

Abstract
This study investigated the role of CRF in the dysphoria-like state associated with alcohol withdrawal in rats. The intracranial self-stimulation procedure was used to assess brain reward thresholds. Cessation of chronic alcohol administration lead to an elevation in brain reward thresholds in the alcohol dependent rats. The CRF receptor antagonist D-Phe CRF(12-41) dose-dependently prevented the elevations in brain reward thresholds associated with alcohol withdrawal. This indicates that the dysphoria associated with alcohol withdrawal is at least partly mediated by the activation of central CRF receptors.
Keywords: Alcohol, CRF, withdrawal, ICSS, brain reward function, dysphoria, rats
1. Introduction
Alcohol addiction is a chronic disorder that is characterized by loss of control over alcohol intake, withdrawal symptoms upon cessation of alcohol use, and relapse after periods of abstinence [1]. Abrupt cessation of chronic excessive alcohol intake in humans is associated with negative mood states as well as somatic signs such as tremors and convulsions [10]. The negative emotional state of alcohol withdrawal has been suggested to play an important role in the maintenance of alcohol addiction (i.e., alcohol self-administration to prevent withdrawal) [3,13]. Benzodiazepines are the treatment of choice for acute and protracted alcohol withdrawal. However, new treatments for alcohol withdrawal are needed because recovering alcoholics are a high-risk group for benzodiazepine abuse and for developing a benzodiazepine addiction [6].
Extensive evidence indicates that a hyperactivity of brain corticotropin-releasing factor (CRF) systems contributes to negative mood states. This is supported by the observation that CRF levels are elevated in patients with depression and CRF levels decrease upon successful treatment with antidepressants [7,20]. In addition, specific CRF1 receptor antagonists have been shown to decrease immobility in the rodent forced swim test, which is indicative of an antidepressant-like effect [9]. Drug withdrawal is characterized by a severe negative mood state that may lead to drug craving and relapse [13]. In recent studies it has been demonstrated that the negative mood state associated with nicotine withdrawal is at least party mediated by the activation of central CRF receptors. The intraventricular administration of the nonspecific CRF1/CRF2 receptor antagonist D-Phe CRF(12-41) or the specific CRF1 receptor antagonist R278995/CRA0450 diminishes the dysphoria-like state associated with nicotine withdrawal in rats [4,5].
Increased CRF transmission systems may also play a role in alcohol addiction. CRF receptor antagonists have been reported to decrease alcohol intake in alcohol dependent rats but do not affect alcohol intake in nondependent animals [8]. Funk and colleagues hypothesized that CRF receptor antagonists decrease alcohol intake in alcohol dependent animals by attenuating the negative emotional state associated with alcohol withdrawal [8]. The aim of the present study was to investigate if a hyperactivity of brain CRF systems mediates the dysphoria-like state associated with alcohol withdrawal in rats. The effect of alcohol withdrawal on brain reward function was investigated with a discrete trial intracranial self-stimulation (ICSS) procedure. Elevations in brain reward thresholds are indicative of a decreased sensitivity to rewarding electrical stimuli and reflect a dysphoric state [2]. Previous research has demonstrated that withdrawal from an alcohol liquid diet or alcohol vapor leads to elevations in brain reward thresholds in alcohol dependent animals [24,25]. The present experiment investigated whether blockade of CRF receptors with the nonspecific CRF1/CRF2 receptor antagonist D-Phe CRF(12-41) prevents the elevations in brain reward thresholds associated with alcohol withdrawal. It was hypothesized that blockade of CRF receptors prevents the elevations in brain reward thresholds associated with alcohol withdrawal. The present study may lead to a better understanding of the neuronal mechanisms that mediate the negative mood state associated with alcohol withdrawal in humans.
2. Materials and Methods
2.1 Animals
Male Wistar rats (Charles River, Raleigh, NC, n = 40), weighing 150-175 grams at the beginning of the experiment, were used. Animals were single-housed in a temperature and humidity controlled vivarium and maintained on a 12-hour reversed light/dark cycle (lights off 9 AM). All animals were treated in accordance with the NIH guidelines regarding the principles of animal care. The experiments were approved by the University of Florida IACUC committee.
2.2 Liquid Diet Procedure
After the rats were habituated to the vivarium for one week, regular lab chow was gradually replaced with the Lieber-DeCarli liquid diet [15]. The alcohol (ethanol) liquid-diet (LD-101A, TestDiet, Richmond, IN) had an energy density of 1 Kcal/g and 40% of the calories were derived from alcohol (5.7% weight/volume [w/v]). Prior to the onset of the liquid-diet procedures the control rats and alcohol rats were pair-matched by weight. The control animals were pair-fed an identical amount of an alcohol-free isocaloric liquid diet. In the control diet, maltodextrin was isocalorically substituted for alcohol. The amount of liquid diet that the control rats received was based on the liquid diet intake of the alcohol rats during the previous day. Fresh diet was provided daily at the onset of the dark cycle. The body weights of the rats and the amount of liquid diet consumed were recorded daily.
2.3 Blood alcohol levels
Tail blood (200 μl) was collected from anesthetized animals (1-3% isoflurane) approximately four hours after the onset of the dark cycle. Samples were centrifuged at 3000 rpm for 5 min and plasma was collected. The plasma samples were then frozen at -70 °C until further processing. Alcohol levels were determined using an NAD-Alcohol dehydrogenase kit (Sigma, St Louis, MO), 200 mg/dl ethanol standards (Analox Instruments, Lunenburg, MA), and a Genesys 20 spectrophotometer (Thermo Scientific, Waltham, MA). For each time point, one sample per rat was analyzed.
2.4 Surgical procedures
The rats were prepared with an 11 mm electrode in the medial forebrain bundle and an 11 mm cannula above the lateral ventricle during the same surgery [5]. At the beginning of the intracranial surgeries, the rats were anesthetized with an isoflurane/oxygen vapor mixture (1-3% isoflurane) and placed in a Kopf stereotaxic frame (David Kopf Instruments, Tujunga, CA) with the incisor bar set 3.3 mm below the interaural line (flat skull). The cannulae were implanted above the lateral ventricle using the following coordinates: anterior posterior (AP) -0.9 mm, medial lateral (ML) ±1.4 mm, dorsal ventral (DV) -3.0 mm from skull. Then the incisor bar was set 5 mm above the interaural line and the rats were prepared with stainless steel bipolar electrodes (model MS303/2 Plastics One, Roanoke, VA) in the medial forebrain bundle (AP -0.5 mm; ML ±1.7 mm; DV -8.3 mm from dura). The cannulae and electrodes were permanently secured to the skull using dental cement anchored with four skull screws.
2.5 Intracranial self-stimulation
The experimental apparatus consisted of twelve Plexiglas chambers (30.5 × 30 × 17 cm; Med Associates, Georgia, VT), each housed in a sound-attenuating melamine chamber (Med Associates, Georgia, VT). The operant conditioning chambers consisted of a metal grid floor and a metal wheel (5 cm wide) centered on a sidewall. A photobeam detector was attached next to the response wheel and recorded every 90 degrees of rotation. Brain stimulation was delivered by constant current stimulators (Model 1200C, Stimtek, Acton, MA). Subjects were connected to the stimulation circuit through bipolar leads (Plastics One, Roanoke, VA) attached to gold-contact swivel commutators (model SL2C Plastics One, Roanoke, VA). A computer controlled the stimulation parameters, data collection, and all test session functions. Rats were trained on a modified discrete-trial ICSS procedure [14] as described previously [17]. First the rats were trained to turn the response wheel on a fixed ratio 1 (FR1) schedule of reinforcement. Each quarter turn resulted in the delivery of a 0.5 second train of 0.1 millisecond cathodal square-wave pulses at a frequency of 100 Hz. After the successful acquisition of responding, the rats were gradually trained on a discrete-trial current-threshold procedure. The rats were subsequently tested on a current-threshold procedure in which stimulation intensities varied according to the classical psychophysical method of limits. A test session consisted of four alternating series of descending and ascending current intensities starting with a descending series. Each test session typically lasted 30 minutes and provided two variables: brain reward thresholds and response latencies. The brain reward threshold was defined as the midpoint between stimulation intensities that supported responding and current intensities that failed to support responding. The response latency was defined as the time interval between the beginning of the non-contingent stimulus and a positive response.
2.6 Experimental design
At the beginning of the experiment the rats were divided into 2 groups; an alcohol group (n = 20) and a control group (n = 20). See Fig. 1 for a timeline of the experimental procedures. During the experiment, 2 of the alcohol rats died from seizures and 2 alcohol rats had to be excluded because of dysfunctional electrodes. After the rats had been fed the liquid diets for about one month, they were prepared with an electrode in the medial forebrain bundle. The ICSS training and test sessions were conducted 2 hours after the onset of the dark cycle. The alcohol diet was introduced prior to the onset of the ICSS training sessions because high levels of alcohol intake per unit of body weight can be more easily obtained in young animals than in older animals. In order to determine blood alcohol levels, tail blood samples were collected 4 hours after the onset of the dark cycle during week 4, 7, 10, and 12 of alcohol intake. The alcohol withdrawal sessions started after the rats had been exposed to the alcohol liquid diet for 12 weeks. During each withdrawal session, the alcohol diet was replaced with the control diet at the onset of the dark cycle and the animals were tested in the ICSS procedure 8 hours after alcohol removal. The alcohol liquid diet was reintroduced immediately after the ICSS test sessions. Therefore, the rats were about 9 hours without alcohol. The control animals remained on the control liquid diet during withdrawal testing. Three withdrawal sessions were conducted and there were at least 7 days between subsequent withdrawal sessions. The CRF receptor antagonist D-Phe CRF(12-41) (0, 10, 20 μg, icv) was dissolved in distilled water and administered in a volume of 5 μl according to a Latin-square design 15 minutes before the rats were placed in the ICSS test chambers. The doses of D-Phe CRF(12-41) were based on a previous study in which the role of CRF in nicotine withdrawal and stress-induced relapse was investigated [5,26]
Fig. 1. Timeline experimental procedures.
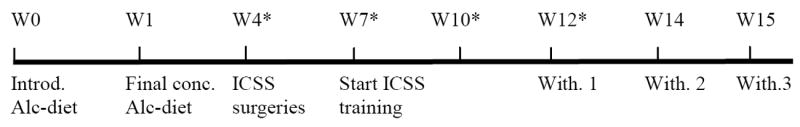
Experimental protocol. Rats were exposed to the alcohol diet from week 0 to week 15 with the exception of the 9-hour withdrawal sessions. Abbreviations: Alc, alcohol; Conc., concentration; ICSS, intracranial self-stimulation; Introd., introduction; W, week; With, withdrawal session. Asterisks (*) indicate when blood samples were collected to determine blood alcohol levels.
2.7 Statistical analyses
ICSS parameters were analyzed by two-way repeated-measures ANOVA with the dose of D-Phe CRF(12-41) as the within-subjects factor and liquid diet content (alcohol vs. control) as the between-subjects factor. One-way ANOVA’s were used to compare the ICSS parameters between control rats and alcohol rats on pretest days. Body weight gain during the liquid diet exposure period was analyzed by two-way repeated-measures ANOVA with time as the within-subjects factor and liquid diet content as the between-subjects factor. The average weekly body weight of each rat was used for the statistical analysis. Statistically significant results in the ANOVA’s were followed by the Newman-Keuls post-hoc test.
3. Results
The average blood alcohol concentrations varied between 198 and 256 mg/dl (Table 1). These blood alcohol concentrations are in line with previous studies in which rats were exposed to alcohol vapor or an alcohol liquid diet (7.9% w/v) [24,25]. The rats that were exposed to the alcohol diet gained less weight than the control rats during the 16-week liquid diet exposure period (Time: F15,510=1563.78, P<0.0001; Treatment: F1,34=23.92, P<0.0001; Time × Treatment: F15,510=40.05, P<0.0001). The body weights of the control rats prior to the onset of the liquid diet procedure and prior to the last withdrawal session were 158 ± 3 and 432 ± 6 grams, respectively. The body weights of the alcohol rats prior to the onset of the liquid diet procedure and prior to the last withdrawal session were 157 ± 3 and 370 ± 9 grams, respectively. There were no significant differences in absolute brain reward thresholds of the alcohol dependent rats and the control rats on the test days prior to any of the withdrawal sessions. The absolute response latencies of the alcohol rats were slightly, but significantly, longer than those of the control rats on the test day prior to the first withdrawal session (alcohol 3.3 ± 0.10 vs. control 3.0 ± 0.07 seconds, F1,35= 6.30, P<0.02). There were no significant differences in response latencies between the alcohol group and the control group on the test-day prior to the second or third withdrawal session. Discontinuation of alcohol administration lead to an elevation in brain reward thresholds in the alcohol rats (Fig. 2A; Treatment: F1,34=26.84, P<0.0001). Pretreatment with D-Phe CRF(12-41) dose-dependently attenuated the elevations in brain reward thresholds associated with alcohol withdrawal and did not affect the brain reward thresholds of the control rats (Dose × Treatment: F2,68=5.27, P<0.007). Post hoc analysis indicated that 10 μg (P<0.05) and 20 μg of D-Phe CRF(12-41) (P<0.01) attenuated the elevations in brain reward thresholds associated with alcohol withdrawal. Furthermore, the 20 μg dose was more effective in attenuating the elevations in brain reward thresholds associated with alcohol withdrawal than the 10 μg dose (P<0.01). Alcohol withdrawal also lead to a small but significant increase in the response latencies (Fig. 2B; Treatment: F1,34=35.49, P<0.0001). Pretreatment with D-Phe CRF(12-41) did not affect the response latencies of the control rats or the alcohol withdrawing rats.
Table 1.
Effect of the alcohol liquid diet (5.7% weight/volume) on blood alcohol levels.
| Time | Blood alcohol (mg/dl) |
|---|---|
| Week 4 | 229.8 ± 8.7 |
| Week 7 | 256.3 ± 16.5 |
| Week 10 | 198.0 ± 10.0 |
| Week 12 | 234.7 ± 8.3 |
Fig. 2.
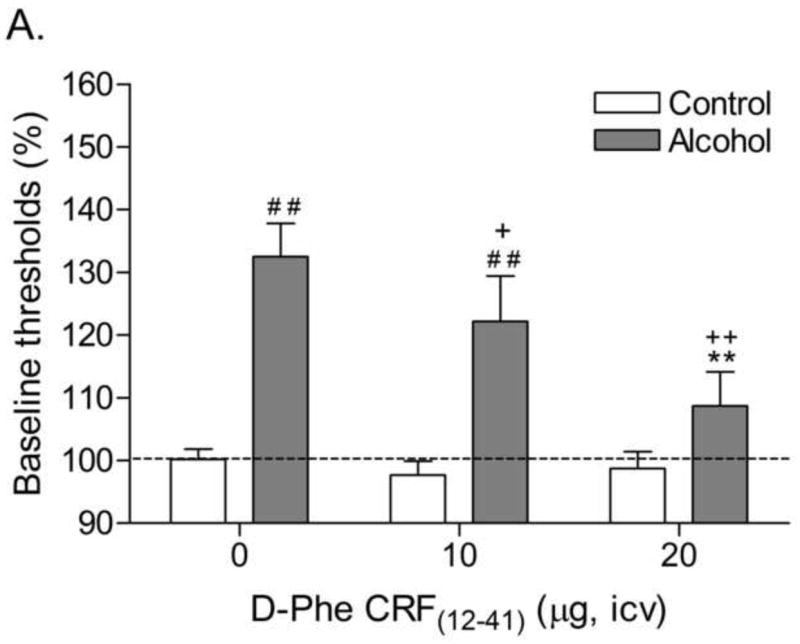
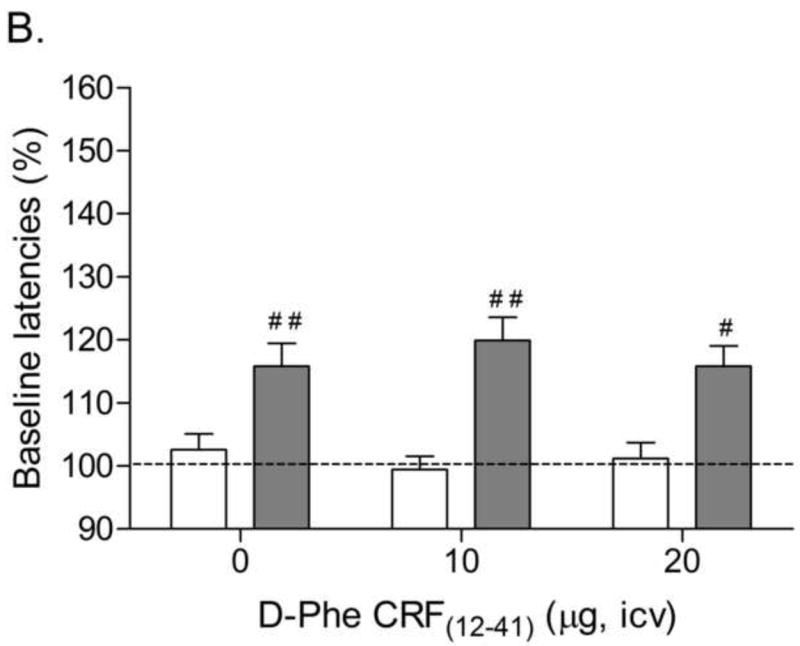
Effect of the CRF receptor antagonist D-Phe CRF(12-41) (control, n = 20; alcohol, n = 16) on the elevations in brain reward thresholds (A) and increased response latencies (B) associated with alcohol withdrawal. Brain reward thresholds and response latencies are expressed as a percentage of the pretest day values. Crosses (# P<0.05, ## P<0.01) indicate elevations in brain reward thresholds or increased response latencies compared to those of the corresponding control group. Plus signs (+ P<0.05, ++ P<0.01) indicate lower brain reward thresholds compared to those of rats withdrawing from alcohol and acutely treated with vehicle. Asterisks (** P<0.01) indicate lower brain reward thresholds compared to those of rats withdrawing from alcohol and acutely treated with 10 μg of D-Phe CRF(12-41).
4. Discussion
This study demonstrates that blockade of central CRF receptors with the nonspecific CRF1/CRF2 receptor antagonist D-Phe CRF(12-41) prevents the elevations in brain reward thresholds associated with alcohol withdrawal. This study provides support for the hypothesis that a hyperactivity of brain CRF systems mediates the dysphoria associated with alcohol withdrawal in humans [11]. To our knowledge, this is the first study to report that the endogenous release of CRF mediates the deficit in brain reward function associated with alcohol withdrawal. Although the peptide CRF receptor antagonist D-Phe CRF(12-41) does not cross the blood brain barrier, numerous nonpeptide CRF receptor antagonists have been developed that readily cross the blood brain barrier [18]. Therefore, CRF receptor antagonists may be potential novel, non-addictive, treatments for the dysphoric state associated with alcohol withdrawal in humans. Drugs that attenuate the dysphoria associated with alcohol withdrawal may diminish alcohol craving and prevent relapse to alcohol abuse.
Alcohol withdrawal was associated with a small, but significant, increase in response latencies and D-Phe CRF(12-41) did not prevent the withdrawal-induced increase in response latencies. Alcohol withdrawal increased the response latencies by approximately 15%. The average response latency of the alcohol rats was 3.3 seconds and therefore the alcohol withdrawal-induced increase in response latencies was less than 1 second. This indicates that alcohol withdrawal did not lead to major motor impairments, but alcohol withdrawal may have caused minor performance deficits. This is in line with a previous study that reported that mice withdrawing from alcohol performed slightly less well than control mice on an accelerating rotarod test [22]. Alcohol withdrawal is associated with tremors and rigidity which may have contributed to the increase in response latencies.
In the present study, D-Phe CRF(12-41) was administered into the lateral ventricles and therefore it can only be speculated via which brain sites the CRF receptor antagonist mediated its effects. Experimental evidence points toward a pivotal role for CRF in the central nucleus of the amygdala and the lateral bed nucleus of the stria terminalis in the dysphoria-like state associated with alcohol withdrawal. Withdrawal from alcohol has been shown to elevate extracellular CRF levels in the central nucleus of the amygdala and the bed nucleus of the stria terminalis [19,21]. In addition, CRF levels in the bed nucleus of the stria terminalis return to baseline levels after the resumption of alcohol intake [21]. At this point in time, only a few studies have investigated the role of increased CRF transmission in the central nucleus of the amygdala and the bed nucleus of the stria terminalis in the behavioral changes associated with drug withdrawal. The administration of CRF receptor antagonists into the central nucleus of the amygdala has been reported to decrease the anxiety-like effects of alcohol withdrawal and morphine withdrawal-induced place aversion [12,23]. Furthermore, in a recent study we reported that blockade of CRF receptors in the central nucleus of the amygdala, but not in the bed nucleus of the stria terminalis, prevents the elevations in brain reward thresholds associated with nicotine withdrawal [16]. Thus, these studies suggest that in the present study D-Phe CRF(12-41) may have prevented the elevations in brain reward thresholds associated with alcohol withdrawal by blockade of CRF transmission in the central nucleus of the amygdala. Future studies are needed to test the hypothesis that increased CRF transmission in the central nucleus of the amygdala and the bed nucleus of the stria terminalis mediates the dysphoria-like state associated with alcohol withdrawal.
Acknowledgments
This work was funded by grant AA016088 to Adrie Bruijnzeel. The authors would like to thank Dr. Jean Rivier (The Salk Institute for Biological Studies, San Diego, CA) for providing D-Phe CRF(12-41).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. American Psychiatric Press; Washington, DC: 2000. [Google Scholar]
- 2.Barr AM, Markou A, Phillips AG. A ‘crash’ course on psychostimulant withdrawal as a model of depression. Trends Pharmacol Sci. 2002;23:475–482. doi: 10.1016/s0165-6147(02)02086-2. [DOI] [PubMed] [Google Scholar]
- 3.Bruijnzeel AW, Gold MS. The role of corticotropin-releasing factor-like peptides in cannabis, nicotine, and alcohol dependence. Brain Res Brain Res Rev. 2005;49:505–528. doi: 10.1016/j.brainresrev.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 4.Bruijnzeel AW, Prado M, Isaac S. Corticotropin-Releasing Factor-1 Receptor Activation Mediates Nicotine Withdrawal-Induced Deficit in Brain Reward Function and Stress-Induced Relapse. Biol Psychiatry. 2009;66:110–117. doi: 10.1016/j.biopsych.2009.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bruijnzeel AW, Zislis G, Wilson C, Gold MS. Antagonism of CRF receptors prevents the deficit in brain reward function associated with precipitated nicotine withdrawal in rats. Neuropsychopharmacology. 2007;32:955–963. doi: 10.1038/sj.npp.1301192. [DOI] [PubMed] [Google Scholar]
- 6.Ciraulo DA, Sands BF, Shader RI. Critical review of liability for benzodiazepine abuse among alcoholics. Am J Psychiatry. 1988;145:1501–1506. doi: 10.1176/ajp.145.12.1501. [DOI] [PubMed] [Google Scholar]
- 7.De Bellis MD, Gold PW, Geracioti TD, Jr, Listwak SJ, Kling MA. Association of fluoxetine treatment with reductions in CSF concentrations of corticotropin-releasing hormone and arginine vasopressin in patients with major depression. Am J Psychiatry. 1993;150:656–657. doi: 10.1176/ajp.150.4.656. [DOI] [PubMed] [Google Scholar]
- 8.Funk CK, O’Dell LE, Crawford EF, Koob GF. Corticotropin-releasing factor within the central nucleus of the amygdala mediates enhanced ethanol self-administration in withdrawn, ethanol-dependent rats. J Neurosci. 2006;26:11324–11332. doi: 10.1523/JNEUROSCI.3096-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Griebel G, Simiand J, Steinberg R, Jung M, Gully D, Roger P, Geslin M, Scatton B, Maffrand JP, Soubrie P. 4-(2-Chloro-4-methoxy-5-methylphenyl)-N-[(1S)-2-cyclopropyl-1-(3-fluoro-4- methylphenyl)ethyl]5-methyl-N-(2-propynyl)-1, 3-thiazol-2-amine hydrochloride (SSR125543A), a potent and selective corticotrophin-releasing factor(1) receptor antagonist. II. Characterization in rodent models of stress-related disorders. J Pharmacol Exp Ther. 2002;301:333–345. doi: 10.1124/jpet.301.1.333. [DOI] [PubMed] [Google Scholar]
- 10.Hall W, Zador D. The alcohol withdrawal syndrome. Lancet. 1997;349:1897–1900. doi: 10.1016/S0140-6736(97)04572-8. [DOI] [PubMed] [Google Scholar]
- 11.Heilig M, Koob GF. A key role for corticotropin-releasing factor in alcohol dependence. Trends Neurosci. 2007;30:399–406. doi: 10.1016/j.tins.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heinrichs SC, Menzaghi F, Schulteis G, Koob GF, Stinus L. Suppression of corticotropin-releasing factor in the amygdala attenuates aversive consequences of morphine withdrawal. Behav Pharmacol. 1995;6:74–80. [PubMed] [Google Scholar]
- 13.Koob GF. A role for brain stress systems in addiction. Neuron. 2008;59:11–34. doi: 10.1016/j.neuron.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kornetsky C, Esposito RU. Euphorigenic drugs: effects on the reward pathways of the brain. Fed Proc. 1979;38:2473–2476. [PubMed] [Google Scholar]
- 15.Lieber CS, DeCarli LM. Alcoholic liver injury: experimental models in rats and baboons. Adv Exp Med Biol. 1975;59:379–393. doi: 10.1007/978-1-4757-0632-1_27. [DOI] [PubMed] [Google Scholar]
- 16.Marcinkiewcz CA, Prado MM, Isaac SK, Marshall A, Rylkova D, Bruijnzeel AW. Corticotropin-releasing factor within the central nucleus of the amygdala and the nucleus accumbens shell mediates the negative affective state of nicotine withdrawal in rats. Neuropsychopharmacology. 2009;34:1743–1752. doi: 10.1038/npp.2008.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Markou A, Koob GF. Construct validity of a self-stimulation threshold paradigm: effects of reward and performance manipulations. Physiol Behav. 1992;51:111–119. doi: 10.1016/0031-9384(92)90211-j. [DOI] [PubMed] [Google Scholar]
- 18.McCarthy JR, Heinrichs SC, Grigoriadis DE. Recent advances with the CRF1 receptor: design of small molecule inhibitors, receptor subtypes and clinical indications. Curr Pharm Des. 1999;5:289–315. [PubMed] [Google Scholar]
- 19.Merlo Pich E, Lorang M, Yeganeh M, Rodriguez de Fonseca F, Raber J, Koob GF, Weiss F. Increase of extracellular corticotropin-releasing factor-like immunoreactivity levels in the amygdala of awake rats during restraint stress and ethanol withdrawal as measured by microdialysis. J Neurosci. 1995;15:5439–5447. doi: 10.1523/JNEUROSCI.15-08-05439.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nemeroff CB, Widerlov E, Bissette G, Walleus H, Karlsson I, Eklund K, Kilts CD, Loosen PT, Vale W. Elevated concentrations of CSF corticotropin-releasing factor-like immunoreactivity in depressed patients. Science. 1984;226:1342–1344. doi: 10.1126/science.6334362. [DOI] [PubMed] [Google Scholar]
- 21.Olive MF, Koenig HN, Nannini MA, Hodge CW. Elevated extracellular CRF levels in the bed nucleus of the stria terminalis during ethanol withdrawal and reduction by subsequent ethanol intake. Pharmacol Biochem Behav. 2002;72:213–220. doi: 10.1016/s0091-3057(01)00748-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Philibin SD, Cameron AJ, Metten P, Crabbe JC. Motor impairment: a new ethanol withdrawal phenotype in mice. Behav Pharmacol. 2008;19:604–614. doi: 10.1097/FBP.0b013e32830ded27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rassnick S, Heinrichs SC, Britton KT, Koob GF. Microinjection of a corticotropin-releasing factor antagonist into the central nucleus of the amygdala reverses anxiogenic-like effects of ethanol withdrawal. Brain Res. 1993;605:25–32. doi: 10.1016/0006-8993(93)91352-s. [DOI] [PubMed] [Google Scholar]
- 24.Rylkova D, Shah HP, Small E, Bruijnzeel AW. Deficit in brain reward function and acute and protracted anxiety-like behavior after discontinuation of a chronic alcohol liquid diet in rats. Psychopharmacology (Berl) 2009;203:629–640. doi: 10.1007/s00213-008-1409-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schulteis G, Markou A, Cole M, Koob GF. Decreased brain reward produced by ethanol withdrawal. Proc Natl Acad Sci U S A. 1995;92:5880–5884. doi: 10.1073/pnas.92.13.5880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zislis G, Desai TV, Prado M, Shah HP, Bruijnzeel AW. Effects of the CRF receptor antagonist D-Phe CRF(12-41) and the alpha2-adrenergic receptor agonist clonidine on stress-induced reinstatement of nicotine-seeking behavior in rats. Neuropharmacology. 2007;58:958–966. doi: 10.1016/j.neuropharm.2007.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]