

Abstract
Vibrio harveyi siphophage 1 (VHS1) is a tailed phage with an icosahedral head of approximately 66 nm in diameter and an unornamented, flexible tail of approximately 153 nm in length. When Vibrio harveyi 1114GL is lysogenized with VHS1, its virulence for the black tiger shrimp (Penaeus monodon) increases by more than 100 times, and this coincides with production of a toxin(s) associated with shrimp hemocyte agglutination. Curiously, the lysogen does not show increased virulence for the whiteleg shrimp (Penaeus [Litopenaeus] vannamei). Here we present and annotate the complete, circular genome of VHS1 (81,509 kbp; GenBank accession number JF713456). By software analysis, the genome contains 125 putative open reading frames (ORFs), all of which appear to be located on the same DNA strand, similar to the case for many other bacteriophages. Most of the putative ORFs show no significant homology to known sequences in GenBank. Notable exceptions are ORFs for a putative DNA polymerase and putative phage structural proteins, including a portal protein, a phage tail tape measure protein, and a phage head protein. The last protein was identified as a component of the species-specific toxin mixture described above as being associated with agglutination of hemocytes from P. monodon.
INTRODUCTION
Vibrio harveyi siphophage 1 (VHS1) is a tailed bacteriophage with an icosahedral head of approximately 66 nm in diameter and an unornamented tail of approximately 153 nm in length (26). It has a circular double-stranded DNA genome of approximately 80 kbp (26). It produces initially clear lytic plaques on lawns of Vibrio harveyi 1114GL. However, after several days of incubation, colonies of lysogens appear within the formerly clear plaques (14). These lysogens produce VHS1 spontaneously upon subculture, without the need for induction of a lytic cycle. Since the phage is carried in the lysogens as an episome, cured isolates can be obtained at high rates by isolation of single colonies upon subculture (14). Random clones representing approximately 25% of the VHS1 genome were previously sequenced and deposited in GenBank (26).
Vibrio harveyi 1114GL (VH0) that has been lysogenized by VHS1 (VH1) is >100 times more lethal to the black tiger shrimp than VH0 (13, 14). In addition, culture supernatant solutions from VH1 are highly toxic for the black tiger shrimp (Penaeus monodon) but not for the whiteleg shrimp (Penaeus [Litopenaeus] vannamei) (13). Comparison of mass spectrometry data from protein bands originating from semipurified fractions of these toxic supernatant solutions showed no significant homology to protein data (direct and deduced) in existing databases (13), particularly for Vibrio species. Since no significant homology was found in the Vibrio database, it was suggested that the toxins might have originated from the VHS1 genome. A number of other toxins are known to originate from phage genomes in lysogenized bacteria, including a toxin proposed to originate from a Vibrio harveyi Myoviridae-like (VHML) phage reported to lysogenize a Vibrio harveyi isolate from Australia (9, 20, 22, 23). The complete genome sequence of VHML (approximately 40 kbp) has been reported (22). Here we present the complete genome sequence of VHS1, the second known bacteriophage that enhances the virulence of Vibrio harveyi for giant tiger shrimp.
MATERIALS AND METHODS
VHS1 propagation and preparation.
Vibrio harveyi 1114GL type 1 (VH1) infected with VHS1 spontaneously produced VHS1 in the supernatant culture medium after overnight incubation at 30°C with shaking at 250 rpm (26). Cultures were centrifuged to remove bacterial cells and cell debris. The supernatant solution was filtered sequentially through 0.45-μm and 0.2-μm disposable membrane filters (Sartorius), and the presence of viable VHS1 particles was confirmed by dot plaque assays on lawns of strain 1114GL. As previously described (14, 26), the solution was precipitated by addition of polyethylene glycol 6000 (PEG 6000) and then subjected to ultracentrifugation at 100,000 × g for 4 h to pellet VHS1. The pellet was resuspended in phosphate-buffered saline (PBS) and layered over a discontinuous Urografin gradient (10 to 40%), followed by centrifugation at 100,000 × g for 4 h at 4°C. VHS1 was located at the 20 to 30% interface. Purified intact VHS1 phage particles were treated with DNase I and RNase before washing and extraction of DNA with QIAamp DNA minikits (Qiagen, Hilden, Germany) in preparation for genome sequencing. Purified virions were negatively stained as previously described (26) and examined by transmission electron microscopy (TEM) using a Hitachi H-7100 electron microscope equipped with a Gatan ES500W Orius model 782 charge-coupled device (CCD) camera that had been calibrated by the installation engineer. Using this setup with negatively stained T7 phage at 100 kV, the mean head diameter was 59 ± 3 nm, compared to that of approximately 60 nm given in the VIIIth Report of the ICTV (10).
DNA sequencing.
DNA sequencing was carried out by Macrogen Inc. (Seoul, South Korea), using Roche 454 technology. The seven resulting contigs were joined by primer walking and PCR amplification with primers designed from the ends of the various contigs. All postcontig sequencing work was also done by Macrogen and was carried out on both strands of the submitted DNA fragments. In summary, all final sequences were based on complete agreement between sequences of cDNA strands. In cases of any disagreement between the two sequences or between new sequences and VHS1 sequences previously deposited in GenBank, additional sequencing reactions were carried out, again on both strands, to obtain the final consensus sequence (i.e., at least three of four sequences were identical).
After obtaining the full sequence, the sequence was subjected to analysis of predicted restriction enzyme digest fragments, using NEBcutter V2.0 (http://tools.neb.com/NEBcutter2/), for EcoRI relative to the lambda DNA-HindIII digest marker for comparison to actual agarose gels of digests obtained previously using this enzyme with the same marker (26).
Sequence analysis. (i) Putative ORFs.
After gap closing and assembly using CAP3 software (12), the VHS1 sequence was annotated ab initio by three Web server predictors, specifically Zcurve (8), GeneMarkS (1), and EasyGene (17), and by three locally run predictors, specifically MetaGene (21), Genewise v. 2 (2), and Glimmer3 (7), using default parameters. All polypeptides from 2,427 viral genomes (downloaded on 14 June 2010 from GenBank) were used as the protein database for Genewise, while a minimal length of 90 bp and GenBank genetic code table 11 were used for Glimmer3. Glimmer3 also requires a probability model of coding sequences produced from previously characterized genes (26) and the long, nonoverlapping open reading frames (ORFs) in the genome, produced by a program in the Glimmer3 package. RNAmmer (16) and tRNAScan (18) were used to predict rRNAs and tRNAs, respectively. These predicted ORFs were examined in Argo2 (http://www.broadinstitute.org/annotation/argo2/) to determine the putative VHS1 ORF set. This putative VHS1 ORF set contained the ORFs predicted by at least two predictors or by one predictor with support from expressed sequence tags (EST) produced by VHS1-lysogenized V. harveyi, from a protein domain predicted by Pfamscan (19), or from a homologue in another organism. Homology with the NCBI nonredundant protein (nr) database was defined by BLASTx, using an E value cutoff of <10−10, for either the whole VHS1 sequence or individual ORFs determined by the predictors described above. To reduce the number of overlapping ORFs, the start codon of such ORFs, if applicable, was moved to the nearest starting site. By CODONW (27), the codon usage (determined by two measures, i.e., the relative frequency of synonymous codons and the effective number of codons [Nc]) (34) of these VHS1 ORFs was compared to that of the Vibrio harveyi ATCC BAA-1116 genome, which was downloaded from GenBank on 16 July 2010. The preferred codons for each amino acid were defined as synonymous codons with relative frequencies within a range of 3% from that for the codon with the highest frequency.
(ii) Homologues in other organisms.
To find homologous sequences in other species, the putative predicted polypeptides were subjected to BLAST searches (either BLASTp or PSI-BLAST, with five iterations) against either the NCBI nr database (downloaded on 6 July 2010) or the protein database constructed from all completely sequenced genomes of tailed phages in the order Caudovirales, using an E value cutoff of <10−3. A similar result was obtained for other E value cutoffs (e.g., E < 10−10, E < 10−20, and E < 10−30).
Construction of suppression-subtractive hybridization libraries.
Extraction of total RNA was carried out using RNeasy minikits from Qiagen, Hilden, Germany. To generate differential EST between parental (VH0) and lysogenic (VH1) bacteria, total RNA extracts were used as the starting material. Suppression-subtractive hybridization was carried out as recommended by the supplier, using a BD PCR-Select cDNA subtraction kit (BD Biosciences-Clontech) with some modification, because the kit was designed for eukaryotic mRNA and was based on the presence of poly(A) tails. Thus, at an early step, the bacterial total RNAs were subjected to polyadenylation using yeast poly(A) polymerase (Ambion). The differentially expressed enriched cDNAs (i.e., PCR products generated from each library) were cloned into pGEM-T Easy vector systems (Promega) and transformed into Escherichia coli DH5α. Resulting transformants were grown individually overnight in LB medium with ampicillin (100 μg/ml) at 37°C in 96-well microtiter plates and stored in 20% glycerol at −70°C. Inserts were confirmed by colony PCR with specific primers located in the vector sequence. PCR products were run in 1% agarose gels to identify clones with inserts. Subtracted libraries were screened for specific fragments by dot blot hybridization. To select true inserts, plasmids were extracted and quantified so that equal amounts of cDNA could be used to generate PCR products for the screening step, as described in the user manual for PCR-Select differential screening kits (Clontech Laboratories, Palo Alto, CA). Inserts were sequenced at Macrogen Inc. (Seoul, South Korea), using standard primers for the T7 and SP6 promoters. Typically, DNA inserts of 500 to 1,500 bp were obtained. After vector sequence removal, the locations of EST sequences best matched to the VHS1 genomic sequence were obtained by BLASTn searches. The EST reported here were those corresponding to putative ORFs supported by only one predicting algorithm plus those that corresponded to ORFs with homology to known viral structural proteins and DNA-related enzymes.
Nucleotide sequence accession number.
The final consensus sequence for the VHS1 genome has been deposited in GenBank under accession number JF713456.
RESULTS AND DISCUSSION
TEM of purified VHS1 virions.
Several rounds of viral purification were carried out with VHS1, and for each round, the virions were examined by TEM with negative staining. In every case, the virions showed heads of 66 ± 3 nm (n = 10) in diameter and flexible, unornamented tails of 153 ± 10 nm (Fig. 1). These sizes were somewhat larger than those reported previously for VHS1 (60- to 62-nm head and 100- to 120-nm tail) (26). In addition, the phage tails in the original description were described as rigid, while long, flexible tails are a common characteristic of the family Siphoviridae (11). In the first publication about VHS1 (26), the electron micrograph presented is analog, and based on the magnification bar of 30 nm, the head and tail of the illustrated phage are 58 and 133 nm, respectively. If the bar instead represents 33 nm (i.e., 10% error), then the head and tail sizes are 66 nm and 151 nm, respectively, which are within the range of those measured in our digital images.
Fig 1.
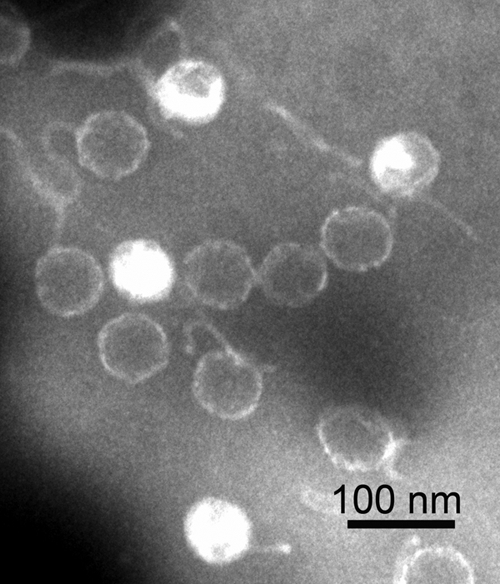
Transmission electron micrograph of negatively stained phage particles of VHS1.
The sizes of the VHS1 genome, head, and tail are consistent with those previously reported for 20 isometric-head marine siphophages of Vibrio parahaemolyticus (i.e., mean sizes for genomes, heads, and tails of 88 ± 36 kbp, 78 ± 16 nm, and 164 ± 26 nm, respectively) (5). The ratio of genome size (kbp) to head diameter (nm) ranged from 0.54 to 1.89 (mean = 1.01 ± 0.37) for these phages, with the lowest ratio for isolate LH3a (genome size of 120 kbp and head size of 65 nm). This range correlated with the ratio of 0.83 for VHS1, which is similar to those of five other phages (0.78 to 0.83) with genome sizes ranging from 120 to 122 kbp. In summary, the morphology of VHS1 is similar to that of other siphophages of marine Vibrio species and distinct from other V. harveyi siphophages recently reported from Thailand (28) and India (31, 32), based on the combination of morphology and genome size. The relationship of VHS1 to V. harveyi siphophages recently reported from Australia (6) is uncertain because their genome sizes were not given.
VHS1 sequence.
Seven contigs were obtained by pyrosequencing (Macrogen). These were combined with previously existing records of VHS1 genome sequences (approximately 20 kbp) in GenBank (accession no. AF465603, AF480606, AF480607, AF480608, AF480609, AF480611, AY579218, AY579219, AY579221, and AY579222), using CAP3 software. The initial process yielded 79,780 bp from the estimated total VHS1 genome of approximately 80 kbp. The remaining gaps between contigs were filled by primer walking until a single long contig was obtained. Using primers designed from each end of this continuous fragment, a 2.5-kb fragment was amplified. After cloning and sequencing, it linked the two ends and closed the DNA circle, yielding a molecule of exactly 81,509 bp (GenBank accession no. JF713456). Except for the seven contigs obtained by pyrosequencing, all other segments were sequenced at least twice on both strands. When this sequence was subjected to NEBcutter V2.0 for prediction of restriction enzyme digest fragments obtained using EcoRI, good agreement was obtained between the predicted gel and the actual gel obtained (Fig. 2). This supported the correctness of the sequencing results. A complete list of the EcoRI digestion fragments and their locations is given in Table S1 in the supplemental material.
Fig 2.

Predicted gel and actual gel of restriction enzyme digest fragments obtained using EcoRI and a lambda DNA-HindIII digest marker. A table of all predicted fragments is given in Table S1 in the supplemental material.
VHS1 nucleic acid sequence analysis.
The whole nucleotide sequence of VHS1 was used for a BLASTn search of GenBank. This yielded 10 hits for existing VHS1 sequences in GenBank (see the introduction), 3 hits for bacterial pyruvate phosphate dikinase (e.g., E = 3 × 10−11 for 132/186 identities for Bacteroides fragilis CR626927), and 93 hits for RecA proteins from many sources (e.g., E = 6 × 10−7 for 63/79 identities of Desulfococcus oleovorans CP000859). There were no other significant hits, even with VHML (GenBank accession no. AY133112), the only other fully sequenced bacteriophage reported for shrimp-pathogenic Vibrio harveyi (22).
RNA genes.
Neither rRNA nor tRNA genes were detected using RNAmmer (16) and tRNAScan (18), respectively.
Origin of replication.
In order to number the putative ORFs of VHS1 in a nonarbitrary manner, we identified the putative area of the origin of replication as indicated in Fig. 3. This was predicted based on the presence of an unusually high AT sequence bias across 350-bp frames, on predicted secondary structure formation similar to origins of replication for some other bacteriophages (predicted by the Vienna RNA Web server) (29), and on the lack of putative ORFs in that AT-rich area.
Fig 3.

Diagrammatic map of the circular genome of VHS1.
Protein-encoding ORFs.
Altogether, the six predictors used gave a total of 147 unique predicted ORFs, but only 123 ORFs were supported by at least two predictors. Of the 24 single-predictor-supported ORFs, we found two additional ORFs (047 and 060) with EST support (Table 1). Also shown in Table 1 are seven ORFs encoding putative phage structural proteins or DNA-related enzymes (see Table 2). Sequence identity between EST and the VHS1 genome sequence was 98 to 100%. The presence of EST that spanned more than one ORF suggested that VHS1 produces polycistronic mRNA. Altogether, we concluded that the total number of putative VHS1 ORFs was 125, all transcribed from the positive strand (Fig. 3; see Table S2 in the supplemental material). Among these 125 ORFs, 27 were supported by the presence of homologues in other genomes, and 16 ORFs were supported by the presence of Pfam domains (see Table S2). The start codons of 14 ORFs were moved to the nearest putative starting site to reduce the number of overlapping ORFs. The ORFs were numbered in order from 001 to 125, beginning with the first ORF after the putative origin of replication.
Table 1.
Preliminary EST support for some putative ORFs of VHS1a
| EST ID | Start (nt) | End (nt) | ORF | ORF start (nt) | ORF end (nt) |
|---|---|---|---|---|---|
| N-231-ESTS | 22836 | 23615 | 046 | 22344 | 22868 |
| 047* | 22868 | 23140 | |||
| 048 | 23211 | 23999 | |||
| N-194-ESTS | 28025 | 28443 | 052 | 26851 | 28032 |
| 053 | 28119 | 29069 | |||
| N-74-ESTS | 28026 | 28443 | 052 | 26851 | 28032 |
| 053 | 28119 | 29069 | |||
| N-117-ESTS | 32041 | 32500 | 059 | 31969 | 32391 |
| 060* | 32469 | 32591 | |||
| N-199-ESTS | 35822 | 36660 | 061 | 32599 | 36738 |
| N-93-ESTS | 46893 | 47236 | 074 | 46252 | 47697 |
| N-209-ESTS | 49182 | 49666 | 077 | 48360 | 49331 |
| 078 | 49342 | 50271 | |||
| N-71-ESTS | 59054 | 59924 | 090 | 58745 | 59554 |
| 091 | 59607 | 61523 | |||
| N-88-ESTS | 68325 | 69032 | 103 | 67904 | 70249 |
| N-148-ESTS | 69186 | 69728 | 103 | 67904 | 70249 |
| N-99-ESTS | 69727 | 70500 | 103 | 67904 | 70249 |
| 104 | 70251 | 72128 | |||
| N-249-ESTS | 70819 | 71201 | 104 | 70251 | 72128 |
| N-237-ESTS | 71422 | 71794 | 104 | 70251 | 72128 |
The ORFs shown include those encoding putative homologues for phage structural proteins (ORF 053, 061, 091, and 104) and DNA-related enzymes (ORFs 174, 077, and 103), as well as two ORFs (047 and 060 [marked with asterisks]) for which there was only a single software predictor. See Table 2 for further details on ORFs.
Table 2.
Top homologs (i.e., those with lowest E values) of 17 VHS1 ORFs found in other Caudovirales species, with E values of <10−3a
| VHS1 ORF | Homolog/species | Conserved domain | Phage family | GenBank accession no. | E value |
|---|---|---|---|---|---|
| 006 | Hypothetical protein/Vibrio phage KVP40 | NA | M | NP_899398 | 7 × 10−19 |
| 008 | Hypothetical protein/Mycobacterium phage Kostya | DUF550 | S | YP_002014578 | 3 × 10−11 |
| 048 | Hypothetical protein/Shigella phage phiSboM-AG3 | NA | M (unclassified) | YP_003358579 | 6 × 10−19 |
| 051 | Putative structural protein/Methanothermobacter phage psiM100 | NA | S (unclassified) | NP_071821 | 6 × 10−11 |
| 053* | Structural protein (capsid protein)/Methanobacterium phage psiM2 | Phage capsid (PF05065) | S | NP_046968 | 3 × 10−23 |
| 058 | Hypothetical protein/Pseudomonas phage DMS3 | NA | S | YP_950463 | 6 × 10−25 |
| 061* | Putative phage tail tape measure protein/Clostridium phage phi3626 | PhageMin_Tail (PF10145), tape_meas_TP901 (TIGR01760), SMC_prok_B (TIGR02168) | S | NP_612842 | 3 × 10−26 |
| 071 | Hypothetical protein/Mycobacterium phage Corndog | AAA (PF00004), P-loop NTPase superfamily | S | NP_817947 | 1 × 10−20 |
| 074 | DNA helicase DnaB/Thermus phage phiYS40 | DnaB_C (PF03796), phycobilisome, P-loop NTPase superfamily | M (unclassified) | YP_874027 | 9 × 10−10 |
| 077 | DNA primase/cyanophage PSS2 | zf-CHC2 (PF01807) | S (unclassified) | YP_003084253 | 9 × 10−4 |
| 079 | Helicase/Corynebacterium phage BFK20 | DEXDc, DEAD (PF00270), ResIII (PF04851) | S (unclassified) | YP_001456771 | 5 × 10−12 |
| 082 | Hypothetical protein (putative RecA)/Mycobacterium phage Cjw1 | RecA (PF00154), P-loop NTPase superfamily | S (unclassified) | NP_817564 | 5 × 10−32 |
| 085 | Conserved protein of unknown function/Mycobacterium phage Orion | RuvC_resolvase | S (unclassified) | YP_655102 | 3 × 10−4 |
| 091* | TerL/Rhodococcus phage ReqiDocB7 | NA | S (unclassified) | YP_874075 | 2 × 10−57 |
| 103 | Putative DNA polymerase/Bacillus phage SPO1 | DNA_Pol_A superfamily | M | YP_002300417 | 1 × 10−45 |
| 104* | Putative portal protein/Lactococcus phage ul36.k1t1 | portal_PBSX | S (unclassified) | ABD63728 | 2 × 10−22 |
| 107 | Putative adenine methylase/Enterobacterium phage phiV10 | MT_A70 superfamily | P | YP_512288 | 1 × 10−24 |
Putative virion structural protein genes are marked with asterisks. The families Myoviridae, Podoviridae, and Siphoviridae are denoted by M, P, and S, respectively. DUF550, protein of unknown function; NA, not found.
Comparison of codon usage between VHS1 and Vibrio harveyi ATCC BAA-1116.
Since the full genome sequence of V. harveyi 1114GL is not known, we chose the full sequence of V. harveyi ATCC BA-1116 to make a codon usage comparison. The VHS1 genome had a G+C content of 46.87%, which was ∼1.4% different from that of Vibrio harveyi ATCC BAA-116 (G+C content = 45.44%). Small differences in G+C content of other host-phage pairs have also been reported (30). Despite the small difference in G+C content between the two genomes, the majority of the preferred codons in the VHS1 genome had G or C at the third position (G/C ending), while the majority in the V. harveyi BAA genome had A or T at the third position (A/T ending). In addition, it was found that 8 of 18 amino acids (aa) (44%) (excluding nondegenerate codons for Met and Trp) were different between the two genomes. Although this suggests that VHS1 and its host may have quite different codon usage, it can be argued that codon usage for Vibrio harveyi ATCC BAA-116 and V. harveyi 1114GL may not be similar. To counter this argument, a recent publication suggests that codon usage among Vibrio species is quite similar (33). Since it has been suggested that the similarity of codon usage patterns is relatively high between well-adapted phages and their Vibrio hosts and relatively lower for less-well-adapted phages (3, 30), our analysis suggested that V. harveyi 1114GL might be a relatively recently acquired host of VHS1.
Functional classification of putative ORFs.
Of the total of 125 putative ORFs, only 27 gave significant homology to known protein sequences in GenBank, and 17 of these had homologues in at least one member of the order Caudovirales (E < 10−3) (Table 2; see Table S1 in the supplemental material). Of these 27 ORFs, 12 encoded hypothetical proteins of unknown function. Of the remaining 15, there were 4 encoding putative phage structural proteins, including ORFs 053 (head protein), 061 (phage tail tape measure protein), 091 (terminase), and 104 (portal protein). Four similar proteins have been reported for VHML, another phage of V. harveyi that is virulent for shrimp (22, 24), but they shared no significant homology with their counterparts in VHS1. There were no hits for other structural proteins, such as tail proteins. The DNA polymerase (ORF 103) was identified previously (26) and was used for phylogenetic comparison among phages (25). Other putative proteins associated with nucleic acids were encoded by ORFs 107 (DNA methyltransferase [DAM]), 071 (nucleoside triphosphatase [NTPase]), 074 and 079 (helicases), 077 (DNA primase), and 082 (RecA). The methyltransferase may be associated with phage defense against the host DNA restriction enzyme system (15). A DAM has also been reported for VHML (24), but it was suggested that it might be associated with toxicity of its lysogenized V. harveyi host due to the presence of a unique putative ADP-ribosylating toxin active site in its deduced amino acid sequence. This active site was absent from the deduced amino acid sequence encoded by VHS1 ORF 107, which also showed a low degree of identity (14%) at low coverage (16%) to the much larger VHML protein (359 aa), with a high E value (1.1). In contrast, the VHS1 DAM showed a much higher identity (27 to 30%), at around 80% coverage, with similarly sized DAMs (approximately 200 aa) from phages of enterobacteria (GenBank accession no. ADU03681, YP512288, YP794086, and NP848237), with low E values (1 × 10−24 to 4 ×10−24).
With respect to potential toxin genes in VHS1, a previous report on extracellular toxins produced by VHS1 lysogens (13) described three major protein bands in SDS-PAGE gels containing semipurified toxin fractions. In that publication, mass spectrometry results for these bands gave no significant hits to known proteins or deduced protein sequences in public databases, including those for Vibrio species. Because of this, it was suggested that the toxin(s) probably originated from the VHS1 genome. That suggestion was confirmed here, when the same mass spectra from the earlier study were used to screen deduced proteins from putative ORFs in the VHS1 genome. This gave highly significant matches for two of the protein bands, to ORFs 053 and 058 (Fig. 4), indicating that the proteins in the toxin extract did arise from the bacteriophage genome rather than the genome of its V. harveyi host. Curiously, ORF 053 encodes the putative head protein of VHS1. Although it has long been known in human medicine that toxins and other bacterial virulence factors can be carried by bacteriophages and result in lysogenic conversion of nonpathogenic bacteria into lethal pathogens (4), we know of no phage structural protein that has been reported to be toxic for a vertebrate or invertebrate. Since we believe that ORF 053 encodes the VHS1 head protein, it may be more likely that ORF 058 is a toxin gene. However, until ORFs 053 and 058 are expressed heterologously and tested in bioassays to show that either one or both are lethal for the black tiger shrimp (Penaeus monodon) but not the whiteleg shrimp (Penaeus vannamei), their designation as the gene(s) for the previously described shrimp toxin will remain open to question.
Fig 4.

Details of MASCOT results for mass spectrum data matches to two putative VHS1 ORFs. Peptides from the mass spectrometry analysis that match the deduced amino acid sequence of VHS1 are indicated in bold, gray, underlined type.
Conclusions.
The complete genome of VHS1, the second bacteriophage of Vibrio harveyi, has been sequenced and annotated. This opens the way for functional studies on host-phage interaction and especially on the two phage ORFs that have been identified as potential sources of a toxin(s) associated with hemocyte agglutination and mortality in the giant tiger shrimp but not the whiteleg shrimp. There was no significant sequence similarity at the nucleic acid or amino acid level between VHS1 and VHML (Myoviridae), the only other phage reported so far from V. harveyi isolates that are lethal for shrimp.
Supplementary Material
ACKNOWLEDGMENTS
This work was partially supported by Mahidol University, the Thai National Center for Genetic Engineering and Biotechnology (BIOTEC), and the Center of Excellence on Agricultural Biotechnology, Science and Technology Postgraduate Education and Research Development Office, Office of Higher Education Commission, Ministry of Education (AG-GIO/PERDO-CHE). Krit Khemayan was supported by a Ph.D. scholarship from Alltech Inc., Nicholasville, KY.
We thank the Biostatistics & Informatics Laboratory at the Genome Institute, BIOTEC, for access to their high-performance computing facilities.
Footnotes
Published ahead of print 3 February 2012
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1. Besemer J, Lomsadze A, Borodovsky M. 2001. GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 29:2607–2618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Birney E, Clamp M, Durbin R. 2004. GeneWise and Genomewise. Genome Res. 14:988–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Carbone A. 2008. Codon bias is a major factor explaining phage evolution in translationally biased hosts. J. Mol. Evol. 66:210–223 [DOI] [PubMed] [Google Scholar]
- 4. Cheetham BF, Katz ME. 1995. A role for bacteriophages in the evolution and transfer of bacterial virulence determinants. Mol. Microbiol. 18:201–208 [DOI] [PubMed] [Google Scholar]
- 5. Comeau AM, Chan AM, Suttle CA. 2006. Genetic richness of vibriophages isolated in a coastal environment. Environ. Microbiol. 8:1164–1176 [DOI] [PubMed] [Google Scholar]
- 6. Crothers-Stomps C, Høj L, Bourne DG, Hall MR, Owens L. 2010. Isolation of lytic bacteriophage against Vibrio harveyi. J. Appl. Microbiol. 108:1744–1750 [DOI] [PubMed] [Google Scholar]
- 7. Delcher AL, Harmon D, Kasif S, White O, Salzberg SL. 1999. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 27:4636–4641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Guo F-B, Ou H-Y, Zhang C-T. 2003. ZCURVE: a new system for recognizing protein-coding genes in bacterial and archaeal genomes. Nucleic Acids Res. 31:1780–1789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Harris L, Owens L. 1999. Production of exotoxins by two luminous Vibrio harveyi strains known to be primary pathogens of Penaeus monodon larvae. Dis. Aquat. Organ. 38:11–22 [Google Scholar]
- 10. Hendrix RW, Casjens SR. 2005. Family Podoviridae, p 71–79 In Fauquet CM, Mayo MA, Maniloff J, Desselberger U, Ball LA. (ed), Virus taxonomy. VIIIth report of the ICTV. Elsevier/Academic Press, London, United Kingdom [Google Scholar]
- 11. Hendrix RW, Casjens SR. 2005. Family Siphoviridae, p 57–70 In Fauquet CM, Mayo MA, Maniloff J, Desselberger U, Ball LA. (ed), Virus taxonomy. VIIIth report of the ICTV. Elsevier/Academic Press, London, United Kingdom [Google Scholar]
- 12. Huang X, Madan A. 1999. CAP3: a DNA sequence assembly program. Genome Res. 9:868–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Intaraprasong A, Khemayan K, Pasharawipas T, Flegel TW. 2009. Species-specific virulence of Vibrio harveyi for black tiger shrimp is associated with bacteriophage-mediated hemocyte agglutination. Aquaculture 296:185–192 [Google Scholar]
- 14. Khemayan K, Pasharawipas T, Puiprom O, Sriurairatana S, Flegel TW. 2006. Unstable lysogeny and pseudolysogeny in VHS1 bacteriophage of Vibrio harveyi. Appl. Environ. Microbiol. 72:1355–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Krüger DH, Bickle TA. 1983. Bacteriophage survival: multiple mechanisms for avoiding the deoxyribonucleic acid restriction systems of their hosts. Microbiol. Rev. 47:345–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lagesen K, et al. 2007. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35:3100–3108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Larsen TS, Krogh A. 2003. EasyGene—a prokaryotic gene finder that ranks ORFs by statistical significance. BMC Bioinformatics 4:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lowe TM, Eddy SR. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25:955–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mistry J, Bateman A, Finn RD. 2007. Predicting active site residue annotations in the Pfam database. BMC Bioinformatics 8:298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Munro J, Oakey J, Bromage E, Owens L. 2003. Experimental bacteriophage-mediated virulence in strains of Vibrio harveyi. Dis. Aquat. Organ. 54:187–194 [DOI] [PubMed] [Google Scholar]
- 21. Noguchi H, Park J, Takagi T. 2006. MetaGene: prokaryotic gene finding from environmental genome shotgun sequences. Nucleic Acids Res. 34:5623–5630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Oakey HJ, Cullen WR, Owens L. 2002. The complete nucleotide sequence of the Vibrio harveyi bacteriophage VHML. J. Appl. Microbiol. 93:1089–1098 [DOI] [PubMed] [Google Scholar]
- 23. Oakey HJ, Owens L. 2000. A new bacteriophage, VHML, isolated from a toxin-producing strain of Vibrio harveyi in tropical Australia. J. Appl. Microbiol. 89:702–709 [DOI] [PubMed] [Google Scholar]
- 24. Oakey J, Cullen B, Owens L. 2005. A hypothetical model for VHML phage conversion of Vibrio harveyi, p 457–464 In Walker P, Lester R, Bondad-Reantaso MG. (ed), Diseases in Asian aquaculture, vol. V Fish Health Section, Asian Fisheries Society, Manila, Philippines [Google Scholar]
- 25. Pasharawipas T, Thaikua S, Flegel TW. 2009. Intriguing phylogenetic arrangement of tailed bacteriophages based on putative DNA polymerase sequences. ScienceAsia 35:125–130 [Google Scholar]
- 26. Pasharawipas T, et al. 2005. Partial characterization of a novel bacteriophage of Vibrio harveyi isolated from shrimp culture ponds in Thailand. Virus Res. 114:63–69 [DOI] [PubMed] [Google Scholar]
- 27. Peden J. 1999. CODONW, p 50–102 In Analysis of codon usage. Ph.D. thesis University of Nottingham, Nottingham, United Kingdom [Google Scholar]
- 28. Phumkhachorn P, Rattanachaikunsopon P. 2010. Isolation and partial characterization of a bacteriophage infecting the shrimp pathogen Vibrio harveyi. Afr. J. Microbiol. Res. 4:1794–1800 [Google Scholar]
- 29. Rakonjac J, et al. 2003. Sequence diversity and functional conservation of the origin of replication in lactococcal prolate phages. Appl. Environ. Microbiol. 69:5104–5114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Seguritan V, Feng I-W, Rohwer F, Swift M, Segall AM. 2003. Genome sequences of two closely related Vibrio parahaemolyticus phages, VP16T and VP16C. J. Bacteriol. 185:6434–6447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shivu MM, et al. 2007. Molecular characterization of Vibrio harveyi bacteriophages isolated from aquaculture environments along the coast of India. Environ. Microbiol. 9:322–331 [DOI] [PubMed] [Google Scholar]
- 32. Thiyagarajan S, et al. 2011. Characterization of four lytic transducing bacteriophages of luminescent Vibrio harveyi isolated from shrimp (Penaeus monodon) hatcheries. FEMS Microbiol. Lett. 325:85–91 [DOI] [PubMed] [Google Scholar]
- 33. Thompson CC, et al. 2009. Genomic taxonomy of vibrios. BMC Evol. Biol. 9:258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wright F. 1990. The ‘effective number of codons’ used in a gene. Gene 87:23–29 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.