

Abstract
Background
One of the goals of personalized medicine is to generate individual risk profiles that could identify individuals in the population that exhibit high risk. The discovery of more than two-dozen independent SNP markers in prostate cancer has raised the possibility for such risk stratification. In this study, we evaluated the discriminative and predictive ability for prostate cancer risk models incorporating 25 common prostate cancer genetic markers, family history of prostate cancer and age.
Methods
We fit a series of risk models and estimated their performance in 7,509 prostate cancer cases and 7,652 controls within the NCI Breast and Prostate Cancer Cohort Consortium (BPC3). We also calculated absolute risks based on SEER incidence data.
Results
The best risk model (C-statistic=0.642) included individual genetic markers and family history of prostate cancer. We observed a decreasing trend in discriminative ability with advancing age (P=0.009), with highest accuracy in men younger than 60 years (C-statistic=0.679). The absolute ten-year risk for 50-year old men with a family history ranged from 1.6% (10th percentile of genetic risk) to 6.7% (90th percentile of genetic risk). For men without family history, the risk ranged from 0.8% (10th percentile) to 3.4% (90th percentile).
Conclusions
Our results indicate that incorporating genetic information and family history in prostate cancer risk models can be particularly useful for identifying younger men that might benefit from PSA screening.
Impact
Although adding genetic risk markers improves model performance, the clinical utility of these genetic risk models is limited.
Keywords: Prostate cancer, polymorphism, risk prediction model
INTRODUCTION
Prostate cancer is estimated to account for a quarter of all new cancer diagnoses and is the second leading cause of cancer-related deaths among men in the United States during 2010 (1). Despite the high prevalence, the etiology of prostate cancer is largely unknown and risk assessment to date has only been based on age, ethnicity and family history of prostate cancer. One of the main goals of personalized medicine is to generate individual risk profiles that would identify individuals in the population that exhibit high risk (c.f. the Gail model in breast cancer). It has been suggested that high-risk groups could be identified based on a profile of genetic predisposition (2). Already, we know that men with a family history of prostate cancer have a two-fold risk of developing prostate cancer and develop prostate cancer at an earlier age of onset (3).
The discovery of more than two-dozen independent SNP markers in genome-wide association studies (GWAS) of prostate cancer has raised the possibility that such genetic profiles could be generated. A recent study suggested that compared to age-threshold screening programs, personalized screening based on genetic risk profiling would improve efficiency by reducing number of individuals eligible for screening while detecting the majority of cancers. For prostate cancer, they estimated that compared with screening men based on age alone, personalized screening at the same risk threshold would result in 16% fewer men being eligible for screening at a cost of 3% fewer screen-detectable cases (4).
In this study, we investigate the discriminative ability of common low-penetrant single nucleotide polymorphisms (SNPs) that have been associated with prostate cancer risk in 7,509 prostate cancer cases and 7,652 controls. We generated a series of statistical models including an aggregated genetic risk score, family history of prostate cancer and interaction effects. We estimated age-specific discriminative performance by calculating C-statistics for the best-fitting models. Previous reports have not estimated stratum-specific discriminative performance and have only included subsets of the genetic variants associated with prostate cancer risk (5,6). Finally, we calculated age-specific absolute risks based on SEER incidence data.
METHODS
Study population
The NCI Breast and Prostate Cancer Cohort Consortium (BPC3) has been described previously (7). In brief, the consortium combines resources from eight well-established cohort studies: the Alpha-Tocopherol, Beta-Carotene Cancer Prevention (ATBC) Study (8), American Cancer Society Cancer Prevention Study II (CPS-II) (9), the European Prospective Investigation into Cancer and Nutrition Cohort (EPIC – comprised of cohorts from Denmark, Great Britain, Germany, Greece, Italy, the Netherlands, Spain, and Sweden) (10), the Health Professionals Follow-up Study (HPFS) (11), the Melbourne Collaborative Cohort Study (MCCS) (12), the Multi-Ethnic Cohort (MEC) (13), the Physicians’ Health Study (PHS) (14), and the Prostate, Lung, Colorectal, and Ovarian (PLCO) Cancer Screening Trial (15). Together, these eight cohorts collectively include over 265,000 men who provided a biospecimen sample. Informed consent was obtained from all subjects and each study was approved by the local Institutional Review Board.
Prostate cancer cases were identified through population-based cancer registries, death certificates or self-reports confirmed by medical records, including pathology reports. Except for the MCCS study, the BPC3 consists of a series of matched nested case-control studies within each cohort; controls were matched to cases on a number of potential confounding factors, such as age, ethnicity, and region of recruitment, depending on the cohort. MCCS used a case-cohort design, with a randomly sampled sub-cohort serving as controls. The current study was restricted to self-reported European-ancestry men. We had genotype data for a total of 10,501 prostate cancer cases and 10,831 controls. Data on disease stage and grade at time of diagnosis were collected from each cohort, wherever possible. A total of 2,641 cases were classified as either high-stage (stage C or D at diagnosis) or high-grade (Gleason grade ≥ 8 or equivalent, i.e. coded as poorly differentiated or undifferentiated). For 15% of the cases, we did not have information about tumor stage or Gleason grade. Family history, which was defined as having at least one first-degree family member diagnosed with prostate cancer, was available for all but two cohort studies (PHS and EPIC). Subject characteristics are displayed in Supplementary Table 1.
Marker Selection
We initially genotyped 39 single nucleotide polymorphisms (SNPs) associated with prostate cancer risk as previously described (16). For this study, we chose 25 of the initial 39 SNPs to obtain a set of independent markers (Supplementary Table 2). To obtain a set of independent markers for this analysis we selected 25 SNPs based on the following criteria: 1) Significant association with prostate cancer risk (P<0.001) (5 SNPs removed). 2) A pair-wise r-sq <0.2 with any of the other SNPs as measured in HapMap CEU individuals. If two or more SNPs had a pair-wise r-sq >0.2, the one with strongest association with prostate cancer risk was selected (6 SNPs); or, 3) A significant association with prostate cancer risk (P<0.001) after adjusting for other risk SNPs at the same locus within a region of 500kb (3 SNPs). To address the potential for over-fitting due to our SNP selection strategy, we recalculated all analyses while randomly replacing the “best” SNPs with other associated SNPs in the same region. This did not appreciably change our results (data not shown).
Genotyping
Genotyping was performed using the TaqMan assay (Applied Biosystems, Foster City, CA) in six different genotyping laboratories: Core Genotyping Facility at National Cancer Institute, Harvard School of Public Health, University of South California, Deutsches Krebsforschungszentrum, UK Cancer Research and the Genetic Epidemiology Laboratory of the University of Melbourne. Blinded duplicated samples indicated high quality genotyping (100% concordance). All autosomal SNPs were consistent with Hardy-Weinberg Equilibrium in the controls (P>0.01).
Imputation
We excluded 42 cases and 41 controls with 20% or more missing genotypes across the 25 selected SNPs, leaving 10,459 cases and 10,790 controls for analysis. We imputed missing genotypes and family history information (yes/no) independently by sampling from the observed distribution of the missing factor conditional on age (in five-year categories) and case-control status. We conducted single SNP imputation by sampling from the observed genotype frequency distribution in all men with non-missing genotype data in the same age category and case-control status (“single conditional draw imputation”) (17). For example, assume that the genotype frequencies of SNP 1 in cases between 60 and 65 years are 0.49 (AA), 0.42 (Aa) and 0.09 (aa) respectively (corresponding to a minor allele frequency of 0.30). To impute a missing genotype for SNP 1 in a case of age 63, we randomly generated a genotype with probabilities 0.49 (AA), 0.42 (Aa) and 0.09 (aa). Genotypes in ATBC, a cohort in Finland, were imputed separately. For family history, we imputed each cohort separately (as prevalence of family history differed across studies, Supplementary Tables 1 and 3). We did not impute family history for EPIC and PHS as this information was completely missing for both. Risk allele frequencies and family history prevalence before and after imputation are displayed in Supplementary Tables 3 and 4.
Variable construction and model selection
First, we calculated the number of risk alleles carried by each individual using genotyped and imputed SNP data from all cohorts (10,459 cases and 10,790 controls). We categorized the number of risk alleles in each carrier by deciles according to the distribution in the controls and calculated decile-specific odds ratios. We compared the goodness-of-fit of various unconditional logistic regression models using Akaike’s Information Criterion (AIC). We constructed various risk models based on 7,509 prostate cancer cases and 7,652 controls from the imputed dataset including all cohorts where family history was available (i.e. excluding EPIC and PHS). Joint effects of the 25 SNPs were incorporated in different ways: by including individual SNPs in an additive main effects model with mutually adjusted log odds ratios estimated for each marker (IndSNP), a simple count of risk alleles as an ordinal (GC) or categorical (GCAT) variable, and a sum of risk alleles weighted by their marginal log odds ratios estimated from the data (GW). We also evaluated risk modification by age and family history by including product terms between the risk allele count (GC), a binary indicator of family history (Yes/No) and an ordinal coding of age in five-year categories. We did not include SNP-SNP interaction terms since the chosen SNPs have not shown evidence of non-additive joint effects (16). All analyses were adjusted for matching factors study and age at diagnosis/selection as control. The qualitative results did not change when we calculated AICs for the complete case datasets (no missing or imputed genotypes).
Discrimination ability and absolute risk estimation
For selected models, we calculated C-statistics and 95% confidence intervals stratified by study and age intervals (≤ 60, 61–65, 66–70, 71–75 and ≥75 years) using estimated linear predictors from each model. The C-statistic (equivalent to the area under the receiver operating characteristic (AUC) curve) measures the discriminative ability of a model; C=1 indicates perfect discrimination whereas C=0.5 indicates no discrimination. Summary C-statistics across studies were calculated using fixed-effects meta-analysis. Confidence intervals were calculated using the “rcorr.cens” command in the ‘Hmisc’ package in R (18). We tested for trends in C-statistics as a function of age using linear regression, treating estimated C-statistics as the dependent variable and age in 5-year interval as the independent variable.
We calculated mortality-adjusted absolute risks based on the distribution of genotypes and family history in controls, the regression parameters from the best-fitting models, five-year average incidence rates for white men based on SEER data for years 1992–2007 (19), and the life tables for U.S. men from 2007 (20,21). All analyses were performed in R (22).
RESULTS
Associations between number of risk alleles carried and prostate cancer risk
The average number of risk alleles carried (max 49) was 23.4 (range 12–35) in cases and 22.0 (range 11–34) in controls (Supplementary Figure 1). Compared to men in the lowest 10th percentile of a simple count of risk alleles, men in the highest 10th percentile had more than 5-fold risk (OR: 5.55, 95% CI, 4.85 – 6.36) of developing prostate cancer (Table 1). Decile-specific odds ratios stratified on disease severity and age of onset (< 65 and ≥ 65 years) are presented in Figure 1. We observed slightly higher decile-specific odds ratios for localized disease compared to aggressive disease. Compared to men in the lowest 10th percentile, men in the highest 10th percentile had a 7-fold risk to develop prostate cancer at age 65 or younger (OR: 7.21, 95% CI, 5.66 – 9.18). Corresponding risk comparison among men older than 65 was somewhat lower (OR: 4.56, 95% CI, 3.86 –5.39). We observed a significant interaction effect between age and number of risk alleles carried (ORInt: 0.982, 95% CI, 0.976 – 0.989, P=1.2×10−7).
Table 1.
Association between decile categories for number of risk alleles carried and prostate cancer risk. Decile-specific odds ratios were estimated based on the imputed dataset (10,459 cases and 10,790 controls). All analyses were adjusted for age and cohort.
| Decile* | Controls (%) | Cases (%) | OR (95% CI) |
|---|---|---|---|
| 1 | 1,079 (10) | 414 (4.0) | 1.00 (Ref.) |
| 2 | 1,079 (10) | 633 (6.1) | 1.53 (1.32–1.78) |
| 3 | 1,079 (10) | 745 (7.1) | 1.79 (1.54–2.08) |
| 4 | 1,079 (10) | 833 (8.0) | 2.03 (1.75–2.35) |
| 5 | 1,079 (10) | 871 (8.3) | 2.14 (1.85–2.48) |
| 6 | 1,079 (10) | 1,011 (9.7) | 2.45 (2.13–2.83) |
| 7 | 1,079 (10) | 1,050 (10.0) | 2.59 (2.24–2.99) |
| 8 | 1,079 (10) | 1,261 (12.1) | 3.12 (2.71–3.59) |
| 9 | 1,079 (10) | 1,436 (13.7) | 3.56 (3.10–4.10) |
| 10 | 1,079 (10) | 2,203 (21.1) | 5.55 (4.85–6.36) |
Numbers of risk alleles were categorized into deciles based on the distribution in controls.
Figure 1.
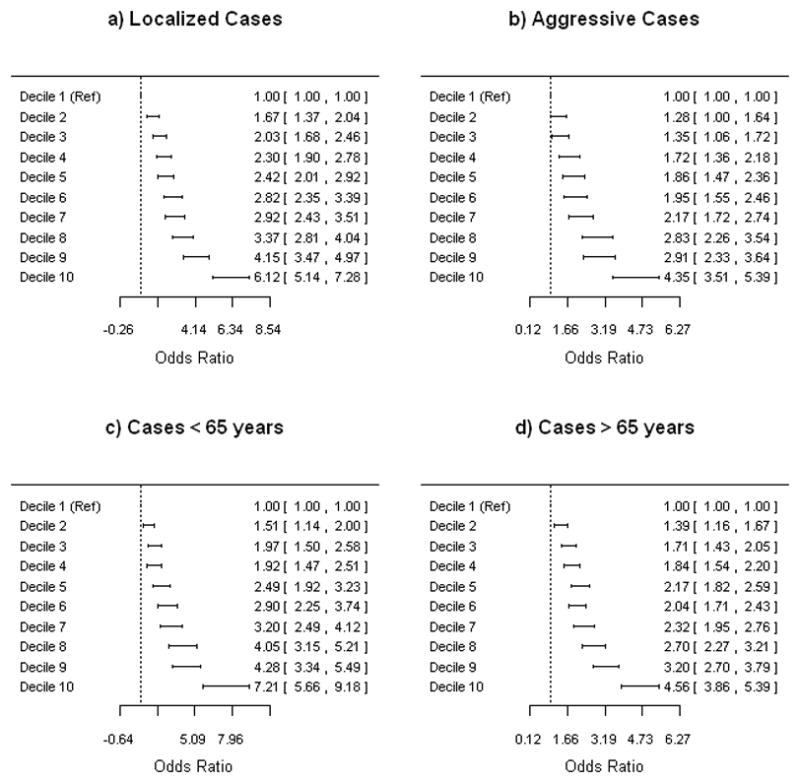
Association between decile categories for number of risk alleles carried and prostate cancer risk stratified by disease aggressiveness and age of onset. Decile-specific odds ratios were estimated based on the imputed dataset (10,459 cases and 10,790 controls). All analyses were adjusted for age and cohort. a) Localized cases (5,721 cases) b) Aggressive cases (2,641 cases) c) ≤65 years (3,315 cases and 4,214 controls) d) >65 years (7,144 cases and 6,576 controls).
Discriminative ability of genetic risk models
We incorporated SNP information, age and family history in various models and calculated goodness-of-fit using imputed genotype and family history data from all cohorts where family history was available (7,509 cases and 7,652 controls, Supplementary Table 5). We chose models that best fit the data from each of three classes: models that used information on family history of prostate cancer and age, models that used information on SNPs and age and finally, models that used information on SNPs, age and family history. The best model from the first class (Model 1) included family history of prostate cancer and an interaction term between family history and age. The best model from the second class (Model 2) included individual SNPs and an interaction term between the sum of risk alleles and age. The best model from the third class (Model 3) included individual SNPs, family history, an interaction term of age and family history and an interaction term between the sum of risk alleles and age. Regression parameters for each chosen model can be found in Supplementary Table 6. We used the regression parameters from each of the three models to estimate linear predictors and calculate C-statistics (Table 2). The inclusion of individual SNPs (average C over all ages=0.634) had much higher discriminatory power than family history only (average C over all ages=0.526). Adding SNP information to family history increased the discriminatory ability (average C over all ages=0.642). For all three models, the discriminative ability decreased with advancing age (Model 1: P=.03; Model 2: P=.009; Model 3: P=.009). Stratifying on aggressive cases did not alter the results (Supplementary Table 7). Figure 2 displays the Receiver Operating Characteristic (ROC) curve for Model 2 (dashed) and Model 3 (solid) for men younger than 65 years and men older than 75 years. We note that both models have higher discriminative probability in younger men. We also calculated cohort-specific C-statistics recognizing our limited power (Supplementary Table 8) across various age ranges. We observe that the cohort-specific estimates are relatively homogenous indicating that this model can be useful for all patients with European Ancestry.
Table 2.
C-statistics for three selected models (see methods) stratified by age. C-statistics were calculated based on estimated regression parameters in the imputed dataset of cohorts where family history was available (7,509 prostate cancer cases and 7,652 controls).
| Age | Model 1: Family History | Model 2: Genetics | Model 3: Genetics + Family History |
|---|---|---|---|
| −60 | 0.547 (0.530–0.564) | 0.663 (0.637–0.689) | 0.679 (0.654–0.705) |
| 61–65 | 0.526 (0.515–0.536) | 0.646 (0.625–0.666) | 0.654 (0.633–0.674) |
| 66–70 | 0.531 (0.523–0.540) | 0.632 (0.616–0.649) | 0.645 (0.628–0.661) |
| 71–75 | 0.523 (0.513–0.533) | 0.630 (0.611–0.648) | 0.636 (0.617–0.655) |
| 75+ | 0.505 (0.489–0.521) | 0.599 (0.571–0.626) | 0.599 (0.571–0.626) |
Figure 2.
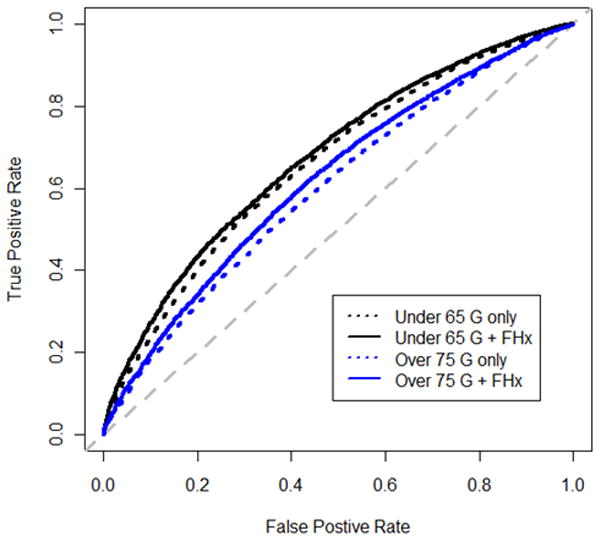
Receiver Operating Characteristic (ROC) curve for genetics only and genetics plus family history.
Estimated absolute risks based on a genetic risk model
We calculated mortality-adjusted 10-year absolute risks based on the estimated regression parameters from Model 2 and age-specific incidence rates from SEER (Table 3). Overall, absolute risks differed widely as a function of genetic risk. For example, the absolute 10-year risk for a 60-year old man with a family history of prostate cancer ranges from 0.06 (10th percentile) to 0.23 (90th percentile) depending on the genetic burden. Of note, a man without a family history of prostate cancer but with high genetic risk (90th percentile), has lower 10-year risk than a man with a family history belonging to the 50th percentile (e.g. 0.033 vs. 0.038 at 50 years).
Table 3.
Age-specific mortality-adjusted 10-year absolute risks of prostate cancer among white US men as a function of family history of prostate cancer (FH) and genetic risk (as estimated by Model 2). Quintiles of genetic risk were based on the distribution in controls. All calculations are based on regression parameters estimated in the imputed dataset. Incidence rates are based on SEER data.
| Age | Family History | No information on genetics | 10th percentile | 30th percentile | 50th percentile | 70th percentile | 90th percentile |
|---|---|---|---|---|---|---|---|
| 50 | Negative FH | 0.020 | 0.008 | 0.012 | 0.017 | 0.023 | 0.034 |
| Positive FH | 0.042 | 0.016 | 0.027 | 0.038 | 0.049 | 0.067 | |
| 60 | Negative FH | 0.064 | 0.029 | 0.043 | 0.056 | 0.075 | 0.109 |
| Positive FH | 0.134 | 0.057 | 0.088 | 0.122 | 0.154 | 0.231 | |
| 70 | Negative FH | 0.089 | 0.046 | 0.065 | 0.081 | 0.102 | 0.139 |
| Positive FH | 0.183 | 0.104 | 0.137 | 0.175 | 0.209 | 0.271 | |
| 80 | Negative FH | 0.063 | 0.039 | 0.049 | 0.060 | 0.071 | 0.089 |
| Positive FH | 0.131 | 0.085 | 0.114 | 0.132 | 0.143 | 0.181 |
The potential utility of incorporating genetic information in a clinical setting can be illustrated by the following example. A 50-year old white man without a family history of prostate cancer sees his physician and wants to know his 10-year risk of developing prostate cancer. By using the information available (family history of prostate cancer, ethnicity and age), the physician will estimate his absolute 10-year risk to be approximately 2%. Incorporating individual-specific genetic information would shift this risk to between 0.8% (10th percentile) and 3.4% (90th percentile) depending on number or risk alleles carried. The mortality-adjusted 10-year risk of developing prostate cancer as a function of the genetic risk burden for a 50-year old man is illustrated in Figure 3.
Figure 3.
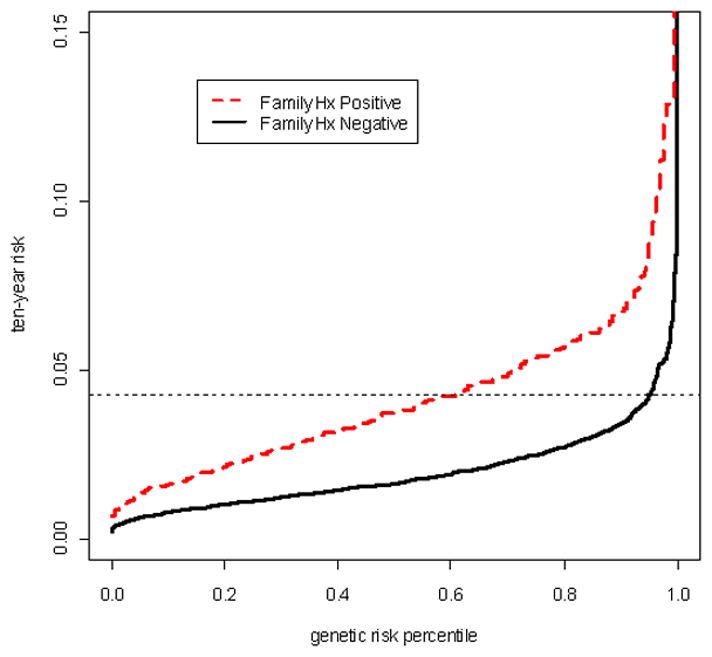
Estimated distribution of ten-year absolute risks of prostate cancer among 50-year–old U.S white men as a function of genetic risk.
DISCUSSION
In this study, we tested the performance of 25 independent SNPs in prostate cancer risk models. We evaluated different models in a series of large prospective nested case-control studies comprising a total of 7,509 prostate cancer cases and 7,652 controls with European ancestry. Our large sample size enabled us to calculate age-specific C-statistics to investigate the performance across age categories. In a model including family history and genetics, the C-statistic shifted from 0.68 for men younger than 60 years, to 0.60 for the subgroup of men older than 75 years. We also observed a statistically significant interaction between number of risk alleles carried and age at diagnosis.
Higher discrimination among younger men has important implications. First, much of the ongoing debate about PSA screening focuses on the age for recommended screening and our results suggest that genetic risk prediction models might have higher clinical utility for younger men. Second, our results illustrate the value of investigating these issues in subgroups that could lead to more effective use of genetics. Estimating population effects by combining clinical strata together may obscure important information. We found a statistically significantly stronger association between a genetic risk score summarizing known prostate cancer risk alleles and prostate cancer risk among younger men. We identified this gene-age interaction by aggregating evidence across risk loci; previous analyses of individual markers provided suggestive but not definitive evidence that many of these markers had larger odds ratios among younger men (16). In addition, some of the SNPs included in our model were identified in a GWAS enriched of younger cases (23). However, we note that the clinical value of genetic risk models can vary by age (or other known risk factors) even if the genetic odds ratio does not vary by age. Thus, the absence of statistical interaction as usually defined for disease outcomes (differences in genetic odds ratios across strata) does not necessarily imply absence of important differences in measures of clinical utility, including discrimination, net reclassification index (24), expected change in adverse events (25, 26), or change in age at recommended screening (2).
Although each genetic variant contributes a very small risk effect, the aggregated sum has a substantial impact on risk. Compared to men in the bottom 10% of risk alleles carried, men in the top 10th percentile had a 5-fold risk of developing prostate cancer during their lifetime and more than 7-fold risk of developing prostate cancer before the age of 65. The 10-year absolute risk of a 60-year old man with a positive family history and high genetic burden (90th percentile) was 23%. We observed similar risk estimates when stratifying on disease aggressiveness, as defined by grade and stage. In agreement with previous studies, our genetic risk model showed equal discriminative ability for aggressive and localized prostate cancer. This is not surprising as the SNPs included in our model have not been associated with disease aggressiveness (16) and it remains to be seen if there exists genetic variants associated with prostate cancer subtypes. Our results are nevertheless disappointing given the widely heterogeneous natural history of prostate cancer and the need to distinguish indolent from aggressive cancer. However, a recent randomized trial showed that active treatment with radical prostatectomy decreases rates of both prostate cancer-specific death and overall death, with the largest benefit for men younger than 65 years (27). Of importance, the authors also observed a significant decrease in overall mortality associated with surgery among men diagnosed with low-grade tumors.
A risk model including only SNPs appears to have higher discriminatory ability than family history alone. This observation can partly be explained by our broad definition of family history as defined by a first-degree relative with prostate cancer. Most likely, a more refined definition would increase the discriminatory power of family history further; other studies will have to address this question, because more detailed data are not available in BPC3. However, detailed family history has inherent limitations as men from small families or men whose fathers died at a young age (of other causes), etc, have less informative history. Of note, men with a family history of prostate cancer experience higher absolute risks than men without a family history regardless of number of risk alleles carried. Thus, family history of prostate cancer is still an important source of information when discussing a patient’s likelihood of developing prostate cancer.
Since it is expected that additional GWAS and next-generation sequencing will discover additional risk variants, the genetic risk models as presented here will require regular updates. However, additional genetic variants will most likely have small risk effects or be rare in the population and thus provide limited increments in discriminative ability. Moreover, it is difficult for new predictors to raise the C-statistic when existing variables (in this case SNPs) discriminate well (28, 29). Nonetheless, Wray and colleagues estimated that a genetic risk model that fully explains the genetic variation of prostate cancer has a maximum C-statistic of 0.90, assuming a heritability of 0.44 (30).
The relatively high performance of our genetic risk model (C=0.68) does not by itself guarantee clinical utility. For comparison, a single PSA test has a better accuracy (C >0.70) (31), yet despite this high C statistic, the public health utility of PSA screening remains controversial. So far, PSA testing has been reported to have low discriminatory power between indolent and clinical aggressive prostate cancer, resulting in substantial over-diagnosis, leading to a debate over the efficacy and cost-effectiveness as a screening tool (32). Monitoring changes in PSA levels over time (known as PSA velocity) has been widely advocated as a more useful marker, but so far, little evidence suggests this provides more clinical information beyond a single PSA test (33). It has been proposed that the combined information on PSA and known genetic architecture (as defined by common genetic variants with modest risk effects) might increase our ability to detect a prostate cancer, but published reports provide only modest support for this hypothesis (5, 34, 35). In the end, the ultimate arbiter of any screening tests utility is its ability to reduce net morbidity and mortality.
In summary, we have constructed genetic risk models for prostate cancer based on 25 SNPs previously identified as well as information on family history and age. Our results indicate that incorporating genetic information and family history in prostate cancer risk models can be particularly useful for identifying younger men that might benefit from PSA screening. Although our model performed reasonably well in terms of discriminatory ability, its clinical utility is still limited.
Supplementary Material
Acknowledgments
Funding/Support
This work was supported by the U.S. National Institutes of Health, National Cancer Institute [cooperative agreements U01-CA98233-07 to David J. Hunter, U01-CA98710-06 to Michael J. Thun, U01-CA98216-06 to Elio Riboli and Rudolf Kaaks, and U01-CA98758-07 to Brian E. Henderson, and Intramural Research Program of NIH/National Cancer Institute, Division of Cancer Epidemiology and Genetics].
References
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60(5):277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Pharoah PD, Antoniou AC, Easton DF, Ponder BA. Polygenes, risk prediction, and targeted prevention of breast cancer. N Engl J Med. 2008;358(26):2796–803. doi: 10.1056/NEJMsa0708739. [DOI] [PubMed] [Google Scholar]
- 3.Bratt O. What should a urologist know about hereditary predisposition to prostate cancer? BJU Int. 2007;99(4):743–7. doi: 10.1111/j.1464-410X.2006.06666.x. discussion 747–8. [DOI] [PubMed] [Google Scholar]
- 4.Pashayan N, Duffy SW, Chowdhury S, Dent T, Burton H, Neal DE, et al. Polygenic susceptibility to prostate and breast cancer: implications for personalised screening. Br J Cancer. 2011;104(10):1656–63. doi: 10.1038/bjc.2011.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johansson M, Holmstrom B, Hinchliffe SR, Bergh A, Stenman UH, Hallmans G, et al. Combining 33 genetic variants with prostate specific antigen for prediction of prostate cancer: Longitudinal study. Int J Cancer. 2011 doi: 10.1002/ijc.25986. [DOI] [PubMed] [Google Scholar]
- 6.Sun J, Kader AK, Hsu FC, Kim ST, Zhu Y, Turner AR, et al. Inherited genetic markers discovered to date are able to identify a significant number of men at considerably elevated risk for prostate cancer. Prostate. 2011;71(4):421–30. doi: 10.1002/pros.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hunter DJ, Riboli E, Haiman CA, Albanes D, Altshuler D, Chanock SJ, et al. A candidate gene approach to searching for low-penetrance breast and prostate cancer genes. Nat Rev Cancer. 2005;5(12):977–85. doi: 10.1038/nrc1754. [DOI] [PubMed] [Google Scholar]
- 8.The effect of vitamin E beta carotene on the incidence of lung cancer and other cancers in male smokers. The Alpha-Tocopherol, Beta Carotene Cancer Prevention Study Group. N Engl J Med. 1994;330(15):1029–35. doi: 10.1056/NEJM199404143301501. [DOI] [PubMed] [Google Scholar]
- 9.Calle EE, Rodriguez C, Jacobs EJ, Almon ML, Chao A, McCullough ML, et al. The American Cancer Society Cancer Prevention Study II Nutrition Cohort: rationale, study design, and baseline characteristics. Cancer. 2002;94(2):500–11. doi: 10.1002/cncr.10197. [DOI] [PubMed] [Google Scholar]
- 10.Riboli E, Hunt KJ, Slimani N, Ferrari P, Norat T, Fahey M, et al. European Prospective Investigation into Cancer and Nutrition (EPIC): study populations and data collection. Public Health Nutr. 2002;5(6B):1113–24. doi: 10.1079/PHN2002394. [DOI] [PubMed] [Google Scholar]
- 11.Giovannucci E, Pollak M, Liu Y, Platz EA, Majeed N, Rimm EB, et al. Nutritional predictors of insulin-like growth factor I and their relationships to cancer in men. Cancer Epidemiol Biomarkers Prev. 2003;12(2):84–9. [PubMed] [Google Scholar]
- 12.Severi G, Morris HA, MacInnis RJ, English DR, Tilley WD, Hopper JL, et al. Circulating insulin-like growth factor-I and binding protein-3 and risk of prostate cancer. Cancer Epidemiol Biomarkers Prev. 2006;15(6):1137–41. doi: 10.1158/1055-9965.EPI-05-0823. [DOI] [PubMed] [Google Scholar]
- 13.Kolonel LN, Altshuler D, Henderson BE. The multiethnic cohort study: exploring genes, lifestyle and cancer risk. Nat Rev Cancer. 2004;4(7):519–27. doi: 10.1038/nrc1389. [DOI] [PubMed] [Google Scholar]
- 14.Chan JM, Stampfer MJ, Ma J, Gann P, Gaziano JM, Pollak M, et al. Insulin-like growth factor-I (IGF-I) and IGF binding protein-3 as predictors of advanced-stage prostate cancer. J Natl Cancer Inst. 2002;94(14):1099–106. doi: 10.1093/jnci/94.14.1099. [DOI] [PubMed] [Google Scholar]
- 15.Hayes RB, Reding D, Kopp W, Subar AF, Bhat N, Rothman N, et al. Etiologic and early marker studies in the prostate, lung, colorectal and ovarian (PLCO) cancer screening trial. Control Clin Trials. 2000;21(6 Suppl):349S–355S. doi: 10.1016/s0197-2456(00)00101-x. [DOI] [PubMed] [Google Scholar]
- 16.Lindstrom S, Schumacher F, Siddiq A, Travis RC, Campa D, Berndt SI, et al. Characterizing Associations and SNP-Environment Interactions for GWAS-Identified Prostate Cancer Risk Markers-Results from BPC3. PLoS One. 2011;6(2):e17142. doi: 10.1371/journal.pone.0017142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Little RJA, Rubin DB. Single imputation methods. In: Little RJA, Rubin DB, editors. Statistical Analysis with Missing Data. New Jersey: Wiley & Sons; 2002. pp. 59–75. [Google Scholar]
- 18.Newson R. Confidence intervals for rank statistics: Somers’ D and extensions. Stata Journal. 2006;6:309–334. [Google Scholar]
- 19.SEER-13. Surveillance, Epidemiology, and End Results (SEER) Program. [Accessed Oct 29, 2010];SEER*Stat Database: Incidence-SEER 13 Regs Research Data. 2009 Nov; ( www.seer.cancer.gov) Sub (1992–2007); http://seer.cancer.gov/
- 20.Arias E. National vital statistics reports. 9. Vol. 59. Hyattsville, MD: National Center for Health Statistics; 2011. United States life tables, 2007. http://www.cdc.gov/nchs/ [PubMed] [Google Scholar]
- 21.Dupont WD. Converting relative risks to absolute risks: a graphical approach. Stat Med. 1989;8(6):641–51. doi: 10.1002/sim.4780080603. [DOI] [PubMed] [Google Scholar]
- 22.R Development Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2009. [Google Scholar]
- 23.Eeles RA, Kote-Jarai Z, Giles GG, Olama AA, Guy M, Jugurnauth SK, et al. Multiple newly identified loci associated with prostate cancer susceptibility. Nat Genet. 2008 Mar;40(3):316–21. doi: 10.1038/ng.90. [DOI] [PubMed] [Google Scholar]
- 24.Pencina MJ, D’Agostino RB, Sr, D’Agostino RB, Jr, Vasan RS. Evaluating the added predictive ability of a new marker: from area under the ROC curve to reclassification and beyond. Stat Med. 2008;27(2):157–72. doi: 10.1002/sim.2929. discussion 207–12. [DOI] [PubMed] [Google Scholar]
- 25.Gail MH. Value of adding single-nucleotide polymorphism genotypes to a breast cancer risk model. J Natl Cancer Inst. 2009;101(13):959–63. doi: 10.1093/jnci/djp130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gail MH. Personalized estimates of breast cancer risk in clinical practice and public health. Stat Med. 2011;30(10):1090–104. doi: 10.1002/sim.4187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bill-Axelson A, Holmberg L, Ruutu M, Garmo H, Stark JR, Busch C, et al. Radical prostatectomy versus watchful waiting in early prostate cancer. N Engl J Med. 2011;364(18):1708–17. doi: 10.1056/NEJMoa1011967. [DOI] [PubMed] [Google Scholar]
- 28.Wang TJ. Assessing the role of circulating, genetic, and imaging biomarkers in cardiovascular risk prediction. Circulation. 2011;123(5):551–65. doi: 10.1161/CIRCULATIONAHA.109.912568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pepe MS, Janes H, Longton G, Leisenring W, Newcomb P. Limitations of the odds ratio in gauging the performance of a diagnostic, prognostic, or screening marker. Am J Epidemiol. 2004;159(9):882–90. doi: 10.1093/aje/kwh101. [DOI] [PubMed] [Google Scholar]
- 30.Wray NR, Yang J, Goddard ME, Visscher PM. The genetic interpretation of area under the ROC curve in genomic profiling. PLoS Genet. 2010;6(2):e1000864. doi: 10.1371/journal.pgen.1000864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schroder F, Kattan MW. The comparability of models for predicting the risk of a positive prostate biopsy with prostate-specific antigen alone: a systematic review. Eur Urol. 2008;54(2):274–90. doi: 10.1016/j.eururo.2008.05.022. [DOI] [PubMed] [Google Scholar]
- 32.Vickers AJ, Lilja H. Prostate cancer: estimating the benefits of PSA screening. Nat Rev Urol. 2009;6(6):301–3. doi: 10.1038/nrurol.2009.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vickers AJ, Savage C, O’Brien MF, Lilja H. Systematic review of pretreatment prostate-specific antigen velocity and doubling time as predictors for prostate cancer. J Clin Oncol. 2009;27(3):398–403. doi: 10.1200/JCO.2008.18.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aly M, Wiklund F, Xu J, Isaacs WB, Eklund M, D’Amato M, et al. Polygenic Risk Score Improves Prostate Cancer Risk Prediction: Results from the Stockholm-1 Cohort Study. Eur Urol. 2011 doi: 10.1016/j.eururo.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gudmundsson J, Besenbacher S, Sulem P, Gudbjartsson DF, Olafsson I, Arinbjarnarson S, et al. Genetic correction of PSA values using sequence variants associated with PSA levels. Sci Transl Med. 2010;2(62):62ra92. doi: 10.1126/scitranslmed.3001513. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.