis, amino acids in Tat from subtype B virus, broadly used in HIV-1 studies and referred to here as wild-type (WT) Tat (supplemental Fig. S1), were replaced with residues of other naturally occurring viral variants or subtypes (Fig. 2A). To test how sites with high MI scores predict Tat function, we studied the gene expression properties of different Tat mutants using a lentiviral vector model of HIV-1, in which the HIV-1 LTR drives expression of green fluorescent protein (GFP) and Tat, separated by an internal ribosome entry sequence (IRES) (LTR-GFP-IRES-Tat or LGIT) (Fig. 2B) (35). GFP expression from single integrations of LGIT in Jurkat cells was used to quantify LTR gene expression, and as previously observed the Tat positive feedback loop results in a bifurcated cell population with either very low or high levels of GFP expression, referred to as the Off and On populations, respectively (supplemental Fig. S3A) (35). Two metrics were used to quantify gene expression (36). First, the “Mean On Peak” indicates the average GFP level of cells above the background threshold of fluorescence (the On gate), a measure of the level of “closed loop” transactivation attained by a particular Tat mutant. Second, the “Percentage Infected but Off” is the fraction of infected cells that are in the Off population, but that can be stimulated to express Tat and GFP via the addition of TNF-α (a strong activator of the NF-κB pathway, which directly activates the LTR) and trichostatin A (an inhibitor of histone deacetylases that also activates HIV gene expression) (36). This metric is a measure of the inability of a particular Tat mutant to activate gene expression from the viral LTR.
When introduced into WT Tat, single point mutations Q35L or I39Q, chosen from the matrix in Fig. 2A, yielded nonexpressing virus (Fig. 2C and supplemental Fig. S3A). The percentage of infected but off cells for these single mutants was ∼70%, nearly three times higher than WT Tat, again indicating that these Tat mutants fail to activate gene expression from the viral LTR (Fig. 2D). Strikingly, however, the introduction of both mutations into the same Tat sequence (henceforth referred to as the double-mutant, DM) rescued Mean On Peak levels close to that of WT Tat, with a similar fraction of silenced cells (Fig. 2, C and D, and supplemental Fig. S3A). Similarly, transfection of the WT, Q35L, I39Q, and DM Tat into a HeLa cell line containing a HIV-1 LTR Luciferase reporter showed analogous trends (supplemental Fig. S3B). From the 917 Tat sequences used in the MI analysis, 124 sequences shared the same Gln35–Ile39 residue pair as WT Tat, and 262 sequences shared the same Leu35–Gln39 residue pair as DM Tat, suggesting that both residue pairs are found in naturally occurring Tat sequences. Furthermore, to show that coevolution between sites 35 and 39 extend to another Tat subtype, we made single point mutants L35Q and Q39I of subtype C Tat. As previously observed for single mutations of Tat B, these mutants resulted in dramatic loss of gene expression and a 3-fold increase in Percentage Infected but Off cells (supplemental Fig. S4, C and D). However, gene expression was restored in the Tat C double mutant, suggesting that sites 35 and 39 alone compensate for one another (supplemental Fig. S4, C and D). Thus, evolutionarily only certain pairs of amino acids at sites 35 and 39 but not their intermediates are tolerated.
In analysis of site 31 in Tat B, the mutation C31S also yielded a slight reduction in gene expression and a statistical increase in the Percentage of Infected but Off cells as compared with WT Tat (p < 0.01); however, coevolution between site 31 and sites 35 or 39 proved difficult to investigate, as mutation at either of the latter two exerted a dominant loss of gene expression. However, the interaction between sites 31, 35, and 39 can be observed statistically. A Gln at site 39 constrains sites 31 and 35 primarily to a Ser and Leu, respectively. Similarly, a Leu, Ile, or Thr at site 39 primarily restricts sites 31 and 35 to a Cys and Gln, respectively (supplemental Fig. S5). These interactions between sites 31, 35, and 39 are discussed from a structural perspective in additional detail in supplemental “Results.”
We next replaced Gln35 and Ile39 in WT Tat with other amino acids based on residues that either appear or do not appear at sites 35 or 39 in the Los Alamos Sequence Database. As anticipated, replacing Gln35 with similar polar residues that are not found (Q35N, Q35E, and Q35K) or rarely found (Q35T) in the database, and are thus not predicted to coevolve with residues at site 39, resulted in nonfunctional Tat proteins (supplemental Fig. S4, A and B). Similarly, replacing Ile39 in WT Tat with nonpolar residues (I39F), or polar residues that occupy similar side chain volumes (I39K) but are not found in the database, gave rise to nonexpressing viruses (supplemental Fig. S4, A and B). In contrast, naturally occurring Tat sequences with a Gln at site 35 have residues such as Leu and Val in addition to Ile at site 39 (Fig. 2A, Val is not shown in this matrix). I39L and I39V Tat mutants yielded similar Mean On Peak and Percentage Infected but Off levels as WT Tat (supplemental Fig. S4, A and B), validating the predictions from MI analysis. These mutations are discussed in greater detail from a structural and energetic perspective in supplemental “Results.”
Compared with sites above the threshold MI value, mutation of positions 7, 12, 17, 19, 24, 29, 32, 40, or 42, which were predicted to be within the background region from the MI analysis, did not show any statistical difference in the level of gene expression or the Percentage of Infected but Off cells compared with WT Tat (p > 0.01, Fig. 2, C and D). These gene expression studies thus strongly support the in silico prediction that sites 35 and 39 strongly coevolve and are critical to ensure the primary function of Tat as a transactivator of the HIV-1 promoter (Fig. 2, C and D).
Finally, WT and DM Tat have different pairs of amino acids at sites 35 and 39 yet induce similar levels of gene expression when present at high levels (Fig. 2C); however, we also wanted to explore their gene activation behavior at lower concentrations. Under these conditions, previous studies have shown that the Tat positive feedback loop is subject to stochastic fluctuations in Tat, one of several factors that may play a role in viral reactivation from latency (35, 36). Because DM Tat has amino acids Leu and Gln at sites 35 and 39, respectively, residues shared by a majority of subtype C Tats at these positions, we included subtype C Tat in these studies to determine the contribution of sites 35 and 39 to this behavior. As described previously, cells were infected with LGIT variants at low multiplicity of infection, and GFP+ cells were sorted by fluorescence activated cell sorting (FACS) 7 days post-infection after stimulation with TNF-α (36). The sorted cells were allowed to relax for 9 days, and GFP− cells infected with the LGIT vector, but not expressing GFP, were sorted (supplemental Fig. S6A). These silent proviruses were then monitored for GFP expression over the course of 12 days (supplemental Fig. S6B).
As anticipated, the functionally inactive Q35L and I39Q Tat variants showed very low levels of gene activation. However, the DM Tat partially restored gene expression to WT Tat levels, and very closely tracked the activation rate of Tat C (supplemental Fig. S6B), a result that suggests that residue pairs at sites 35 and 39 may be important determinants in setting the gene activation levels for different Tat variants.
Coevolving Sites 35 and 39 Impact Both P-TEFb Binding and Phosphorylation at the CTD of RNAPII
To identify potential molecular mechanisms that restore gene expression for the DM Tat, we reasoned that the compromised transactivation of either Tat single mutant may be due to disruption in its binding to one of numerous cellular factors necessary for efficient transactivation. For example, the activation domain of Tat (amino acids 1–48) has previously been shown to interact with P-TEFb, which mediates the critical phosphorylation of the CTD of RNAPII and thus the production of full-length viral transcripts (9, 37). HeLa cells were transfected with plasmids to express FLAG-tagged Tat under the control of the human ubiquitin promoter (Ubiquitin-mCherry-IRES-Tat or UbChIT), and immunoprecipitates of Tat were probed for Cdk9 and CycT1 (Fig. 3, A and B) (18). WT Tat bound P-TEFb; however, the Q35L Tat mutant failed to efficiently bind either Cdk9 or CycT1, suggesting that site 35 is critical for binding P-TEFb (Fig. 3, A and B), and the loss of this binding possibly underlies the defective gene expression for this mutant (Fig. 2, C and D). Similarly, other factors that have recently been shown to interact with the Tat·P-TEFb complex and aid in transcriptional activation, such as ENL, AF9, AFF4, and ELL2, failed to bind to the Q35L Tat mutant (supplemental Fig. S7) (38). Interestingly, the DM Tat partially restores binding with Cdk9 and CycT1, likely the mechanism by which the I39Q mutation rescues the loss of function for the Q35L Tat mutant (Fig. 3, A and B).
FIGURE 3.

Co-immunoprecipitation and homology modeling shows that the Q35L Tat mutant fails to bind P-TEFb. A, immunoprecipitation (IP) of nuclear extracts (NE) with α-FLAG, obtained from HeLa cells transfected with the UbChIT vector, were followed by Western blots (WB) with α-Cdk9 and α-CycT1 antibodies. B, quantification of binding of different Tat mutants with CycT1. CycT1 is normalized to Tat, and its interaction with WT Tat is arbitrarily assigned the value 1. Error bars represent S.D. for two independent α-FLAG IPs and Western blots. ** denotes statistically significant differences (p < 0.05) from WT Tat in CycT1 binding. C, interaction of P-TEFb with WT and DM Tat are shown. Dark green and purple colors represent the ATP+ and ATP− structures for WT Tat, respectively, whereas light green and purple colors represent the ATP+ and ATP− structures for DM Tat. The structure shows hydrogen bonding between Asn180 in CycT1 with Gln35 in WT Tat, as well as hydrogen bonding between Asn180 in CycT1 with Gln39 in DM Tat. Hydrogen bonding is thus critical for Tat·P-TEFb binding.
To gain further insights into the loss of P-TEFb binding, we performed in silico modeling based on a recently solved structure of Tat·P-TEFb (37). These results indicated that Asn180 of CycT1 is positioned between and can form hydrogen bonds with a Gln at either Tat site 35 or 39. The Q35L mutation results in the loss of this hydrogen bonding, whereas the compensating mutation I39Q in the DM Tat enables Gln39 to replace this hydrogen bond with Asn180 in CycT1 (Fig. 3C). Although a previous study predicted that other naturally occurring mutations (except Tyr) could readily be accommodated at site 35 and maintain the structure of the protein complex (37), our analysis and accompanying experimental data suggest that the loss of hydrogen bonding may well be responsible for the drastic loss of function observed in the Q35L Tat mutant.
In contrast to the Q35L Tat mutant, however, the I39Q Tat mutant is able to bind CycT1 at levels close to the DM Tat (Fig. 3B), but lower than WT Tat, suggesting that its inability to activate gene expression (Fig. 2C) arises from reasons other than P-TEFb binding. To probe other transcriptional steps at which I39Q Tat may fail, we quantified viral transcripts. Cells infected with LGIT vectors containing one of the four Tat variants were stimulated with TNF-α 7 days post-infection, and infected, GFP+ cells were isolated by FACS. The sorted cells were allowed to relax for 9 days (Fig. 4A), total cellular RNA was extracted, and the levels of viral transcripts were quantified using RT-qPCR (36). The I39Q and DM Tat both had similar percentages of elongated transcripts (Fig. 4B); however, the I39Q Tat has much lower levels of total transcripts compared with the DM Tat (Fig. 4C), suggesting that it fails to induce transcription at the same efficiency as the DM Tat.
FIGURE 4.

Viral transcript quantification reveals a potential transcriptional step at which I39Q Tat may fail. A, GFP histograms for Jurkat cells infected with wild-type (red), Q35L (green), I39Q (blue), and DM (brown) Tat 9 days post-sorting of TNF-α-stimulated GFP+ cells. B and C, quantification of the percentage of elongated and total viral transcripts obtained from total cellular RNA of infected Jurkat cells. β-Actin is used for normalization. The assay is able to detect and quantify transcripts containing the full transactivation response element RNA but not very short aborted transcripts. All qPCR measurements are in triplicate and error bars represent S.D. * denotes statistically significant differences (p < 0.05) between the indicated pairs of Tat variants.
We explored the possibility that loss of gene expression for I39Q Tat arises due to its inability to interact with an upstream transcription factor such as Sp1 or a chromatin remodeling complex such as SWI/SNF (21, 39). However, co-immunoprecipitation showed no differences in Sp1 binding between I39Q and the DM Tat (Fig. 5A). A change in interaction with SWI/SNF could alter disruption of the nucleosome (Nuc-1) situated at the transcription start site; however, nuclease sensitivity assays showed that both I39Q and the DM Tat had similar effects on Nuc-1 (Fig. 5B).
FIGURE 5.

Mutations do not alter Tat binding with Sp1 or show differences in Nuc-1 disruption. A, immunoprecipitation of nuclear extracts with α-FLAG antibody, obtained from HeLa cells transfected with the UbChIT vector, were followed by Western blots with α-Sp1 antibody. Both I39Q and DM Tat appear to bind Sp1 with similar affinity. B, Jurkat cells infected with different Tat variants were incubated with or without a nuclease DNase I and genomic DNA was extracted using the EpiQ Chromatin Analysis Kit. The human hemoglobin gene (hHBB) was used as an internal control. All qPCR measurements were made in triplicate and error bars represent S.D. None of the Tat variants showed any statistical difference in Nuc-1 disruption from each other (p > 0.05).
At the heart of viral gene expression is the apparent ability of Tat to affect RNAPII phosphorylation. Sequential phosphorylation of serines at positions 5 (Ser5) and 2 (Ser2) within an evolutionarily conserved but unstructured domain in mammalian RNAPII, consisting of 52 repeats of the heptapeptide Y1S2P3T4S5P6S7 at its CTD, is critical for mRNA synthesis and processing (40). Normally, RNAPII recruited to the promoter of a gene is phosphorylated at Ser5 by Cdk7 within the transcription factor complex TFIIH (41). Shortly after transcription initiation, the polymerase briefly stalls ∼30–40 bp downstream of the transcription start site to allow for pre-mRNA processing steps such as capping (42, 43). Phosphorylation at Ser2 by the P-TEFb complex then promotes transcriptional elongation. In HIV-1 gene expression, however, Tat directly recruits and enables P-TEFb to phosphorylate both Ser5 and Ser2, and thereby greatly enhances transcriptional elongation (11, 12, 44).
Consistent with these results, the recently solved crystal structure of the Tat·P-TEFb complex shows that Tat binding induces P-TEFb conformational changes (37). To analyze the potential structural effects of mutations at sites 35 and 39, we performed additional in silico modeling based on this structure of Tat·P-TEFb, either in complex with or without an ATP analog molecule (ATP+ or ATP−). Interestingly, both the ATP+ and ATP− structures of I39Q Tat are slightly energetically stabilized compared with WT Tat. In contrast, for the DM Tat the ATP+ structure was destabilized by 3.42 kcal/mol*, and the ATP− structure was stabilized by 2.38 kcal/mol* compared with WT Tat (Table 1). Based on the energetics of the ATP+ structures, these modeling results suggest that compared with I39Q Tat, P-TEFb associated with the DM Tat may have a higher propensity to transfer the phosphate group from ATP to a substrate and transit to the more stable ATP− state. Moreover, based on the collective evidence from literature, viral transcript data, and in silico modeling results, we hypothesized that the I39Q Tat mutant, unlike the DM, may fail to efficiently induce P-TEFb-mediated phosphorylation of the CTD of RNAPII (Fig. 4C and Table 1) (37, 44).
TABLE 1.
In silico modeling results of the stability (ΔΔG) of the Tat·P-TEFb or Tat·P-TEFb·ATP complex after introducing mutations in Tat
Negative values indicate greater stability of a complex as compared to the complex containing WT Tat. The asterisk over kcal/mol indicates that these values are computational determined. In contrast to the I39Q Tat, the Tat·P-TEFb·ATP complex for the DM Tat is destabilized and hence may have greater propensity to transfer the phosphate group to the CTD of RNAPII and transition into the more stable Tat·P-TEFb complex.
| ΔΔG (kcal/mol*) | Tat·P-TEFb complex (ATP−) (PDB 3MI9) | Tat·P-TEFb·ATP complex (ATP+) (PDB 3MIA) |
|---|---|---|
| Q35L | −3.00 | 4.95 |
| I39Q | −1.01 | −1.99 |
| DM | −2.38 | 3.42 |
To explore this potential phosphorylation defect for I39Q Tat during transcriptional initiation and early elongation involved in efficient escape of RNAPII from the promoter, we performed chromatin immunoprecipitation (ChIP) with qPCR analysis to quantify the levels of total and Ser5 and Ser2 phosphorylated RNAPII associated with the HIV-1 promoter in the presence of different Tat variants. Interestingly, even though similar levels of total RNAPII are recruited to the viral promoter (Fig. 6C), the level of Ser-5-P-CTD of RNAPII close to the transcription start site for the I39Q Tat mutant was dramatically lower than for the DM Tat, and slightly lower than for WT Tat (Fig. 6A). Similarly, the level of Ser-2-P-CTD of RNAPII for I39Q Tat during early elongation was significantly (p < 0.05) lower than both WT and DM Tat (Fig. 6B).
FIGURE 6.

I39Q Tat fails to efficiently induce phosphorylation of the CTD of RNAPII, and the subtypes of Tat have potentially evolved alternate modes of inducing viral gene expression. A–C, ChIP for Ser-5-P-CTD of RNAPII, Ser-2-P-CTD of RNAPII and total RNAPII close to the transcription start site. Although similar levels of RNAPII are recruited to the viral promoter for all Tat variants, the I39Q Tat apparently fails to induce P-TEFb to efficiently phosphorylate the CTD of RNAPII, unlike the DM and WT Tat. Controls were performed without antibody. All qPCR measurements are in triplicate, and error bars represent S.D. * denotes statistically significant differences (p < 0.05) between the indicated pairs of Tat variants. D, plot of Ser-5-P-CTD of RNAPII versus CycT1 binding, and Ser-2-P-CTD of RNAPII versus CycT1 binding, for different Tat variants. The black and red symbols correspond to the levels of Ser-5-P-CTD of RNAPII and Ser-2-P-CTD of RNAPII for different Tat variants, respectively. The blue oval encompasses the Q35L and I39Q Tat variants that fail to activate gene expression, either due to its inability to bind P-TEFb or due to its failure to induce P-TEFb to efficiently phosphorylate the CTD of RNAPII. The green oval shows that the WT and DM Tat activate gene expression, although potentially through different mechanisms. Markers within the green oval show that subtype B Tat (WT Tat) displays high P-TEFb binding affinity and low Ser-5-P-CTD of RNAPII, whereas the DM Tat, mimicking most subtype C Tats at sites 35 and 39, shows moderate P-TEFb binding and high Ser-5-P-CTD of RNAPII, with both Tat variants displaying comparable levels of Ser-2-P-CTD of RNAPII.
Thus, it appears that the combination of weak Ser-5-P-CTD of RNAPII, strong P-TEFb binding affinity, and high Ser-2-P-CTD of RNAPII, for the WT Tat or the combination of high Ser-5-P-CTD of RNAPII, moderate P-TEFb binding affinity, and high Ser-2-P-CTD of RNAPII for the DM Tat, mediates efficient escape of RNAPII from the HIV-1 promoter and activates gene expression for these two variants (Figs. 3B and 6, A and B). Thus, it is possible that WT and DM Tat achieve similar levels of gene expression through orthogonal combinations of P-TEFb binding and Ser-5-P-CTD of RNAPII (Fig. 6D).
In contrast, although the I39Q Tat displays moderate P-TEFb binding affinity, the extremely low levels of Ser-5-P-CTD and Ser-2-P-CTD of RNAPII likely impairs its ability to activate gene expression (Figs. 3B and 6, A and B). Therefore, it appears that a Gln at site 35 (as seen for the WT and I39Q Tat) reduces the ability of Tat to induce P-TEFb-mediated Ser-5-P-CTD of RNAPII, but the presence of a Leu at site 35 (as in the Q35L and DM Tat) dramatically increases this function (Fig. 6A). However, Q35L Tat fails to bind P-TEFb and promote efficient escape of RNAPII from the promoter, as seen from the significantly lower levels (p < 0.05) of Ser-2-P-CTD of RNAPII for this mutant compared with WT and DM Tat. Thus, although the I39Q and DM Tat both have similar, but lower, P-TEFb binding affinity than WT Tat, the high Ser-5-P-CTD and Ser-2-P-CTD of RNAPII observed with the DM but not I39Q Tat apparently rescues gene expression.
DISCUSSION
For such a small protein, Tat shows a surprising diversity of function mediated by interaction with numerous cellular partners. It is post-translationally modified at specific sites by several cellular factors that impact transactivation. In addition to acetylation by PCAF and p300, there is evidence for methylation of Tat at Arg52 and Arg53 by the arginine methyltransferase PRMT6 (15) and methylation at Lys51 by the lysine methyltransferase Set7/9 (KMT7). Similarly, other lysine methyltransferases have been shown to interact with Tat (16, 17). Tat is also phosphorylated by Cdk2 (Ser16, Ser46) and PKR (Ser62, Thr64, and Ser68) (18, 19). Further evidence of the versatility of Tat can be seen in its interaction with other cellular proteins such as SKIP/SNW1 and SWI/SNF (20, 21, 23). Conserved and functionally important individual sites involved in these interactions can often be identified from multiple sequence alignments. However, the identification of mutually dependent coevolving sites, which can readily be missed by simple site conservation, is enabled through the use of statistical measures such as MI.
To date, the experimental discovery of correlated sites within the HIV-1 proteome, such as in Tat, reverse transcriptase, nucleocapsid, and Rev, has involved creation of libraries of viral proteins or long term culture of HIV-1 strains with single, site-directed mutations to reveal potential “suppressor mutations” (45–48). These approaches can sometimes yield either reversion of the introduced mutation or suppressor sites that do not naturally or specifically coevolve but act in a global, independent manner to increase fitness. By comparison, statistical analysis of viral sequence databases can identify positions whose evolution is correlated in a natural or clinical setting, as well as reveal correlations between new, unanticipated amino acid pairs. Here, we have harnessed MI to demonstrate functionally important correlations between pairs of sites in Tat.
In computationally guided experiments using a model lentiviral system mimicking the positive feedback loop in HIV-1, we found that single point mutations Q35L and I39Q yielded Tat variants that failed to activate gene expression from the viral LTR, with a majority of the proviruses existing in a silenced state that was activated only upon stimulation with pharmacological agents. However, introduction of both mutations Q35L and I39Q into the same Tat protein restored gene expression (Fig. 2, C and D). Thus, the Gln35–Ile39 and Leu35–Gln39 residue pairs both result in efficient gene expression from the viral promoter, for two Tat subtypes (Figs. 2C and supplemental Fig. S4C), confirming that sites 35 and 39 are coevolving. Furthermore, co-immunoprecipitation and ChIP studies revealed distinct, complementary mechanisms that constrain amino acid residues at these two sites: effective P-TEFb binding and alteration of P-TEFb substrate specificity to include the phosphorylation of Ser5 and Ser2 residues on the RNAPII CTD. Specifically, we show that the Q35L single mutant fails to bind P-TEFb, whereas the DM partially rescues P-TEFb binding (Fig. 3). In contrast, the I39Q Tat binds P-TEFb at levels close to the DM Tat, yet still suffers from very low gene expression (Figs. 2C and 3B) potentially due to its inability to induce P-TEFb to phosphorylate the CTD of RNAPII (Fig. 6, A and B). It is plausible that the inability of the I39Q Tat to induce efficient phosphorylation of the CTD of RNAPII involves loss of interaction with additional host factors that remain to be discovered. At any rate, unlike most coevolving or suppressor mutations that help restore a single biological function (49), the coevolving sites 35 and 39 each contribute to distinct Tat-mediated mechanisms that are integrated to yield an active protein. That is, mutationally induced deficits in one mechanism can be compensated for by mutations in the coevolving site that affect the other.
Most subtype B Tats pair Gln35 with Ile39/Leu39/Thr39, whereas a majority of subtype C Tats contain the Leu35–Gln39 residue pair, similar to the DM Tat, with a few having a Gln35–Leu39 residue pair. Based on the P-TEFb binding assay and levels of Ser-5-P-CTD of RNAPII, it appears that different subtypes could potentially have evolved alternate modes or “solutions” to inducing gene expression from the viral LTR. Subtype B Tats induce a low level of Ser-5-P-CTD in RNAPII (Fig. 6A), but the strong binding to P-TEFb could at least in part compensate for this deficit (Fig. 3B) and eventually produce efficient elongation, as can been seen from the high levels of Ser-2-P-CTD of RNAPII, a marker for P-TEFb-induced elongation (supplemental Fig. S8). In contrast, the DM Tat, which mimics the majority of subtype C Tats at sites 35 and 39, induces very high levels of Ser-5-P (Fig. 6A) coupled with relatively weaker P-TEFb binding (Fig. 3B) that in combination could ultimately drive comparable levels of Tat-mediated gene expression as measured by GFP expression (Fig. 2C), viral transcript analysis, and Ser-2-P-CTD of RNAPII (supplemental Fig. S8). Thus, the diversification of HIV-1 into different subtypes has apparently resulted in the evolution of compensatory mechanisms to trade off substrate binding and catalytic activity in inducing Tat-mediated gene expression from the viral LTR, such that the overall activity may be determined by the combination or “sum” of contributions from individual positions or functions (Fig. 6D). This novel finding has some parallels with other biological systems. For instance, it has been shown previously that autophosphorylation mutants of the epidermal growth factor (EGF) receptor stimulate similar levels of MAP kinase activation, gene expression, and mitogenesis as the WT EGF receptor though different compensatory mechanisms (50).
We have previously found that gene expression from the LTR is a stochastic process, with bursts of mRNA production separated by long intervals, a feature that could play an important role in the establishment of viral latency (35, 36, 51). Changes to Tat that distinctly affect transcriptional initiation or elongation could differentially impact the frequency and size of mRNA bursts. Gene expression data at low Tat levels indicates that Tat variants from different subtypes, with potentially alternate mechanisms for inducing gene expression, could impact probabilistic gene expression events (supplemental Fig. S6). Future work may explore whether these differences in Tat result in different propensities for viral latency. In addition to its application to HIV-1, such an integrated computational and experimental approach could readily be extended to other pathogens to gain deeper insights into their function and evolution, as well as potentially aid in the rational development of novel therapeutic strategies.
Supplementary Material
Acknowledgments
We thank Prof. Kathryn Miller-Jenson for careful perusal of the manuscript and helpful discussions. We thank Dr. Sharon Aviran and Morgan Price for technical assistance with the MI analysis, Jasper Rine's lab and Laura Lombardi for use of the Branson Sonifier 450 and Steve Okino (Bio-Rad) for the generous gifts of the EqiQ Chromatin SYBR Supermix and Analysis Kit. Cell sorting was performed at the University of California Berkeley Flow Cytometry Core Facility with assistance from Hector Nolla and Alma Valeros.
This work was supported, in whole or in part, by National Institutes of Health Grant R01GM073058 (to A. P. A. and D. V. S.) and Grants R01AI41757-11 and R01AI41757-11S1 (to Q. Z.).

This article contains supplemental “Experimental Procedures,” “Results,” and Figs. S1–S8.
- HIV-1
- human immunodeficiency virus-1
- P-TEFb
- positive transcription-elongation factor b
- CycT1
- cyclin T1
- Cdk9
- cyclin-dependent kinase 9
- CTD
- C-terminal domain
- RNAPII
- RNA polymerase II
- MI
- mutual information
- DM
- double mutant
- IRES
- internal ribosome entry sequence
- LGIT
- LTR-GFP-IRES-Tat
- UbChIT
- ubiquitin-mCherry-IRES-Tat
- GFP
- green fluorescent protein
- qPCR
- quantitative PCR.
REFERENCES
- 1. Negroni M., Buc H. (2001) Mechanisms of retroviral recombination. Annu. Rev. Genet. 35, 275–302 [DOI] [PubMed] [Google Scholar]
- 2. Johnson V. A., Brun-Vezinet F., Clotet B., Gunthard H. F., Kuritzkes D. R., Pillay D., Schapiro J. M., Richman D. D. (2009) Update of the drug resistance mutations in HIV-1: December 2009. Top. HIV Med. 17, 138–145 [PubMed] [Google Scholar]
- 3. Rambaut A., Posada D., Crandall K. A., Holmes E. C. (2004) The causes and consequences of HIV evolution. Nat. Rev. Genet. 5, 52–61 [DOI] [PubMed] [Google Scholar]
- 4. van Opijnen T., Jeeninga R. E., Boerlijst M. C., Pollakis G. P., Zetterberg V., Salminen M., Berkhout B. (2004) Human immunodeficiency virus type 1 subtypes have a distinct long terminal repeat that determines the replication rate in a host cell-specific manner. J. Virol. 78, 3675–3683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Desfosses Y., Solis M., Sun Q., Grandvaux N., Van Lint C., Burny A., Gatignol A., Wainberg M. A., Lin R., Hiscott J. (2005) Regulation of human immunodeficiency virus type 1 gene expression by clade-specific Tat proteins. J. Virol. 79, 9180–9191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kurosu T., Mukai T., Komoto S., Ibrahim M. S., Li Y. G., Kobayashi T., Tsuji S., Ikuta K. (2002) Human immunodeficiency virus type 1 subtype C exhibits higher transactivation activity of Tat than subtypes B and E. Microbiol. Immunol. 46, 787–799 [DOI] [PubMed] [Google Scholar]
- 7. Camps M., Herman A., Loh E., Loeb L. A. (2007) Genetic constraints on protein evolution. Crit. Rev. Biochem. Mol. Biol. 42, 313–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kao S. Y., Calman A. F., Luciw P. A., Peterlin B. M. (1987) Anti-termination of transcription within the long terminal repeat of HIV-1 by Tat gene product. Nature 330, 489–493 [DOI] [PubMed] [Google Scholar]
- 9. Wei P., Garber M. E., Fang S. M., Fischer W. H., Jones K. A. (1998) A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell 92, 451–462 [DOI] [PubMed] [Google Scholar]
- 10. Roy S., Delling U., Chen C. H., Rosen C. A., Sonenberg N. (1990) A bulge structure in HIV-1 TAR RNA is required for Tat binding and Tat-mediated transactivation. Genes Dev. 4, 1365–1373 [DOI] [PubMed] [Google Scholar]
- 11. Zhou Q., Yik J. H. (2006) The Yin and Yang of P-TEFb regulation. Implications for human immunodeficiency virus gene expression and global control of cell growth and differentiation. Microbiol. Mol. Biol. Rev. 70, 646–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. D'Orso I., Frankel A. D. (2010) RNA-mediated displacement of an inhibitory snRNP complex activates transcription elongation. Nat. Struct. Mol. Biol. 17, 815–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kiernan R. E., Vanhulle C., Schiltz L., Adam E., Xiao H., Maudoux F., Calomme C., Burny A., Nakatani Y., Jeang K. T., Benkirane M., Van Lint C. (1999) HIV-1 Tat transcriptional activity is regulated by acetylation. EMBO J. 18, 6106–6118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Marzio G., Tyagi M., Gutierrez M. I., Giacca M. (1998) HIV-1 tat transactivator recruits p300 and CREB-binding protein histone acetyltransferases to the viral promoter. Proc. Natl. Acad. Sci. U.S.A. 95, 13519–13524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xie B., Invernizzi C. F., Richard S., Wainberg M. A. (2007) Arginine methylation of the human immunodeficiency virus type 1 Tat protein by PRMT6 negatively affects Tat interactions with both cyclin T1 and the Tat transactivation region. J. Virol. 81, 4226–4234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pagans S., Kauder S. E., Kaehlcke K., Sakane N., Schroeder S., Dormeyer W., Trievel R. C., Verdin E., Schnolzer M., Ott M. (2010) The cellular lysine methyltransferase Set7/9-KMT7 binds HIV-1 TAR RNA, monomethylates the viral transactivator Tat, and enhances HIV transcription. Cell Host Microbe. 7, 234–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Van Duyne R., Easley R., Wu W., Berro R., Pedati C., Klase Z., Kehn-Hall K., Flynn E. K., Symer D. E., Kashanchi F. (2008) Lysine methylation of HIV-1 Tat regulates transcriptional activity of the viral LTR. Retrovirology 5, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ammosova T., Berro R., Jerebtsova M., Jackson A., Charles S., Klase Z., Southerland W., Gordeuk V. R., Kashanchi F., Nekhai S. (2006) Phosphorylation of HIV-1 Tat by CDK2 in HIV-1 transcription. Retrovirology 3, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Endo-Munoz L., Warby T., Harrich D., McMillan N. A. (2005) Phosphorylation of HIV Tat by PKR increases interaction with TAR RNA and enhances transcription. Virol. J. 2, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brès V., Yoshida T., Pickle L., Jones K. A. (2009) SKIP interacts with c-Myc and Menin to promote HIV-1 Tat transactivation. Mol. Cell 36, 75–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mahmoudi T., Parra M., Vries R. G., Kauder S. E., Verrijzer C. P., Ott M., Verdin E. (2006) The SWI/SNF chromatin-remodeling complex is a cofactor for Tat transactivation of the HIV promoter. J. Biol. Chem. 281, 19960–19968 [DOI] [PubMed] [Google Scholar]
- 22. Deng L., de la Fuente C., Fu P., Wang L., Donnelly R., Wade J. D., Lambert P., Li H., Lee C. G., Kashanchi F. (2000) Acetylation of HIV-1 Tat by CBP/P300 increases transcription of integrated HIV-1 genome and enhances binding to core histones. Virology 277, 278–295 [DOI] [PubMed] [Google Scholar]
- 23. Agbottah E., Deng L., Dannenberg L. O., Pumfery A., Kashanchi F. (2006) Effect of SWI/SNF chromatin remodeling complex on HIV-1 Tat activated transcription. Retrovirology 3, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Martin L. C., Gloor G. B., Dunn S. D., Wahl L. M. (2005) Using information theory to search for co-evolving residues in proteins. Bioinformatics 21, 4116–4124 [DOI] [PubMed] [Google Scholar]
- 25. Kass I., Horovitz A. (2002) Mapping pathways of allosteric communication in GroEL by analysis of correlated mutations. Proteins 48, 611–617 [DOI] [PubMed] [Google Scholar]
- 26. Göbel U., Sander C., Schneider R., Valencia A. (1994) Correlated mutations and residue contacts in proteins. Proteins 18, 309–317 [DOI] [PubMed] [Google Scholar]
- 27. Lockless S. W., Ranganathan R. (1999) Evolutionarily conserved pathways of energetic connectivity in protein families. Science 286, 295–299 [DOI] [PubMed] [Google Scholar]
- 28. Korber B. T., Farber R. M., Wolpert D. H., Lapedes A. S. (1993) Covariation of mutations in the V3 loop of human immunodeficiency virus type 1 envelope protein. An information theoretic analysis. Proc. Natl. Acad. Sci. U.S.A. 90, 7176–7180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fares M. A., Travers S. A. (2006) A novel method for detecting intramolecular coevolution. Adding a further dimension to selective constraints analyses. Genetics 173, 9–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gilbert P. B., Novitsky V., Essex M. (2005) Covariability of selected amino acid positions for HIV type 1 subtypes C and B. AIDS Res. Hum. Retroviruses 21, 1016–1030 [DOI] [PubMed] [Google Scholar]
- 31. Skerker J. M., Perchuk B. S., Siryaporn A., Lubin E. A., Ashenberg O., Goulian M., Laub M. T. (2008) Rewiring the specificity of two-component signal transduction systems. Cell 133, 1043–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dull T., Zufferey R., Kelly M., Mandel R. J., Nguyen M., Trono D., Naldini L. (1998) A third generation lentivirus vector with a conditional packaging system. J. Virol. 72, 8463–8471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dunn S. D., Wahl L. M., Gloor G. B. (2008) Mutual information without the influence of phylogeny or entropy dramatically improves residue contact prediction. Bioinformatics 24, 333–340 [DOI] [PubMed] [Google Scholar]
- 34. Weigt M., White R. A., Szurmant H., Hoch J. A., Hwa T. (2009) Identification of direct residue contacts in protein-protein interaction by message passing. Proc. Natl. Acad. Sci. U.S.A. 106, 67–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Weinberger L. S., Burnett J. C., Toettcher J. E., Arkin A. P., Schaffer D. V. (2005) Stochastic gene expression in a lentiviral positive-feedback loop. HIV-1 Tat fluctuations drive phenotypic diversity. Cell 122, 169–182 [DOI] [PubMed] [Google Scholar]
- 36. Burnett J. C., Miller-Jensen K., Shah P. S., Arkin A. P., Schaffer D. V. (2009) Control of stochastic gene expression by host factors at the HIV promoter. PLoS Pathog. 5, e1000260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tahirov T. H., Babayeva N. D., Varzavand K., Cooper J. J., Sedore S. C., Price D. H. (2010) Crystal structure of HIV-1 Tat complexed with human P-TEFb. Nature 465, 747–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. He N., Liu M., Hsu J., Xue Y., Chou S., Burlingame A., Krogan N. J., Alber T., Zhou Q. (2010) HIV-1 Tat and host AFF4 recruit two transcription elongation factors into a bifunctional complex for coordinated activation of HIV-1 transcription. Mol. Cell 38, 428–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chun R. F., Semmes O. J., Neuveut C., Jeang K. T. (1998) Modulation of Sp1 phosphorylation by human immunodeficiency virus type 1 Tat. J. Virol. 72, 2615–2629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Phatnani H. P., Greenleaf A. L. (2006) Phosphorylation and functions of the RNA polymerase II CTD. Genes Dev. 20, 2922–2936 [DOI] [PubMed] [Google Scholar]
- 41. Serizawa H., Mäkelä T. P., Conaway J. W., Conaway R. C., Weinberg R. A., Young R. A. (1995) Association of Cdk-activating kinase subunits with transcription factor TFIIH. Nature 374, 280–282 [DOI] [PubMed] [Google Scholar]
- 42. Zhang Z., Klatt A., Gilmour D. S., Henderson A. J. (2007) Negative elongation factor NELF represses human immunodeficiency virus transcription by pausing the RNA polymerase II complex. J. Biol. Chem. 282, 16981–16988 [DOI] [PubMed] [Google Scholar]
- 43. Schroeder S. C., Schwer B., Shuman S., Bentley D. (2000) Dynamic association of capping enzymes with transcribing RNA polymerase II. Genes Dev. 14, 2435–2440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhou M., Halanski M. A., Radonovich M. F., Kashanchi F., Peng J., Price D. H., Brady J. N. (2000) Tat modifies the activity of CDK9 to phosphorylate serine 5 of the RNA polymerase II carboxyl-terminal domain during human immunodeficiency virus type 1 transcription. Mol. Cell. Biol. 20, 5077–5086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tachedjian G., Aronson H. E., Goff S. P. (2000) Analysis of mutations and suppressors affecting interactions between the subunits of the HIV type 1 reverse transcriptase. Proc. Natl. Acad. Sci. U.S.A. 97, 6334–6339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Verhoef K., Berkhout B. (1999) A second-site mutation that restores replication of a Tat-defective human immunodeficiency virus. J. Virol. 73, 2781–2789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cimarelli A., Sandin S., Höglund S., Luban J. (2000) Rescue of multiple viral functions by a second-site suppressor of a human immunodeficiency virus type 1 nucleocapsid mutation. J. Virol. 74, 4273–4283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jain C., Belasco J. G. (1996) A structural model for the HIV-1 Rev-RRE complex deduced from altered specificity rev variants isolated by a rapid genetic strategy. Cell 87, 115–125 [DOI] [PubMed] [Google Scholar]
- 49. del Alamo M., Mateu M. G. (2005) Electrostatic repulsion, compensatory mutations, and long range nonadditive effects at the dimerization interface of the HIV capsid protein. J. Mol. Biol. 345, 893–906 [DOI] [PubMed] [Google Scholar]
- 50. Li N., Schlessinger J., Margolis B. (1994) Autophosphorylation mutants of the EGF-receptor signal through auxiliary mechanisms involving SH2 domain proteins. Oncogene 9, 3457–3465 [PubMed] [Google Scholar]
- 51. Skupsky R., Burnett J. C., Foley J. E., Schaffer D. V., Arkin A. P. (2010) HIV promoter integration site primarily modulates transcriptional burst size rather than frequency. PLoS Comput. Biol. 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.