

Abstract
A copper(II)–hydroperoxo complex, [Cu(Me6-tren)(OOH)]+ (2), and a copper(II)–cumylperoxo complex, [Cu(Me6-tren)(OOC(CH3)2Ph)]+ (3), were synthesized by reacting [Cu(Me6-tren)(CH3CN)]2+ (1) with H2O2 and cumyl-OOH, respectively, in the presence of triethylamine. These intermediates, 2 and 3, were successfully characterized by various physicochemical methods such as UV-vis, ESI-MS, resonance Raman and EPR spectroscopies, leading us to propose structures of the Cu(II)–OOR species with a trigonal-bipyramidal geometry. Density functional theory (DFT) calculations provided geometric and electronic configurations of 2 and 3, showing trigonal bipyramidal copper(II)–OOR geometries. These copper(II)–hydroperoxo and –cumylperoxo complexes were inactive in electrophilic and nucleophilic oxidation reactions.
Introduction
The generation and study of metal–peroxo complexes has a considerable history, as the employment of such species is of interest to a number of disciplines, including organic oxidation processes occurring in chemical or biochemical settings.1–3 The latter field captures the interest of a very large (bio)chemical research community, since metalloenzymes, especially those containing heme, non-heme iron and copper, bind molecular oxygen and then utilize metal–peroxo and metal–hydroperoxo species (or species derived from them) to effect biochemical substrate oxidations.1,4–7
Our own interests include coordination chemistry studies on copper–dioxygen complexes and derived species, with the primary goal of establishing fundamental aspects of adduct formation, structure–spectroscopic correlations and substrate–reactivity patterns.8–11 The studies are inspired by the diverse array of reactions mediated at protein active sites, reversible binding of O2, insertion of O-atom(s) into substrates by oxygenases, and dehydrogenation reactions mediated by copper oxidases.12 Protein active-site environments may possess one, two or three proximate copper ions.12–16
Copper–peroxo species are now well established to occur in the blood O2-carrier proteins hemocyanins, which bind O2 via formation of binuclear CuII–(O22−)–CuII complexes, as do monooxygenase tyrosinases.13,14,17 A copper–peroxo species is also strongly implicated at the trinuclear active center in ‘blue’ multicopper oxidases, with a CuICuII2–(O22−) formulation18 as supported by crystallographic studies.19 Protonation to give a hydroperoxo (−OOH) intermediate, or proton-induced peroxo complex reduction to water, follows.18,20
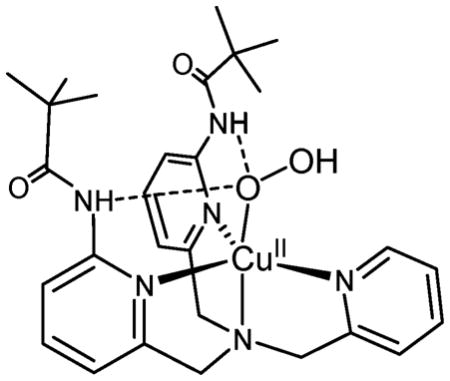
As implied, all oxygenases and oxidases utilize molecular oxygen; thus, O2-derived (i.e., reduced) active species must begin by reaction with a copper(I) center to produce a cupric–superoxo (i.e., CuII–O2•−) initial adduct (Scheme 1).8,21,22 Significantly, only one example of a putative protein CuII–O2•− species is known (see below).23 In fact, advances in synthetic model chemistry have provided considerable information, in that there are now well characterized O2-adducts or derived species representing both mono- (Scheme 1) and dicopper entities. For the former case, both end-on η1-(O2•−) copper(II) complexes21,22 and a side-on η2-(O2•−) copper(II) complex24 have been structurally and spectroscopically characterized. With a strongly donating anionic ligand for copper; Tolman and coworkers25 described a side-on η2-(O22−) copper(III) complex (Scheme 1). Mononuclear CuII–hydroperoxo complexes have been studied;11,26–36 they are formed either from addition of H2O2/base to a precursor copper(II) complex, or in one instance, via reaction of a ligand–CuII–(O2•−) complex with a phenol as an electron/proton donor.35 Masuda and coworkers26 were able to obtain an X-ray structure employing a tripodal tetradentate ligand which also carries groups capable of H-bonding to the coordinated −OOH ligand (see the structure below).
Scheme 1.

Mononuclear copper–O2 adducts and derived species of interest.
Tripodal tetradentate ligands have been very popular in studying dioxygen reactivity by ligand–copper(I) complexes. These include tris(2-pyridylmethyl)amine (TMPA) and analogues or derivatives.9,30 This is also true for the classic tripodal tren ligand, tris(2-aminoethyl)amine; tren derivatives and copper–dioxygen chemistry have been especially studied by the research groups of Schindler21,37–39 and Suzuki,40 providing extensive insights into ligand–CuI/O2 reactivity and kinetics, as well as formation of CuII–(O2•−) species (with an as-mentioned X-ray structure),21 and dicopper(II)–peroxo species, the latter also with an X-ray structure known from Suzuki.40
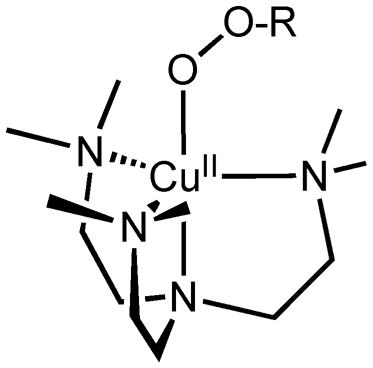
Notably, copper–hydroperoxo or –alkylperoxo species generated using tren derivatives do not appear in the literature. Thus, in this report, we have added to the base of compounds studied for this category of compounds. Using Me6-tren (tris(N,N′-dimethylaminoethyl)amine), we describe the synthesis and detailed characterization of a hydroperoxo and cumylperoxo complex with Me6-tren (see the structure below).
In fact, there is currently an increase of research effort in the community to elucidate reactivity trends for CuII–(OOH) vs. CuII–(O2•−) species. This is due to the considerable literature discussion35,41,42 concerning which entity is responsible for initial hydrogen-atom (H-atom) abstraction reaction occurring in the closely related copper enzymes dopamine β-monooxygenase (DβM) and peptidylglycine α-hydroxylating monooxygenase (PHM). These possess well-separated (~11 Å) copper ions; however, biochemical studies indicate the active site is at one of the copper ions (the so-called CuM site, possessing His2Met N2S ligation).43 An X-ray structure of a PHM derivative displays a presumed CuII–(O2•−) entity with end-on η1-coordination.23 Such a cupric superoxo complex capable of effecting an enzymatic substrate hydroxylation via initial H-atom abstraction has drawn experimental43 and theoretical44 support. Yet, in synthetic chemistry there is so far only one example45 where a CuII–(O2•−) entity is shown to be capable of H-atom abstraction. A CuII–OOH species would derive from an electron–proton downstream reaction, and some of us have published some examples where copper–hydroperoxo complexes appear to initiate H-atom abstraction and oxygen atom transfer reactions.32,35,36,46 In addition, studies of CuII–OOR (R = acyl, alkyl) species and their reactivity have contributed to our basic understanding in the field.47–51 From a CuII–OOR species, a thus far highly elusive high-valent copper–oxo species (“cupryl”, Scheme 1) may form via O–O cleavage chemistry, and this is likely a relevant species somewhere on the path of CuI/O2 reaction to species involved in enzyme-like C–H substrate oxidation chemistry.10,36,41,51–54 Thus, extensive investigations to fully elucidate the chemistry of CuII–(O2•−) and CuII–OOR moieties are required.
Results and discussion
Copper(II) precursor complex
The starting material, [Cu(Me6-tren)(CH3CN)](ClO4)2 (1-(ClO4)2), was prepared by reacting Cu(ClO4)2·6H2O and Me6-tren in CH3CN. The UV-visible spectrum of 1 in CH3CN exhibits one broad band at ~850 nm (ε = 500 M−1 cm−1) (Fig. 2a). The electrospray ionization mass spectrum (ESI-MS) of 1 shows intense isotope envelopes at a mass-to-charge (m/z) of 392.1 for [Cu(Me6-tren)(ClO4)]+ (see Fig. S1†). After numerous attempts to determine the X-ray structure of the starting material, suitable crystals were obtained by the addition of NaBPh4 to the solution of 1-(ClO4)2.
Fig. 2.

(a) UV-vis spectra of 1 (0.4 mM) (black line) and 2 (0.4 mM) (red line) in CH3CN/CH3OH (1:1) at 0 °C. (b) ESI-MS of 2-18O. Inset shows isotope distribution patterns for 2-18O (blue line) and 2-16O (red line). (c) Resonance Raman spectra of 2 (4 mM) obtained upon excitation at 407 nm in CH3CN/CH3OH (1:1) at −20 °C. Red/blue lines: samples prepared with H216O2/H218O2; green line: difference spectrum, (16O–18O). The peak marked with “s” is ascribed to the solvents. (d) X-band EPR spectrum of 2 recorded in CH3CN/CH3OH (1:1) at 4.3 K. Spectroscopic settings: frequency = 9.10 GHz, microwave power = 1 mW, modulation frequency = 100 kHz.
The crystal structure of the cation part of 1-(BPh4)2 is shown in Fig. 1, and selected bond distances and angles are listed in Table 1. Complex 1 has a five-coordinated Cu(II) ion with four nitrogens of the Me6-tren ligand and one nitrogen of CH3CN coordinating to the free axial position. The Cu(II) geometry is best described as trigonal bipyramidal (τ = 0.95), similar to that found in [Cu(Me6-tren)(H2O)]2+.55,56
Fig. 1.
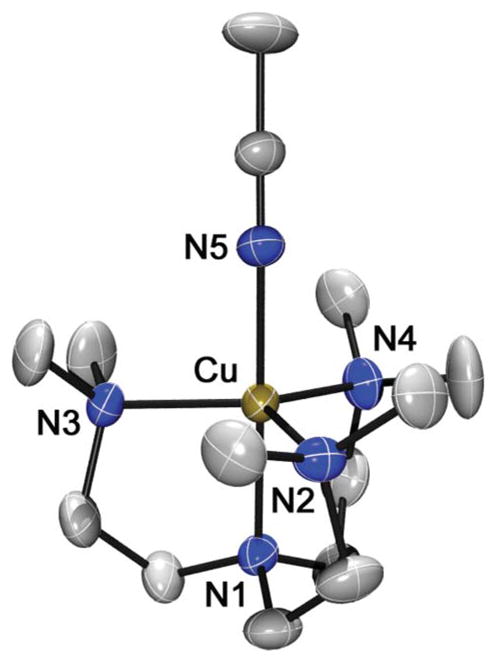
X-ray structure of the [Cu(Me6 -tren)(CH3CN)]2+ cation in 1-(BPh4)2 showing 30% probability thermal ellipsoids. Hydrogen atoms are omitted for clarity.
Table 1.
Selected bond distances (Å) and angles (°) from the obtained crystal structure and the DFT calculations as described in the text
| Exp. | Calcd | Exp. | Calcd | ||
|---|---|---|---|---|---|
| Cu–N1 | 1.995(2) | 2.07 | Cu–N2 | 2.128(2) | 2.21 |
| Cu–N3 | 2.124(2) | 2.22 | Cu–N4 | 2.152(2) | 2.23 |
| Cu–N5 | 1.968(2) | 2.05 | |||
| N1–Cu–N2 | 86.35(9) | 85.24 | N1–Cu–N3 | 85.36(9) | 85.09 |
| N1–Cu–N4 | 85.89(10) | 85.02 | N1–Cu–N5 | 178.22(11) | 179.62 |
| N2–Cu–N3 | 121.25(10) | 119.61 | N2–Cu–N4 | 117.40(10) | 119.42 |
| N2–Cu–N5 | 95.43(10) | 95.13 | N3–Cu–N4 | 119.81(10) | 118.83 |
| N3–Cu–N5 | 93.71(9) | 94.80 | N4–Cu–N5 | 93.27(10) | 94.72 |
Copper(II)–hydroperoxo complex and characterization
The copper(II)–hydroperoxo complex, [Cu(Me6-tren)(OOH)]+ (2), was prepared by adding 10 equiv of H2O2 to a reaction solution containing 1 in the presence of 2 equiv triethylamine (TEA) in a solvent mixture of CH3CN and CH3OH (1:1) at 0 °C; the color of the solution changed from blue to green (Scheme 2). A UV-vis spectrum of 2 shows an intense band at 375 nm (ε = 1250 M−1 cm−1) and two weak bands at 680 (ε = 220 M−1 cm−1) and 840 nm (ε = 270 M−1 cm−1) (Fig. 2a), which is similar to those of the previously reported copper(II)–hydroperoxo complexes.26,29,57,58 The former band has been assigned as the LMCT band of the CuII–OOH species, and the latter two bands have been assigned as the d–d transition bands of the Cu(II) ion with a trigonal-bipyramidal geometry.29
Scheme 2.

The ESI-MS of 2 exhibits a prominent ion peak at m/z of 326.1 (Fig. 2b), whose mass and isotope distribution pattern correspond to [Cu(Me6-tren)(OOH)]+ (2-16O) (calculated m/z of 326.2). When the reaction was carried out with isotopically labeled H218O2, a mass peak corresponding to [Cu(Me6-tren)(18O18OH)]+ (2-18O) appeared at m/z of 330.1 (calculated m/z of 330.2) (Fig. 2b). The shift in four mass units on substitution of 16O with 18O proves that 2 contains an O2 unit.
The resonance Raman spectrum of 2 was collected using 407 nm excitation in a solvent mixture of CH3CN and CH3OH (1:1) at − 20 °C. 2 prepared with H216O2 exhibits two isotopically sensitive bands at 846 and 509 cm−1 that shift to 798 and 484 cm−1, respectively, in samples of 2 prepared with H218O2 (Fig. 2c). The higher-energy feature (846 cm−1) with 16Δ – 18Δ value of 48 cm−1 (16Δ – 18Δ (calcd) = 48 cm−1) is ascribed to the O–O stretching vibration of the hydroperoxo ligand, and the lower-energy feature (509 cm−1) is assigned to the Cu–O stretching vibration (16Δ – 18Δ = 25 cm−1; 16Δ – 18Δ (calcd) = 23 cm−1).
The electron paramagnetic resonance (EPR) spectrum of a frozen solution of 2 measured at 4.3 K is typical of trigonal-bipyramidal Cu(II) complexes, having a “reverse” axial appearance with g⊥ > g|| ~ 2.0 and A⊥ = 60–100 × 10−4 cm−1 (Fig. 2d).59–63 The g⊥ (2.20), g|| (2.00), A⊥ (87 G), and A|| (83 G) values are comparable to those of the previously reported Cu(II) complexes with a trigonal-bipyramidal geometry.
Copper(II)–cumylperoxo complex and characterization
The copper(II)–cumylperoxo complex, [Cu(Me6-tren)(OOC-(CH3)2Ph)]+ (3), was generated by adding 5 equiv of cumene hydroperoxide (cumyl-OOH) to the solution of 1 in the presence of 2 equiv triethylamine in CH3CN at 25 °C; the color of the solution changed from blue to green (Scheme 2). Spectroscopic properties of 3 are similar to those of 2. The UV-visible spectrum of 3 in CH3CN at 25 °C exhibits weak bands at 440 (ε = 280 M−1 cm−1), 680 (ε = 250 M−1 cm−1) and 787 nm (ε = 300 M−1 cm−1) (Fig. 3a).
Fig. 3.

(a) UV-vis spectrum of 3 (2 mM) in CH3CN at 25 °C. (b) ESI-MS of 3-16O. Inset shows isotope distribution patterns for 3-16O (red line) and 3-18O (blue line). Isotopically labeled cumyl-18O18OH (70% 18O-enriched) was used for 3-18O. (c) Resonance Raman spectra of 3 (16 mM) obtained upon excitation at 442 nm in CH3CN at −20 °C. Red/blue lines were obtained with samples prepared with cumyl-16O16OH/Cumyl-18O18OH (90% 18O-enriched); green line: difference spectrum, (16O – 18O). The peaks marked with “s” are ascribed to the solvent. (d) X-band EPR spectrum of 3 recorded in CH3CN at 4.3 K. Spectroscopic settings: frequency = 9.10 GHz, microwave power = 1 mW, modulation frequency = 100 kHz.
The ESI-MS of 3 in CH3CN at 25 °C shows a prominent ion peak at m/z of 444.0 (Fig. 3b), whose mass and isotope distribution pattern correspond to [Cu(Me6-tren)(OOC(CH3)2Ph)]+ (3-16O) (calculated m/z of 444.2). When the reaction was carried out with isotopically labeled cumyl-18O18OH, a mass peak corresponding to [Cu(Me6-tren)(18O18OC(CH3)2Ph)]+ (3-18O) appeared at m/z of 448.0 (calculated m/z of 448.2) (Fig. 3b, inset). The four mass unit increase upon substitution of 16O with 18O indicates that 3 contains an O2 unit.
The resonance Raman spectrum of 3 was collected using 442 nm excitation in CH3CN at −20 °C. 3 prepared with cumyl-OOH exhibits two isotopically sensitive bands at 887 and 839 cm−1, which shift to 805 cm−1 in samples of 3 prepared with cumyl-18O18OH (Fig. 3c). Such a band shift has been observed in copper(II)–alkylperoxo and iron(II)–alkylperoxo complexes,51,64 and these bands have been assigned to the ν(O–O) mode.
The EPR spectrum of a frozen CH3CN solution of 3 measured at 4.3 K shows a “reverse” axial appearance as shown in 2, suggesting a trigonal-bipyramidal geometry for the Cu(II) ion. The g⊥ (2.19), g|| (2.00), A⊥ (77 G), and A|| (87 G) values are comparable to those of 2.
Density Functional Theory results
In order to gauge the reliability of the calculations, structural comparisons were made between the crystal structure of 1 and that calculated by density functional theory (DFT).65 The root mean square (RMS) deviation was calculated to be 0.13 Å (geometries are given in Table 1, Fig. 4 and Fig. S2†). Since some differences are expected when comparing gas-phase calculated structures with crystal forms, the structural quality of the calculations was deemed acceptable, and gave credibility to the subsequent calculations where no crystal structures are available. The Cu–peroxide geometries obtained are, as expected, of the end-on η1 type with the metal ligand atoms in a trigonal bipyramidal fashion, with τ = 0.85 for both 2 and 3 (Fig. 4), in agreement with the EPR spectroscopic results described above. For 3, there are three possible isomers of the structure, where the phenyl ring position can be interchanged with any of the two cumene methyl positions. However, the isomers are virtually degenerate in energy, with very little differences in the core geometrical and spin parameters (hence, we only present one of them here, 3a, and defer the other two to ESI). The O–O bond is calculated to be 1.52 Å in both 2 and 3a. This may be compared to that observed for Masuda’s copper(II)–hydroperoxo complex (vide supra) (1.460 Å)26 and a structurally characterized Fujisawa/Kitajima copper(II)–cumylperoxo complex with pyrazolylborate ligand (1.460 Å for both structures).47,49 It is also in line with what is found in calculations of protonated Compound 0 in P450 system models.66
Fig. 4.

DFT calculated structures for 2 (top) and one of the isomers of 3a (bottom) with the bond distances shown for the Cu–O, O–O, Cu–Nax and (largest) Cu–Neq bonds (gray, C; white, H; red, O; blue, N; yellow, Cu).
Frequency calculations were done to check that the optimized structures were indeed in a ground state. These calculations, however, also produce Raman frequencies that can be compared to the experimentally observed ones. The nearest calculated frequencies to the experimentally determined 846 and 509 cm−1 for 2 (vide supra) were found to be at 854 and 489 cm−1, indeed corresponding to the O–O and O–Cu bond stretching frequencies, respectively. For 3, experiments found two bands at 887 and 839 cm−1, that shift to 805 cm−1 when using 18O. The calculations indeed found a frequency at 845 cm−1 in 3a (the isomer shown on Fig. 4) that corresponds to the O–O bond stretching, while in the other two isomers, this frequency has shifted to 857 and 855 cm−1. No other frequencies near 887 cm−1 exhibited vibrations involving the oxygens. Hence, recognizing the crude level of calculations for spectroscopic assignments, we tentatively suggest that the experimentally observed two peaks are due to the existence of structural isomers, which collapses to one peak when the heavier (and less sensitive) 18O is used. Further, more specialized, calculations may be necessary to identify the origin of the two bands more accurately.
Spin density distribution calculations show that the Cu ion retains about half a radical in the state, while the rest is distributed between the coordinating ligand atoms (Table 2). This is indicative of an unpaired electron occupying the σ*z2 orbital, which is mainly distributed along the axial direction, as visualized in Fig. 5.
Table 2.
Mulliken spin density distribution
| Complex | Cu | Oinner | Oouter | 4 × N | Rest |
|---|---|---|---|---|---|
| 1 | 0.53 | — | — | 0.43 | 0.05 |
| 2 | 0.51 | 0.25 | 0.00 | 0.24 | 0.00 |
| 3a | 0.51 | 0.26 | −0.01 | 0.23 | 0.01 |
Fig. 5.

The singly occupied molecular orbital (SOMO) for 2 (and also for 3) is a σ*z2 orbital.
Reactivity
The reactivity of 2 and 3 was investigated with respective to potential electrophilic and nucleophilic reactions. First, the electrophilic character of 2 and 3 was tested in the oxidation of thioanisole and cyclohexene. Upon addition of substrates to a solution of 2 in a solvent mixture of CH3CN and CH3OH (1:1) at 0 °C and 3 in CH3CN at 25 °C, 2 and 3 remained intact without showing any absorption spectral changes. The nucleophilic character of 2 and 3 was also investigated in aldehyde deformylation with cyclohexane carboxaldehyde and 2-phenylpropionaldehyde, and no spectral changes were observed. Further, product analysis of the reaction solutions revealed that no oxidized products were formed in these reactions. These results demonstrate that 2 and 3 are not capable of conducting electrophilic and nucleophilic oxidation reactions under the reaction conditions.
Conclusions
A copper(II)–hydroperoxo complex, [Cu(Me6-tren)(OOH)]+ (2), and a copper(II)–cumylperoxo complex, [Cu(Me6-tren)(OOC(CH3)2Ph)]+ (3), were generated and characterized by various spectroscopic methods. DFT calculations successfully reproduced the structure of the obtained crystal structure of [Cu(Me6-tren)(CH3CN)]2+ (1), lending credibility to the calculated structure of 2 and 3. Calculated Raman frequencies also show good agreement with experiments. The intermediates, 2 and 3, were inactive in electrophilic and nucleophilic oxidation reactions. These findings from the combined experimental and theoretical studies provide important fundamental information in the understanding of the nature of intermediates possibly involved in O2-activating Cu enzymes such as DβM and PHM.
Experimental
General
All chemicals obtained from Aldrich Chemical Co. were the best available purity and used without further purification unless otherwise indicated. Solvents were dried according to published procedures and distilled under Ar prior to use.67 H218O2 (90% 18O-enriched, 2% H218O2 in water) and 18O2 (95% 18O-enriched) were purchased from ICON Services Inc. (Summit, NJ, USA). 18O-Labeled cumene hydroperoxide (70% and 90% 18O-enriched) was prepared by the reported method68 and used in ESI-MS (70% 18O-enriched) and resonance Raman (90% 18O-enriched) studies. Me6-tren (tris(N,N′-dimethylaminoethyl)amine) was prepared according to a published procedure.69 [Cu(Me6-tren)(CH3CN)](ClO4)2 was prepared by reacting Cu(ClO4)2·6H2O (0.19 g, 0.50 mmol) with Me6-tren (0.12 g, 0.50 mmol) in CH3CN (3 mL). The mixture was stirred for several hours, and then Et2O (20 mL) was added to the resulting solution to yield a blue powder, which was collected by filtration, washed with Et2O, and dried in vacuo. Yield: 0.17 g (63%). UV-vis (λmax (nm) [ε (M−1 cm−1)]) in CH3CN: ~850 (500). ESI-MS in CH3CN: m/z 167.1 for [Cu(Me6-tren)(CH3CN)]2+ and m/z 392.1 for [Cu(Me6-tren)(ClO4)]+. The BPh4 salt of this complex was obtained by dissolution of this solid in CH3CN and addition of 4 equiv of NaBPh4 in CH3CN. Vapor diffusion of Et2O into this solution yielded blue crystals suitable for X-ray diffraction. Caution: Perchlorate salts are potentially explosive and should be handled with care!
UV-vis spectra were recorded on a Hewlett Packard 8453 diode array spectrophotometer equipped with a UNISOKU Scientific Instruments for low-temperature experiments or with a circulating water bath. Electrospray ionization mass spectra (ESI-MS) were collected on a Thermo Finnigan (San Jose, CA, USA) LCQ™ Advantage MAX quadrupole ion trap instrument, by infusing samples directly into the source using a manual method. The spray voltage was set at 4.2 kV and the capillary temperature at 80 °C. Resonance Raman spectra were obtained using a liquid nitrogen cooled CCD detector (CCD-1024 × 256-OPEN-1LS, HORIBA Jobin Yvon) attached to a 1-m single polychromator (MC-100DG, Ritsu Oyo Kogaku) with a holographic grating (1200 grooves mm−1). Excitation wavelengths of 406.7 nm and 441.6 nm were provided by a Kr+ laser (Spectra Physics, BeamLok 2060) and a He–Cd laser (Kimmon Koha, IK5651R-G and KR1801C), respectively, with 15 mW power at the sample point. All measurements were carried out with a spinning cell (1000 rpm) at −20 °C. Raman shifts were calibrated with indene, and the accuracy of the peak positions of the Raman bands was ±1 cm−1. Product analysis was performed with an Agilent Technologies 6890 N gas chromatograph (GC) and Thermo Finnigan (Austin, Texas, USA) FOCUS DSQ (dual stage quadrupole) mass spectrometer interfaced with Finnigan FOCUS gas chromatograph (GC-MS). EPR spectra were taken at 4 K using a X-band Bruker EMX-plus spectrometer equipped with a dual-mode cavity (ER 4116DM). Low temperature was achieved and controlled with an Oxford Instruments ESR900 liquid He quartz cryostat with an Oxford Instruments ITC503 temperature and gas flow controller.
Generation and characterization of [Cu(Me6-tren)(OOH)]+ (2)
Treatment of [Cu(Me6-tren)(CH3CN)](ClO4)2 (0.4 mM) with 10 equiv H2O2 in the presence of 2 equiv triethylamine (TEA) in CH3CN/CH3OH (1:1) at 0 °C afforded a green solution. [Cu(Me6-tren)(18O18OH)]+ was prepared by adding 10 equiv H218O2 (6 μL, 90% 18O-enriched, 2% H218O2 in water) to a solution containing [Cu(Me6-tren)(CH3CN)](ClO4)2 (0.4 mM) and 2 equiv TEA in CH3CN/CH3OH (1:1) at 0 °C. UV-vis (λmax (nm) [ε (M−1 cm−1)]) in CH3CN: 375 (1250), 680 (220), 840 (270). ESI-MS in CH3CN/CH3OH (1:1) at 0 °C: m/z 326.1 for [Cu(Me6-tren)(OOH)]+.
Generation and characterization of [Cu(Me6-tren)(OOC(CH3)2Ph)]+ (3)
Treatment of [Cu(Me6-tren)(CH3CN)](ClO4)2 (2 mM) with 5 equiv cumene hydroperoxide (cumyl-OOH) in the presence of 2 equiv TEA in CH3CN afforded a green solution. [Cu(Me6-tren)(18O18OC(CH3)2Ph)]+ was prepared by adding 5 equiv cumyl-18O18OH to a solution containing [Cu(Me6-tren)(CH3CN)](ClO4)2 (0.4 mM) and 2 equiv TEA in CH3CN at ambient temperature. UV-vis (λmax (nm) [ε (M−1 cm−1)]) in CH3CN: 440 (280), 680 (250), 737 (300). ESI-MS in CH3CN at 25 °C: m/z 444.0 for [Cu(Me6-tren)(OOC(CH3)2Ph)]+.
X-ray crystallography
A single crystal of [Cu(Me6-tren)(CH3CN)](BPh4)2 (1-(BPh4)2) was picked from solutions by a nylon loop (Hampton Research Co.) on a hand-made copper plate mounted inside a liquid N2 Dewar vessel at ca. −40 °C and mounted on a goniometer head in a N2 cryostream. Data collections were carried out on a Bruker SMART APEX CCD equipped with a monochromator in the Mo Kα (λ = 0.71073 Å) incident beam. The CCD data were integrated and scaled using the Bruker-SAINT software package, and the structure was solved and refined using SHEXTL V 6.12.70 Hydrogen atoms were located in the calculated positions. Crystal data for 1-(BPh4)2: C62H73B2N5Cu, triclinic, P1̄, Z = 2, a = 11.9513(17), b = 12.3535(18), c = 8.160(3) Å, α = 90.838(3), β = 91.333(3), γ = 90.275(3), V = 2680.1(7) Å3, μ = 0.451 mm−1, ρcalcd = 1.206 g cm−3, R1 = 0.0440, wR2 = 0.0871 for 10 297 unique reflections, 630 variables. The crystallographic data for 1-(BPh4)2 are listed in Table 3, and Table 1 lists the selected bond distances and angles.
Table 3.
Crystal data and structure refinements for 1-(BPh4)2
| 1-(BPh4)2 | |
|---|---|
| Empirical formula | C62 H73 B2 CuN5 |
| Formular weight | 973.41 |
| Temperature (K) | 298(2) |
| Wavelength (Å) | 0.71073 |
| Crystal system, space group | Triclinic, P1̄ |
| a (Å) | 11.9513(17) |
| b (Å) | 12.3535(18) |
| c (Å) | 18.160(3) |
| α (°) | 90.838(3) |
| β (°) | 91.333(3) |
| γ (°) | 90.275(3) |
| Volume (Å3) | 2680.1(7) |
| Z | 2 |
| Calculated density (g cm−3) | 1.206 |
| Absorption coefficient (mm−1) | 0.451 |
| Reflections collected | 15 072 |
| Independent reflections [R(int)] | 10 297 [0.0367] |
| Refinement method | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 10297/0/638 |
| Goodness-of-fit on F2 | 0.735 |
| Final R indices [I > 2σ(I)] | R1 = 0.0440, wR2 = 0.0803 |
| Largest difference peak and hole (e Å−3) | 0.269 and −0.341 |
Computational details
The DFT calculations were done using Jaguar 7.771 at unrestricted B3LYP level72–76 using LACVP basis set71,77 as defined in Jaguar. This basis set is of double-ζ quality (6-31G) with an effective core potential on the transition metals, and is widely used for geometry optimizations. An extension of this basis set at triple-ζ quality with polarization and diffuse functions (LACV3P*+)71 was used for single-point energy calculation to compare the different isomers of 3. Due to their computationally demanding nature, the frequency calculations were done on the LACVP level. A scaling factor of 0.9613 for the calculated vibration frequencies is sometimes recommended for B3LYP due to systematic overestimation originating from inaccurately calculated electron correlation interactions as well as the harmonic approximation.78 However, this scaling factor is based on calculations with a slightly different basis set (6-31G(d)), and in our systems, the scaling in fact gave worse agreement with the experimental results. The calculations were done in the gas-phase, and the validity of the chosen method for structural calculations was confirmed by calculating 1, with total charge/mutiplicity set to +2/2 respectively, and compare it to the crystal structure as described in the main text. Using the same method, the structures of 2 and 3 (both +1/2) were subsequently calculated.
Reactivity
All reactions were run in a 1-cm UV cuvette by monitoring UV-vis spectral changes of reaction solutions, and rate constants were determined by fitting the changes in absorbance at 375 nm for 2 and 440 nm for 3. Reactions were run at least in triplicate, and the data reported represent the average of these reactions. The purity of substrates was checked with GC and GC–MS prior to use. After completion of the reactions, products were analyzed by injecting reaction solutions directly into GC and GC–MS. Products were identified by comparing with authentic samples, and product yields were determined by comparison against standard curves prepared with authentic samples and using decane as an internal standard.
Supplementary Material
Acknowledgments
The research was supported by NRF/MEST of Korea through CRI (W.N.) and WCU (R31-2008-000-10010-0) (W.N. and K.D.K.) Programs, the Ministry of Education, Culture, Sports, Science and Technology of Japan through the Global COE program and Priority Area (No. 20050029) (T.O.), NIH grant GM-28962 (K.D.K.), and NRF (NRF-2010-1054-1-2) (J.C.).
Footnotes
Electronic supplementary information (ESI) available: ESI-MS of 1 and xyz coordinates, selected geometrical parameters and overlay figure from the computational calculations. CCDC reference number 775689. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0dt01036g
Contributor Information
Kenneth D. Karlin, Email: karlin@jhu.edu.
Jaeheung Cho, Email: jaeheung@ewha.ac.kr.
Wonwoo Nam, Email: wwnam@ewha.ac.kr.
References
- 1.Sheldon RA, Kochi JK. Metal-Catalyzed Oxidations of Organic Compounds. Academic Press; New York: 1981. [Google Scholar]
- 2.Mimoun H. In: The Chemistry of Functional Groups Peroxides. Patai S, editor. Wiley; New York: 1983. pp. 463–482. [Google Scholar]
- 3.Martell AE, Sawyer DT. Oxygen Complexes and Oxygen Activation by Transition Metals. Plenum; New York: 1988. [Google Scholar]
- 4.Spiro TG. Metal ion activation of dioxygen: Metal ions in biology. Wiley-interscience; New York: 1981. [Google Scholar]
- 5.Holm RH, Solomon EI. Chem Rev. 2004;104:347–348. doi: 10.1021/cr0206364. and review articles in the special issue. [DOI] [PubMed] [Google Scholar]
- 6.Nam W. Acc Chem Res. 2007;40:465. doi: 10.1021/ar700027f. and review articles in the special issue. [DOI] [PubMed] [Google Scholar]
- 7.Karlin KD. Nature. 2010;463:168–169. doi: 10.1038/463168a. [DOI] [PubMed] [Google Scholar]
- 8.Karlin KD, Kaderli S, Zuberbühler AD. Acc Chem Res. 1997;30:139–147. [Google Scholar]
- 9.Hatcher LQ, Karlin KD. Adv Inorg Chem. 2006;58:131–184. [Google Scholar]
- 10.Himes RA, Karlin KD. Curr Opin Chem Biol. 2009;13:119–131. doi: 10.1016/j.cbpa.2009.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kamachi T, Lee YM, Nishimi T, Cho J, Yoshizawa K, Nam W. J Phys Chem A. 2008;112:13102–13108. doi: 10.1021/jp804804j. [DOI] [PubMed] [Google Scholar]
- 12.Solomon EI, Sundaram UM, Machonkin TE. Chem Rev. 1996;96:2563–2605. doi: 10.1021/cr950046o. [DOI] [PubMed] [Google Scholar]
- 13.Solomon EI, Chen P, Metz M, Lee SK, Palmer AE. Angew Chem, Int Ed. 2001;40:4570–4590. doi: 10.1002/1521-3773(20011217)40:24<4570::aid-anie4570>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 14.Mirica LM, Ottenwaelder X, Stack TDP. Chem Rev. 2004;104:1013–1045. doi: 10.1021/cr020632z. [DOI] [PubMed] [Google Scholar]
- 15.Rosenzweig AC, Sazinsky MH. Curr Opin Struct Biol. 2006;16:729–735. doi: 10.1016/j.sbi.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 16.Kosman DJ. JBIC, J Biol Inorg Chem. 2010;15:15–28. doi: 10.1007/s00775-009-0590-9. [DOI] [PubMed] [Google Scholar]
- 17.Matoba Y, Kumagai T, Yamamoto A, Yoshitsu H, Sugiyama M. J Biol Chem. 2006;281:8981–8990. doi: 10.1074/jbc.M509785200. [DOI] [PubMed] [Google Scholar]
- 18.Solomon EI, Augustine AJ, Yoon J. Dalton Trans. 2008:3921–3932. doi: 10.1039/b800799c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bento I, Martins LO, Lopes GG, Carrondo MA, Lindley PF. Dalton Trans. 2005:3507–3513. doi: 10.1039/b504806k. [DOI] [PubMed] [Google Scholar]
- 20.Chen Z, Durao P, Silva CS, Pereira MM, Todorovic S, Hildebrandt P, Bento I, Lindley PF, Martins LO. Dalton Trans. 2010;39:2875–2882. doi: 10.1039/b922734b. [DOI] [PubMed] [Google Scholar]
- 21.Würtele C, Gaoutchenova E, Harms K, Holthausen MC, Sundermeyer J, Schindler S. Angew Chem, Int Ed. 2006;45:3867–3869. doi: 10.1002/anie.200600351. [DOI] [PubMed] [Google Scholar]
- 22.Maiti D, Fry HC, Woertink JS, Vance MA, Solomon EI, Karlin KD. J Am Chem Soc. 2007;129:264–265. doi: 10.1021/ja067411l. [DOI] [PubMed] [Google Scholar]
- 23.Prigge ST, Eipper BA, Mains RE, Amzel LM. Science. 2004;304:864–867. doi: 10.1126/science.1094583. [DOI] [PubMed] [Google Scholar]
- 24.Chen P, Root DE, Campochiaro C, Fujisawa K, Solomon EI. J Am Chem Soc. 2003;125:466–474. doi: 10.1021/ja020969i. [DOI] [PubMed] [Google Scholar]
- 25.Aboelella NW, Lewis EA, Reynolds AM, Brennessel WW, Cramer CJ, Tolman WB. J Am Chem Soc. 2002;124:10660–10661. doi: 10.1021/ja027164v. [DOI] [PubMed] [Google Scholar]
- 26.Wada A, Harata M, Hasegawa K, Jitsukawa K, Masuda H, Mukai M, Kitagawa T, Einaga H. Angew Chem, Int Ed. 1998;37:798–799. doi: 10.1002/(SICI)1521-3773(19980403)37:6<798::AID-ANIE798>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 27.Ohta T, Tachiyama T, Yoshizawa K, Yamabe T, Uchida T, Kitagawa T. Inorg Chem. 2000;39:4358–4369. doi: 10.1021/ic000018a. [DOI] [PubMed] [Google Scholar]
- 28.Kodera M, Kita T, Miura I, Nakayama N, Kawata T, Kano K, Hirota S. J Am Chem Soc. 2001;123:7715–7716. doi: 10.1021/ja010689n. [DOI] [PubMed] [Google Scholar]
- 29.Fujii T, Yamaguchi S, Funahashi Y, Ozawa T, Tosha T, Kitagawa T, Masuda H. Chem Commun. 2006:4428–4430. doi: 10.1039/b609673e. [DOI] [PubMed] [Google Scholar]
- 30.Yamaguchi S, Masuda H. Sci Technol Adv Mater. 2005;6:34–47. [Google Scholar]
- 31.Maiti D, Lucas HR, Sarjeant AAN, Karlin KD. J Am Chem Soc. 2007;129:6998–6999. doi: 10.1021/ja071704c. [DOI] [PubMed] [Google Scholar]
- 32.Maiti D, Sarjeant AAN, Karlin KD. J Am Chem Soc. 2007;129:6720–6721. doi: 10.1021/ja0719024. [DOI] [PubMed] [Google Scholar]
- 33.Kunishita A, Scanlon JD, Ishimaru H, Honda K, Ogura T, Suzuki M, Cramer CJ, Itoh S. Inorg Chem. 2008;47:8222–8232. doi: 10.1021/ic800845h. [DOI] [PubMed] [Google Scholar]
- 34.Kunishita A, Kubo M, Ishimaru H, Ogura T, Sugimoto H, Itoh S. Inorg Chem. 2008;47:12032–12039. doi: 10.1021/ic801568g. [DOI] [PubMed] [Google Scholar]
- 35.Maiti D, Lee DH, Gaoutchenova K, Würtele C, Holthausen MC, Sarjeant AAN, Sundermeyer J, Schindler S, Karlin KD. Angew Chem, Int Ed. 2008;47:82–85. doi: 10.1002/anie.200704389. [DOI] [PubMed] [Google Scholar]
- 36.Maiti D, Sarjeant AAN, Karlin KD. Inorg Chem. 2008;47:8736–8747. doi: 10.1021/ic800617m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schindler S. Eur J Inorg Chem. 2000:2311–2326. [Google Scholar]
- 38.Würtele C, Sander O, Lutz V, Waitz T, Tuczek F, Schindler S. J Am Chem Soc. 2009;131:7544–7545. doi: 10.1021/ja902327s. [DOI] [PubMed] [Google Scholar]
- 39.Schatz M, Becker M, Walter O, Liehr G, Schindler S. Inorg Chim Acta. 2001;324:173–179. [Google Scholar]
- 40.Komiyama K, Furutachi H, Nagatomo S, Hashimoto A, Hayashi H, Fujinami S, Suzuki M, Kitagawa T. Bull Chem Soc Jpn. 2004;77:59–72. [Google Scholar]
- 41.Itoh S. Curr Opin Chem Biol. 2006;10:115–122. doi: 10.1016/j.cbpa.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 42.Cramer CJ, Tolman WB. Acc Chem Res. 2007;40:601–608. doi: 10.1021/ar700008c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Klinman JP. J Biol Chem. 2006;281:3013–3016. doi: 10.1074/jbc.R500011200. [DOI] [PubMed] [Google Scholar]
- 44.Chen P, Solomon EI. Proc Natl Acad Sci U S A. 2004;101:13105–13110. doi: 10.1073/pnas.0402114101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kunishita A, Kubo M, Sugimoto H, Ogura T, Sato K, Takui T, Itoh S. J Am Chem Soc. 2009;131:2788–2789. doi: 10.1021/ja809464e. [DOI] [PubMed] [Google Scholar]
- 46.Poater A, Cavallo L. Inorg Chem. 2009;48:4062–4066. doi: 10.1021/ic802269v. [DOI] [PubMed] [Google Scholar]
- 47.Kitajima N, Katayama T, Fujisawa K, Iwata Y, Moro-oka Y. J Am Chem Soc. 1993;115:7872–7873. [Google Scholar]
- 48.Sanyal I, Ghosh P, Karlin KD. Inorg Chem. 1995;34:3050–3056. [Google Scholar]
- 49.Chen P, Fujisawa K, Solomon EI. J Am Chem Soc. 2000;122:10177–10193. [Google Scholar]
- 50.Kunishita A, Teraoka J, Scanlon JD, Matsumoto T, Suzuki M, Cramer CJ, Itoh S. J Am Chem Soc. 2007;129:7248–7249. doi: 10.1021/ja071623g. [DOI] [PubMed] [Google Scholar]
- 51.Kunishita A, Ishimaru H, Nakashima S, Ogura T, Itoh S. J Am Chem Soc. 2008;130:4244–4245. doi: 10.1021/ja800443s. [DOI] [PubMed] [Google Scholar]
- 52.Yoshizawa K, Kihara N, Kamachi T, Shiota Y. Inorg Chem. 2006;45:3034–3041. doi: 10.1021/ic0521168. [DOI] [PubMed] [Google Scholar]
- 53.Gherman BF, Tolman WB, Cramer CJ. J Comput Chem. 2006;27:1950–1961. doi: 10.1002/jcc.20502. [DOI] [PubMed] [Google Scholar]
- 54.Hong S, Huber SM, Gagliardi L, Cramer CC, Tolman WB. J Am Chem Soc. 2007;129:14190–14192. doi: 10.1021/ja0760426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee SC, Holm RH. J Am Chem Soc. 1993;115:11789–11798. [Google Scholar]
- 56.The parameter τ is defined as τ = (β − α)/60, where α and β are the two largest L–M–L angles with β ≥ α. τ = 0 for square-pyramidal and τ = 1 for trigonal-bipyramidal.
- 57.Yamaguchi S, Wada A, Nagatomo S, Kitagawa T, Jitsukawa K, Masuda H. Chem Lett. 2004;33:1556–1557. doi: 10.1021/ic0496572. [DOI] [PubMed] [Google Scholar]
- 58.Yamaguchi S, Nagatomo S, Kitagawa T, Funahashi Y, Ozawa T, Jitsukawa K, Masuda H. Inorg Chem. 2003;42:6968–6970. doi: 10.1021/ic035080x. [DOI] [PubMed] [Google Scholar]
- 59.Lucchese B, Humphreys KJ, Lee DH, Incarvito CD, Sommer RD, Rheingold AL, Karlin KD. Inorg Chem. 2004;43:5987–5998. doi: 10.1021/ic0497477. [DOI] [PubMed] [Google Scholar]
- 60.Karlin KD, Hayes JC, Juen S, Hutchinson JP, Zubieta J. Inorg Chem. 1982;21:4106–4108. [Google Scholar]
- 61.Barbucci R, Bencini A, Gatteschi D. Inorg Chem. 1977;16:2117–2120. [Google Scholar]
- 62.Jiang F, Karlin KD, Peisach J. Inorg Chem. 1993;32:2576–2582. [Google Scholar]
- 63.Lee Y, Park GY, Lucas HR, Vajda PL, Kamaraj K, Vance MA, Milligan AE, Woertink JS, Siegler MA, Sarjeant AAN, Zakharov LN, Rheingold AL, Solomon EI, Karlin KD. Inorg Chem. 2009;48:11297–11309. doi: 10.1021/ic9017695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Namuswe F, Hayashi T, Jiang Y, Kasper GD, Sarjeant AAN, Moënne-Loccoz P, Goldberg DP. J Am Chem Soc. 2010;132:157–167. doi: 10.1021/ja904818z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kohn W, Sham LJ. Phys Rev. 1965;140:A1133–A1138. [Google Scholar]
- 66.Shaik S, Kumar D, de Visser SP, Altun A, Thiel W. Chem Rev. 2005;105:2279–2328. doi: 10.1021/cr030722j. [DOI] [PubMed] [Google Scholar]
- 67.Armarego WLF, Perrin DD. Purification of Laboratory Chemicals. Pergamon Press; Oxford: 1997. [Google Scholar]
- 68.Finn MG, Sharpless KB. J Am Chem Soc. 1991;113:113–126. [Google Scholar]
- 69.Britovsek GJP, England J, White AJP. Inorg Chem. 2005;44:8125–8134. doi: 10.1021/ic0509229. [DOI] [PubMed] [Google Scholar]
- 70.Sheldrick GM. SHELXTL/PC Version 6.12 for Windows XP. Bruker AXS Inc; Madison, WI: 2001. [Google Scholar]
- 71.Jaguar, version 7.7. Schrödinger, LLC; New York, NY: 2010. [Google Scholar]
- 72.Becke AD. Phys Rev A: At, Mol, Opt Phys. 1988;38:3098–3100. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- 73.Becke AD. J Chem Phys. 1993;98:1372–1377. [Google Scholar]
- 74.Becke AD. J Chem Phys. 1993;98:5648–5652. [Google Scholar]
- 75.Lee C, Yang W, Parr RG. Phys Rev B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 76.Perdew JP. Phys Rev B. 1986;33:8822–8824. doi: 10.1103/physrevb.33.8822. [DOI] [PubMed] [Google Scholar]
- 77.Hay PJ, Wadt WR. J Chem Phys. 1985;82:299–310. [Google Scholar]
- 78.Wong MW. Chem Phys Lett. 1996;256:391–399. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.