

Abstract
Four cysteine residues (Cys866, Cys917, Cysl094, and Cys1105) have direct roles in cooperatively regulating Janus kinase 2 (JAK2) catalytic activity. Additional site-directed mutagenesis experiments now provide evidence that two of these residues (Cys866 and Cys917) act together as a redox-sensitive switch, allowing JAK2's catalytic activity to be directly regulated by the redox state of the cell. We created several variants of the truncated JAK2 (GST/(NΔ661)rJAK2), which incorporated cysteine-to-serine or cysteine-to-alanine mutations. The catalytic activities of these mutant enzymes were evaluated by in vitro autokinase assays and by in situ autophosphorylation and transphosphorylation assays. Cysteine-to-alanine mutagenesis revealed that the mechanistic role of Cys866 and Cys917 is functionally distinct from that of Cysl094 and Cys1105. Most notable is the observation that the robust activity of the CC866.917AA mutant is unaltered by pretreatment with dithiothreitol or o-iodosobenzoate, unlike all other JAK2 variants previously examined. This work provides the first direct evidence for a cysteine-based redox-sensitive switch that regulates JAK2 catalytic activity. The presence of this redox-sensitive switch predicts that reactive oxygen species can impair the cell's response to JAK-coupled cytokines under conditions of oxidative stress, which we confirm in a murine pancreatic β-islet cell line.
Keywords: Janus kinase, Cysteine, Redox, β-Islet, Diabetes, Cytokines, Free radicals
Biological redox (reduction/oxidation) reactions remain poorly understood despite their importance to most normal physiological and many pathophysiological processes [1,2]. Redox reactions normally regulate vascular tone [3], platelet activation [4], and the immune response [5]. When redox homeostasis is compromised and chronic oxidative stress persists, the pathophysiological consequences can include hypertension [6], chronic obstructive pulmonary disease [7], and diabetic nephropathy [8]; oxidative damage is also an important component of the free radical theory of aging [9,10]. Given that redox regulation is central to cellular homeostasis and given the broad role of Janus protein-tyrosine kinase 2 (JAK2)3 in signal transduction, our lab has sought to ascertain the molecular basis for JAK2's response to the cellular redox state. JAK2 is one of four mammalian JAK isoforms, the others being TYK2 (tyrosine kinase 2), JAK1, and JAK3. JAK2 plays a profound role in the biology of cells of hematopoietic and epithelial origin [11,12] and is essential to normal physiological development [13,14], based largely on its essential role in transducing signals initiated by numerous cytokines including interleukin-3, erythropoietin, growth hormone, leptin, and prolactin.
Redox reactions affect JAK2, yet there remains disagreement as to whether JAK catalysis is inhibited or enhanced under oxidized conditions. Reactive oxygen species (most commonly hydrogen peroxide) appear to stimulate JAK/STAT activity in some cells [15-18]. In contrast, oxidative stress inhibits JAK activity in hematopoietin receptor systems, including leptin [19], IL-3 [20], IL-2 [21,22], CNTF [23,24], and interferon-α [25]. Interestingly, the JAK2-mediated signaling outcome after hydrogen peroxide exposure is dependent on the cell line used [23]. This raises the question of whether redox regulation of JAK2 activity depends on any intrinsic structural elements of JAK2 or whether redox regulation is solely due to indirect control exerted through other redox-sensitive molecules such as protein-tyrosine phosphatases [26–29]. Site-directed mutagenesis experiments allowed us to show that four cysteines in the JAK2 kinase domain (Cys866, Cys917, Cysl094, and Cys1105) cooperatively maintained JAK2's catalytic competency [30]. Within the bilobate architecture of the catalytic domain, Cys866 and Cys917 are located on separate β-sheets near the ATP-binding site in the N lobe, whereas Cysl094 and Cys1105 are located on opposite ends of an α-helix in the C lobe [31]. The significant physical distance between these two cysteine pairs suggests that the N-lobe and C-lobe cysteines may serve different functions. However, the functions of these two pairs of cysteines are unknown, and it is unknown if either or both of these cysteine pairs function as the elusive redox switch.
As a follow-up to earlier experiments involving cysteine-to-serine substitutions that conserved the polar characteristics of cysteine [30], we now report on experiments involving site-directed substitution of apolar alanine residues for the four critical cysteine residues (Csy866, Cys917, Cysl094, and Cys1105). Originally, we expected that such conservative substitutions (with respect to steric considerations) would provide a way to assess the functional importance of polarity at these sites. Our unexpected results show that the N-lobe residues Cys866 and Cys917 function together as a redox-sensitive activity switch that could directly couple the cellular redox state to control of enzymatic activity. Given the many biological roles of JAK2 and the range of diseases in which chronic oxidative stress is of reputed significance, it would be interesting to test this prediction in an unexamined system. Although the oxidative inhibition of JAK signaling has already been examined in several cytokine systems [19–25], it has not yet been examined in either the growth hormone or the prolactin systems.
Growth hormone (GH) and prolactin, two hormones essential for normal development of pancreatic islet function and insulin secretion [32,33], require JAK2 as an essential signal transducer to activate STAT5 in the pancreas [34]. Transgenic animal models demonstrate the need for functional prolactin receptors [35], growth hormone receptors [36], and STAT5 [37] in preserving functional pancreatic β-islet cell mass. Given that the progressive loss of functional pancreatic β-islet mass is the central pathogenic event in type II diabetes mellitus, and given the consensus that chronic oxidative stress is a major contributing factor to the demise of the pancreatic β-islet cell [38], it is reasonable to assume that if JAK2 were inhibited under the cellular conditions associated with type II diabetes, this would help to explain how chronic oxidative stress leads to the loss of functional pancreatic β-islet mass. In this article we therefore conduct a preliminary test of the prediction that oxidative pretreatment of pancreatic β-islet cells blocks the ability of either growth hormone or prolactin to stimulate STAT5 phosphorylation, consistent with the result expected if oxidative stress places the redox switch in the “inhibitory position” in these cells. This finding would have significance for type II diabetes and other diseases involving chronic oxidative stress.
Material and methods
Construction of recombinant baculoviruses
The recombinant baculoviruses producing the hyperactive GST/ (NΔ661)rJAK2 enzyme, the inactive GST/(NΔ661)rJAK2(K882E) and GST/rJAK2(K882E) enzymes, and the four cysteine-to-serine GST/ (NΔ661)rJAK2 mutants have been previously described [30,39]. The four cysteine-to-alanine mutants used in this study were constructed via site-directed mutagenesis of the pAcGHLT-A:(NΔ661)rJAK2 transfer vector using the QuikChange site-directed mutagenesis kit (Stratagene) as described below. After DNA sequence analysis to verify the mutations, these transfer vectors were used to generate recombinant baculoviruses, as previously described [39].
Site-directed mutations were cumulatively introduced into a baculoviral transfer vector until all targeted cysteine codons were converted to serine or alanine codons. After DNA sequence verification, recombinant baculoviruses were generated, isolated, and amplified, and the infectious titers were ascertained as described [40].
Site-directed mutagenesis
The pAcGHLT-A:(NΔ661)rJAK2 transfer vector was used as a template and the following primer sets were used: 5′-CGGGTCATAGCGGGCCATCTCCACACTCC-3′ and 5′-GGAGTGTGGAGATGGCCCGCTATGACCCG-3′ to convert Cys866 to Ala, 5′-GAAGTACAAGGGAGTGGCCTA-CAGTGCTGGTCG-3′ and 5′-CGACCAGGACTGTAGGCCACTCCCTTGTACTTC-3′ to convert Cys917 to Ala, 5′-CTGCCGAGACCAGAAGGGGCCCCAGACGA-GATTTATG-3′ and 5′-CATAAATCTCGTCCTGGTCTCGGCAGTGGGGCCCCTT-3′ to convert Cys1094 to Ala, and 5′-GTGATCATGACAGAAGCCTGGAA-CAACAATGTCAACCAACGTCCC-3′ and 5′-GGGACGTTGGTTGACATTGT-TGTTCGAGGCTTCTGTCATGATCAC-3′ to convert Cys1105 to Ala.
Mutagenesis was performed with the QuikChange site-directed mutagenesis kit (Stratagene) according to the manufacturer's protocol, except that the extension steps were 21 min long. After the bacterial transformation step, DNA was isolated from a single bacterial colony, and the desired point mutation was verified via DNA sequencing (Davis Sequencing, Davis, CA, USA) before generating the recombinant baculovirus as described above.
Nomenclature of mutants described in this article
We use an abbreviated nomenclature to indicate the nature and location of the cysteine mutants (e.g., CC866,917AA or CC1094,1105AA for pairwise substitutions; 4C:4A for substitution of all four cysteines). To emphasize that all of these mutants were derived from a GST-tagged, amino-terminally truncated, hyperactive JAK2, we refer to the parental form as GST/(NΔ661)rJAK2. We refer to the inactive substrate protein as GST/rJAK2(K882E) to avoid confusion with the inactive kinase K882E mutant derived from the parental GST/(NΔ661)rJAK2.
Production and immunoprecipitation of recombinant JAK2 proteins
Sf21 cells were counted, seeded onto tissue culture dishes, and allowed 20 min to attach before infection with recombinant baculoviruses. Recombinant baculoviruses were used singly or in combination to infect cells at a multiplicity of infection of 10. Cells were then incubated at 27 °C for 72 h, harvested via centrifugation, and stored at − 80 °C until lysis.
At a ratio of 1 ml to 1 × 107 cells, infected cells were lysed with insect cell lysis buffer (1% Triton X-100, 130 mM NaCl, 10 mM NaF, 10 mM sodium pyrophosphate, 10 mM sodium phosphate, 16 μg/ml benzamidine HCl, 10 μg/ml phenanthroline, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 10 μg/ml pepstatin A, 1 mM phenylmethanesul-fonyl fluoride, 10 mM Tris–Cl, pH 8.0) at 4 °C for 2 h [41]. Cell lysates were centrifuged at 10,000 g for 15 min and the insoluble fraction was removed. The JAK2-rich lysate was incubated overnight at 4 °C with anti-JAK2 antiserum followed by a 2-h incubation with protein A–Sepharose. The immobilized JAK2/antibody/protein A–Sepharose complexes were recovered by centrifugation (3000 g for 5 min at 4 °C) and washed three times with insect cell lysis buffer.
Redox pretreatment and in vitro radiolabeling autokinase assay
The immobilized JAK2/antibody/protein A–Sepharose complexes were incubated for 1 h at 4 °C with 1 ml of the reducing agent dithio-threitol (DTT; 10 mM) or the oxidizing agent ortho-iodosobenzoate (o-IBZ; 2.5 mM). Samples were then centrifuged (500 g for 5 min), the supernatant was discarded, and immune complexes were washed three times with insect cell lysis buffer. Before the final wash, each sample was divided into two equal aliquots. One aliquot was prepared for Western blot analysis (stored at − 20 °C if necessary). The other aliquot was incubated with mixing for 20 min at room temperature in 100 μl of radioactive autokinase cocktail (250 μCi/ml [γ-32P] ATP, 5mM MgCl2, 5 mM MnCl2, 50 mM NaCl, 100 μM Na2VO3, 10 mM Hepes, pH 7.6). After 20 min, samples were centrifuged, washed three times, and then prepared for SDS–PAGE and Phosphor-Imager (Molecular Dynamics/GE Healthcare) analysis. For most experiments, the intensity of each of the radioactive autokinase signals was calculated as a percentage of the GST/(NΔ661)rJAK2 autokinase signal and then normalized to the anti-JAK2 signal intensity relative to GST/(NΔ661)rJAK2, as previously described [30].
The mouse pancreatic β-islet cell line βTC-6 was purchased from the ATCC (Catalog No. CRL-11506). These cells were routinely maintained in glucose-free RPM1 1640 (Mediatech, Catalog No. 15-043-CV) supplemented with 5.6 mM D-glucose, 10% heat-inactivated fetal bovine serum (FBS), 10 mM Hepes, 100 IU/ml penicillin, 100 μg/ml streptomycin, 2.5 μg/ml amphotericin B in a 37 °C, 5% CO2 incubator. Before cytokine stimulation, βTC-6 cells were incubated 21–22 h in quiescence medium (supplemented RPMI 1640 containing 5.6 mM glucose, but only 0.3% heat-inactivated FBS), hyperglycemia medium (quiescence medium containing 25 mM glucose), or AG490 medium (quiescence medium containing 20 μM AG490). Where indicated, cells were then preincubated with varying concentrations of hydrogen peroxide for 30 min before cytokine stimulation. Cells were then stimulated for 15 min with or without 15 nM recombinant mouse prolactin or 15 nM recombinant mouse growth hormone; recombinant hormones were obtained from Dr. A.F. Parlow (National Hormone & Peptide Program, Torrance, CA, USA).
Cells were immediately placed on ice, washed with cold phosphate-buffered saline, and then lysed by scraping in the presence of RIPA (1.0% v/v NP-40, 0.5% w/v sodium deoxycholate, 0.1% w/v sodium dodecyl sulfate, 150 mM NaCl, 50 mM Tris–Cl, pH 7.6) containing protease (Millipore, Catalog No. 20-201) and phosphatase inhibitors (Sigma, Catalog No. P5726), followed by transfer to pre-chilled tubes, vigorous pipetting, and end-over-end rotation for 1 h at 4 °C. Clarified cell lysates were precleared with protein A–Sepharose and then immunoprecipitated with anti-STAT5 antibodies and processed for SDS–PAGE and Western blot analysis essentially as described elsewhere, substituting RIPA buffer for insect cell lysis buffer.
SDS–PAGE and Western blotting
Immobilized JAK2/antibody complexes were boiled for 3 min in 30 μl SDS–PAGE sample buffer and resolved by 7.5% SDS–PAGE (1 × 106cells/lane). Gels were transblotted onto Immobilon-P transfer membranes (Millipore) and probed with anti-JAK2 (Upstate Biotechnology, No. 06-255) or anti-phosphoSTAT5(Tyr694) (Cell Signaling, No. 9351S), washed three times, probed with peroxidase-conjugated secondary antibodies, and washed three times again, and the proteins were visualized using an enhanced chemiluminescence system (ECL; GE Healthcare). The relative intensity of the anti-JAK2 or anti-STAT5 signal was estimated from digital scans of the film using ImageQuant 5.2 software (Molecular Dynamics/GE Healthcare). In some experiments the membrane was then “stripped” via 30 min incubation in 100 mM β-mercaptoethanol, 2% SDS, 62.5 mM Tris–HCl, pH 6.7, at 50 °C. The membrane was then reprobed with anti-phosphotyrosine antibody (4 G 10; Upstate Biotechnology, No. 05-321) or anti-STAT5 (Santa Cruz Biotechnology, No. sc-835), washed three times, probed with peroxidase-conjugated secondary antibodies, washed three times again, and visualized using ECL. In some experiments the membranes were stripped a second time and reprobed with anti-phosphoJAK2 antibody (Upstate Biotechnology, No. 07-606) to assess the phosphorylation of the activation loop tyrosines Tyr1007/1008. Selection of peroxidase-conjugated secondary antibodies was based on whether the primary antibodies were raised in rabbit (KPL, No. 074-1516) or as a mouse monoclonal antibody (KPL, No. 074-1806).
Statistical analyses
All experiments were repeated three or more times. ANOVA was used for comparative analysis of STAT5 phosphorylation data. Student–Newman–Keuls test was used for pairwise comparison of radiolabeling autokinase activity. The statistical software package used was SigmaStat version 2.03.
Results
In our earlier study, we created several recombinant rat JAK2 (rJAK2) proteins containing cysteine-to-serine substitutions and then overexpressed these proteins via the baculovirus expression vector system [30]. To determine the effect of cysteine-to-alanine mutagenesis on in vitro radiolabeling autokinase activity, recombinant proteins were recovered from baculovirus-infected Sf21 cell lysates via immunoprecipitation with polyclonal antibodies that recognized JAK2. These samples were subdivided into two aliquots. One aliquot was assayed for in vitro radiolabeling autokinase activity and the other aliquot was assayed for relative JAK2 abundance via Western immunoblot. As shown in Fig. 1, the four cysteine-to-alanine single mutants exhibit a substantial decrease in normalized in vitro radiolabeling autokinase activity compared to the nonmutated GST/(NΔ661)rJAK2 (P<0.001). The in vitro autokinase activities of the C866A, C917A, C1094A, and C1105A mutants, normalized to the nonmutated control, were 36% (±11% SE, n = 3), 59% (±11% SE, n = 3), 3% (± 1% SE, n = 3), and 11% (±3% SE, n = 3), respectively, and the statistical significance of pairwise differences between N-lobe and C-lobe mutants (P ranging from <0.001 to 0.018) was much greater than either of the pairwise differences between the C-lobe mutants (P= 0.368) or between the N-lobe mutants (P = 0.024) (Fig. 1). The loss of autokinase activity was greater when a single alanine substitution occurred in the C lobe of the kinase domain (e.g., at position 1094 or 1105) than when it occurred in the N lobe (e.g., at position 866 or 917). These data are compared to the normalized in vitro radiolabeling autokinase activities of corresponding Cys-to-Ser mutants [30] in Table 1. Mutation of cysteine to serine at each of these four sites resulted in comparable lower levels of autokinase activity for all four mutants. In contrast, individual Cys-to-Ala mutants appeared to have caused smaller losses of autokinase activities when occurring in the N lobe and greater losses of autokinase activities when occurring in the C lobe.
Fig. 1.
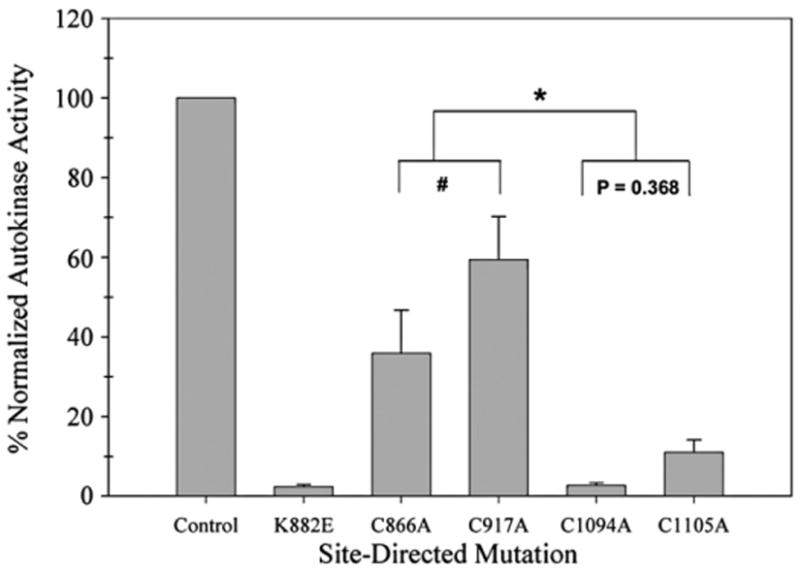
In vitro autokinase activities of four cysteine-to-alanine mutants expressed as a normalized percentage of GST/(NΔ661)rJAK2 activity. Sf21 cells were infected separately with baculoviruses expressing recombinant enzyme variants. Each JAK2-immunoprecipitated sample was divided into two aliquots. One aliquot was analyzed via anti-JAK2 immunoblot. The other aliquot was treated with 10 mM DTT and then assayed for radiolabeling autokinase activity. The intensity of each of the autokinase signals was calculated as a percentage of the nonmutated GST/(NΔ661)r]AK2 autokinase signal and then normalized for the anti-JAK2 signal intensity relative to the nonmutated GST/(NA661)rJAK2. The normalized autokinase activities of nonmutated GST/ (NΔ661)rJAK2, kinase-inactive K882E, and C866A, C917A C1094A and C1105A mutants are plotted left to right (n = 3; bars indicate mean±standard error). *P from <0.001 to 0.018; #P = 0.024.
Table 1.
In vitro radiolabeling autokinase activity changes due to alanine vs serine substitutions of critical cysteines in GST/(NΔ661)rJAK2.
| Site-directed mutation (% normalized autokinase activity ±SE) | |
|---|---|
| C866A (36±11) | C866S (15±8) |
| C917A (59±11) | C917S (28±7) |
| C1094A (3±1) | C1094S (32±7) |
| C1105A (11±3) | C1105S (19±4) |
To better understand the functional consequences of Cys-to-Ala substitution at these four critical sites and to verify that the recombinant proteins are catalytically active inside the cell, their ability to phosphorylate an inactive substrate was assessed using an in situ trans-phosphorylation assay. In this assay, Sf21 cells were co-infected with baculovirus expressing the inactive 140-kDa GST/rJAK2(K882E) and one of the eight 84-kDa GST/(NΔ661)rJAK2 cysteine mutants (Fig. 2). The upper arrow (Fig. 2A) points to the GST/rJAK2(K882E) substrate proteins, and the lower arrow points to the truncated GST/(NΔ661) rJAK2 variants. All of the GST/(NΔ661)rJAK2 variants, including the positive and negative control variants, were expressed at comparable levels (Fig. 2A, lanes 1–10). Analysis of the anti-phosphotyrosine Western blot confirmed that the inactive K882E mutant was incapable of phosphorylating the GST/rJAK2(K882E) substrate, as expected (Fig. 2B, lane 2). In contrast, all of the other JAK2 variants tested, namely, the GST/(NΔ661)rJAK2, C866S, C917S, C1094S, C1105S, C866A, C917A, C1094A, and C1105A mutants, exhibited measurable in situ kinase activity as indicated by their ability to phosphorylate the inactive substrate GST/rJAK2(K882E) (Fig. 2B, lane 1 and lanes 3–10, respectively). As shown by this in situ assay, none of the eight GST/ (NΔ661)rJAK2 cysteine mutants exhibited a gross loss of transphosphorylation activity.
Fig. 2.

In situ exokinase activities of GST/(NA661)rJAK2 mutants containing serine or alanine substitutions at four critical cysteine residues. Sf21 cells were co-infected with the inactive GST/rJAK2(K882E) substrate and GST/(NA661 )rJAK2, K882E, C866S, C917S, C1094S, C1105S, C866A, C917A C1094A or C1105A mutants (lanes 1–10, respectively). (A) Immunoprecipitated proteins were analyzed via Western immunoblot with anti-JAK2, then (B) the PVDF membranes were “stripped” and reprobed with anti-phosphotyrosine antibodies. The upper arrows indicate the 140-kDa GST/ rJAK2(K882E) substrate and the lower arrows indicate the various 84-kDa GST/ (NΔ661)rJAK2 variants.
The in vitro radiolabeling autokinase assay results shown in Fig. 1 were obtained after pretreatment with the reducing agent DTT, and all eight GST/(NΔ661)rJAK2 cysteine mutants exhibited some autokinase activity, which revealed differences among the mutants. The in vitro radiolabeling autokinase assay was used once again to determine whether the autocatalytic activities were sensitive to redox reagents in a reversible manner. Each of the eight GST/(NΔ661)rJAK2 cysteine mutants was recovered from baculovirus-infected Sf21 cells via immunoprecipitation and then pretreated with either 2.5 mM oxidant o-IBZ or 10 mM reductant DTT. Half of the o-IBZ-pretreated enzyme aliquots were re-treated with DTT, and half of the DTT-pretreated enzyme aliquots were re-treated with o-lBZ; the immunoprecipitated enzymes were removed from the redox buffers before initiating the in vitro radiolabeling autokinase assays.
As previously reported [30], the in vitro radiolabeling autokinase activity of GST/(NΔ661)rJAK2 was maximal when pretreated with reductant and nearly abolished when pretreated with oxidant, and these activities were fully reversible by re-treatment with the reciprocal redox reagent. Here we demonstrate that the reversible response to redox reagents was also preserved in each of the four cysteine-to-serine mutants, C866S, C917S, C1094S, and C1105S, as shown in Fig. 3A–D (lanes 1–4). The activity of each of these four mutants was inhibited by oxidative pretreatment (Fig. 3A–D, lane 2) and the kinase activity was restored by reductive re-treatment (Fig. 3A–D, compare lanes 2 and 4). Conversely, the activity of each of these four mutants was highest after reductive pretreatment (Fig. 3A–D, lane 1) and the kinase activity was severely inhibited by oxidative re-treatment (Fig. 3A–D, compare lanes 1 and 3).
Fig. 3.
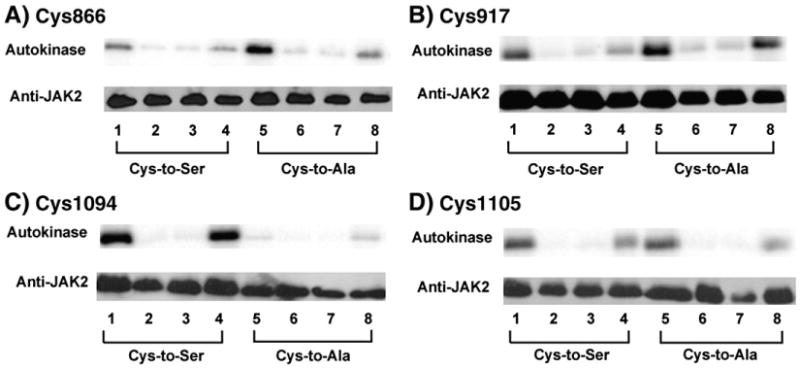
Redox-reversible in vitro radiolabeling autokinase activity of GST/(NΔ661)rJAK2 mutants containing serine or alanine substitutions at the critical cysteine residues. Using immunoprecipitated recombinant GST/(NA661)rJAI<2 variants produced in Sf21 cells, the reversible effects of redox pretreatments on in vitro radiolabeling activities were assessed on mutants in which cysteines at residues (A) 866, (B) 917, (C) 1094, and (D) 1105 were mutated to serines (lanes 1–4) or alanines (lanes 5–8). Before assay, proteins were treated with DTT (lanes 1 and 5), o-IBZ (lanes 2 and 6), DTT and then o-IBZ (lanes 3 and 7), or o-IBZ and then DTT (lanes 4 and 8). Autokinase activities are shown above, and Western immunoblots with anti-JAK2 are shown below.
Similarly, the reversible response to redox reagents was preserved in all four cysteine-to-alanine mutants (Fig. 3A–D, lanes 5–8). As before, the in vitro radiolabeling autokinase activity was inhibited by oxidative pretreatment (Fig. 3A–D, lane 6) and restored by reductive re-treatment (Fig. 3A–D, compare lanes 6 and 8). As before, maximal activity was observed after reductive pretreatment (Fig. 3A–D, lane 5) and it was severely inhibited by oxidative re-treatment (Fig. 3A–D, compare lanes 5 and 7). The reversible sensitivity to redox modulators is easiest to demonstrate with mutants possessing high autokinase activities, such as C917A or C1094S, which exhibit 59 and 32% of the nonmutated GST/(NΔ661)rJAK2's activity, respectively (Fig. 1 and [30]). The reversible response to redox reagents is much less apparent in mutants such as C1094A or C866S, whose low intrinsic autokinase activities are only 3 and 15% relative to the nonmutated GST/(NΔ661)rJAK2, respectively (Fig. 1 and [30]).
It is helpful to refer to the three-dimensional structure of JAK2's catalytic domain [31] to find clues about the possible functional role(s) of these residues. In this structure, the sulfur centers of Cys866 and Cys917 are approximately 9 Å apart, and these cysteines are located in the (β2- and β4-sheets, respectively, of the N lobe. Their spatial proximity, the conformational flexibility afforded by the arrangement of the β-sheets in the N lobe, and the absence of any significant steric obstacles in the space between Cys866 and Cys917 make it plausible to think that an intramolecular disulfide could reversibly crosslink these two residues. Intramolecular disulfide formation seems less plausible for Cys1094 and Cys1105, which are approximately 12 Å apart and at the opposite ends of the H helix in the C lobe.
Based on the structural evidence indicating that the N-lobe critical cysteines and the C-lobe critical cysteines have independent functional roles, a set of pairwise mutants was created. These mutants allow an examination of the consequences of eliminating one set of cysteines while retaining the other set on enzymatic activity. We also created GST/(NΔ661)rJAK2(4C:4S) and GST/(NΔ661)rJAK2(4C:4A) variants in which all four critical cysteines were substituted with serines or alanines, respectively, for comparison.
The in vitro radiolabeling autokinase activities of the “positive control” GST/(NΔ661)rJAK2 and “negative control” K882E were compared to those of CC866,917SS, CC1094,1105SS, 4C:4S, CC866,917AA, CC1094,1105AA, and 4C:4A (Fig. 4). When all four critical cysteines were substituted with alanines, no detectable in vitro radiolabeling autokinase activity remained (Fig. 4, lane 8). This was also the case when all critical cysteines were substituted with serines (Fig. 4, lane 5 [30]). When the serine mutations were made in pairwise fashion in either the N lobe (CC866,917SS) or the C lobe (CC1094,1105SS), the activities were nearly undetectable (Fig. 4A, lanes 3 and 4, respectively). The kinase activities of these variants were virtually indistinguishable from that of the inactive K882E control (Fig. 4, lane 2). Because these activities were so low, it was not technically feasible to determine whether these mutants retained a reversible response to redox reagents. Interestingly, the alanine-substituted mutants, CC866,917AA and CC1094,1105AA, behaved quite different from their serine-substituted counterparts (Fig. 4, lanes 3 and 4 vs 6 and 7). Both of these cysteine-to-alanine pairwise mutants exhibited significant levels of in vitro radiolabeling autokinase activity.
Fig. 4.
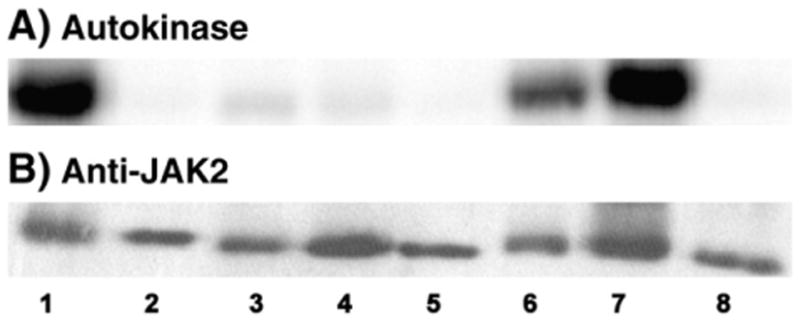
In vitro radiolabeling autokinase activities of GST/(NΔ661)rJAK2 containing combinatorial cysteine-to-serine and cysteine-to-alanine mutations. Using immunoprecipitated recombinant GST/(NA661)rJAK2 variants produced in Sf21 cells, the autokinase activities of DTT-pretreated GST/(NA661)rJAK2, K882E, CC866,917SS, CC1094,1105SS, 4C:4S, CC866,917AA CC1094,1105AA and 4C:4A are shown in (A) lanes 1–8, respectively, and corresponding Western immunoblots probed with anti-JAK2 are shown in (B).
Although the in vitro radiolabeling autokinase assay provides distinct experimental advantages, it is not as sensitive for the detection of very low levels of activity as the in situ transphosphorylation assay [39]. To determine whether the CC866,917SS, CC1094,1105SS, or 4C:4A mutants were truly inactive, as well as to verify that the alanine-substituted mutants were functional kinases within cells, in situ transphosphorylation assays were performed using coproduced inactive GST/rJAK(K882E) as the phosphorylation substrate (Fig. 5). The “positive control” GST/(NΔ661)rJAK2 exhibited robust autophosphorylation and transphosphorylation activities (Fig. 5, lane 1), whereas the “negative control” K882E exhibited no sign of either activity (Fig. 5, lane 2). These experiments clearly show that pairwise serine substitutions in the N lobe (CC866,917SS; Fig. 5, lane 3) and the C lobe (CC1094,1105SS; Fig. 5, lane 4) may impair in situ autokinase and exokinase activities, but they do not abolish them. Furthermore, mutation of all four critical cysteines to either serines (Fig. 5, lane 5 [30]) or alanines (Fig. 5, lane 8) did not truly abolish either the autokinase or the exokinase activities, although these were diminished. Consistent with the results of the in situ radiolabeling autokinase assay (Fig. 4), pairwise cysteine-to-alanine substitutions in the C lobe (CC1094,1105AA) had almost no effect on either the autokinase or the exokinase activity (Fig. 5, lane 7). Pairwise cysteine-to-alanine substitutions in the N lobe caused CC866,917AA to have slightly impaired autokinase and exokinase activities (Fig. 5, lane 6), but these were much higher than those of the serine-substituted counterpart. Based on the evidence shown in Figs. 4 and 5, it seems that mutation of cysteines in the N lobe has a somewhat greater impact on activity than mutation of cysteines in the C lobe.
Fig. 5.
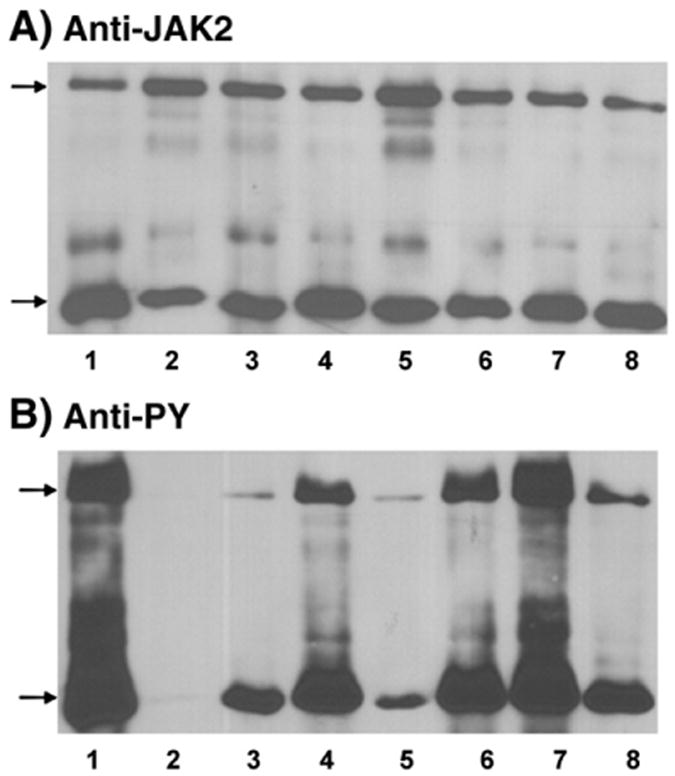
In situ exokinase activities of GST/(NΔ661)rJAK2 containing combinatorial cysteine-to-serine and cysteine-to-alanine mutations. Immunoprecipitated proteins were recovered from Sf21 cells co-infected with GST/rJAK2(K882E) and GST/(NΔ661) rJAK2, K882E, CC866,917SS, CC1094,1105SS, 4C:4S, CC866,917AA, CC1094,1105AA, or 4C:4A (in lanes 1–8, respectively). (A) Proteins were analyzed via Western immuno-blot with anti-JAK2 and then (B) the PVDF membrane was “stripped” and reprobed with anti-phosphotyrosine. The upper arrows indicate the 140-kDa GST/rJAK2(K882E) substrate and the lower arrows indicate the various 84-kDa GST/(NΔ661)rJAK2 mutants.
Because of the high levels of catalytic activity of both CC866,917AA and CC1094,1105AA, these two variants could be used to examine whether the N-lobe cysteines or the C-lobe cysteines are responsible for the redox-reversible responses of JAK2 to oxidants and reductants. These experiments are not feasible with the serine-substituted counterparts CC866,917SS and CC1094,1105SS. As is the case with nonmutated JAK2 variants, maximal activity was observed after reductive pretreatment of CC1094,1105AA (Fig. 6A, lane 1) and activity was severely inhibited by oxidative re-treatment (Fig. 6A, compare lanes 1 and 3). Conversely, autokinase activity was inhibited by oxidative pretreatment (Fig. 6A, lane 2) and restored by reductive re-treatment (Fig. 6A, compare lanes 2 and 4). In this respect, the CC1094,1105AA mutant behaved as all other JAK2 variants previously examined in the redox-reversibility assay (Fig. 3 [30]), and the evidence did not implicate the C-lobe cysteines in the reversible redox-responsiveness of the enzyme.
Fig. 6.
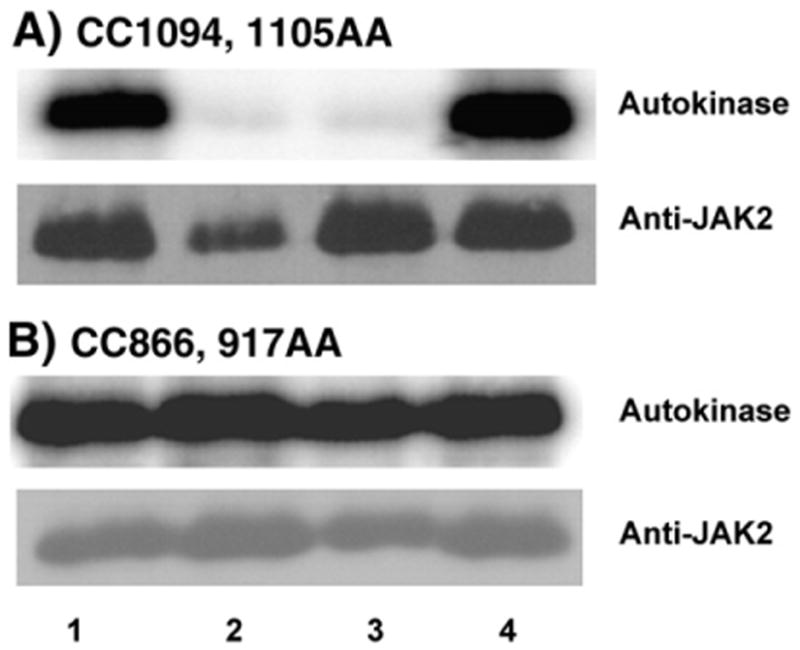
Redox-reversible in vitro radiolabeling autokinase activity of CC866,917AA and CC1094,1105AA. (A) The CC1094,1105AA mutant or (B) the CC866,917AA mutant was recovered from Sf21 cells via immunoprecipitation. Before assay, proteins were treated with DTT (lane 1), o-IBZ (lane 2), DTT and then o-IBZ (lane 3), or o-IBZ and then DTT (lane 4). Autokinase activities are shown above, and Western immunoblots with anti-JAK2 are shown below.
This typical pattern of behavior was not observed when the role(s) of the N-lobe cysteines was examined after converting these cysteines to alanines. The high level of autokinase activity exhibited by CC866,917AA after reductive pretreatment was not affected by oxidative re-treatment (Fig. 6B, lanes 1 vs 3). Oxidative pretreatment of CC866,917AA did not perceptibly inhibit the enzyme's autokinase activity (Fig. 6B, lane 2). The autokinase activity of CC866,917AA, unlike all other JAK2 variants examined in similar fashion, was completely refractive to redox reagents. This redox-refractive behavior demonstrates that Cys866 and Cys917 cooperatively function as a redox-sensitive “switch” or “sensor” in the N lobe of JAK2's catalytic domain (Fig. 7A).
Fig. 7.
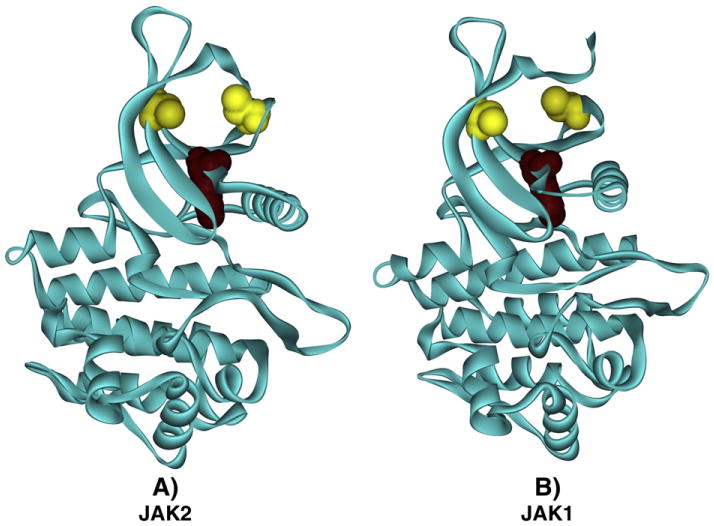
Simplified representation of the redox sensor switch in JAK2 and JAK1. The kinase domains of (A) JAK2 and (B) JAK1 are shown as ribbon diagrams in which the N lobe is positioned above the C lobe. Gold space-filling elements indicate the locations of Cys866 (A) and Cys892 (B) on the left and Cys917 (A) and Cys944 (B) on the right. The location of the essential lysine, Lys882 in (A) and Lys908 in (B), is indicated by a Deep Purple space-filling element.
This observation is of potentially broad biological significance, given the many biological roles of JAK2 and the range of diseases in which chronic oxidative stress is of reputed significance. As discussed in the introduction, JAK2 is the essential signal transducer linking growth hormone and prolactin receptor stimulation to STAT5 phosphorylation, and this signaling network is essential for the development and preservation of functional pancreatic β-islet cell mass [32-37]. The progressive loss of functional pancreatic β-islet mass is the hallmark event in the pathogenesis of type II diabetes mellitus, and chronic oxidative stress contributes to its demise [38].
We therefore examined the effect of ROS pretreatment on prolactin-stimulated STAT5 phosphorylation in the glucose-responsive, insulin-secreting pancreatic β-islet cell line βTC-6 [42], As expected, brief stimulation of quiescent βTC-6 cells with 15 nM prolactin resulted in the phosphorylation of STAT5 (Fig. 8). Overnight pretreatment of these cells with 20 μM AG490, an inhibitor of JAK2, substantially inhibited the ability of prolactin to stimulate the phosphorylation of STAT5. Intriguingly, quiescent cells treated with 0.5 mM hydrogen peroxide for 30 min before prolactin stimulation exhibited a comparably low level of STAT5 phosphorylation, indicating that JAK2 was unable to transduce signals under these conditions. Further, overnight incubation under hyperglycemic conditions (25 mM glucose) resulted in statistically significant inhibition of STAT5 tyrosine phosphorylation. Hyperglycemia contributes to chronic oxidative stress in type II diabetes [43], and high glucose medium results in a more oxidizing intracellular environment in βTC-6 cells, compared to cells grown in low-glucose medium (data not shown). Thus, oxidative stress arising from physiologically relevant sources can impair cytokine/JAK/STAT signal transduction in a manner consistent with oxidation of the redox switch.
Fig. 8.

Effects of hydrogen peroxide, hyperglycemia, and AG-490 on prolactin-stimulated STAT5 tyrosine phosphorylation in βTC-6 cells. βTC-6 cells were incubated 21–22 h in quiescence medium, hyperglycemia medium, or AG490 medium; a set of quiescent cells was then incubated with 500 μM hydrogen peroxide for 30 min before cytokine stimulation. Cells were then stimulated for 15 min without or with 15 nM recombinant mouse prolactin (mPRL). The cells were then lysed, and STAT5 was immunoprecipitated from the PAS-precleared lysate and analyzed via SDS–PAGE and Western immunoblot as described under Material and methods to measure the ratio of phosphorylated STAT5 to total STAT5; these ratios were expressed as the mean (n = 3) ± standard deviation. *P<0.001 relative to mPRL-stimulated, nonoxidized quiescent cells; #P<0.05 relative to mPRL-stimulated cells grown under hyperglycemic conditions.
Given that 500 μM H2O2 nearly obliterated the ability of JAK2 to phosphorylate STAT5 in response to prolactin, we elected to employ lower doses of H2O2 when examining the effects of ROS on growth hormone-initiated signal transduction. As shown in Fig. 9, brief stimulation of quiescent βTC-6 cells with 15 nM growth hormone resulted in robust phosphorylation of STAT5. Pretreatment of quiescent cells with 25, 50, and 100 μM H2O2 for 30 min before GH stimulation progressively inhibited GH-stimulated STAT5 phosphorylation, such that GH stimulation after pretreatment with 100 μM H2O2 resulted in half of the response observed in the control (untreated) sample. The data shown in Fig. 9 provide preliminary support for a simple yet important concept, which is that theJAK2 redox switch is capable of providing a graduated regulatory response to incremental changes in the intracellular redox state.
Fig. 9.
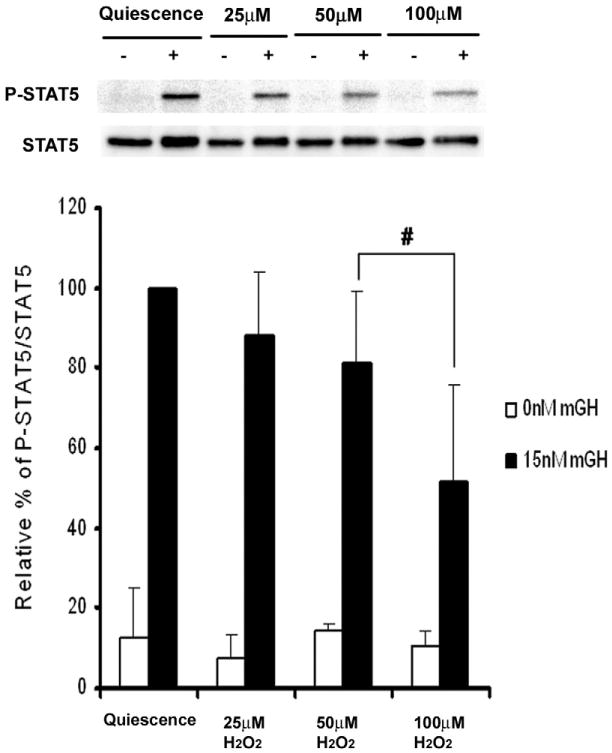
Effect of hydrogen peroxide on growth hormone-stimulated STAT5 tyrosine phosphorylation in βTC-6 cells. βTC-6 cells were incubated 21–22 h in quiescence medium and then incubated with 0, 25, 50, or 100 μM hydrogen peroxide for 30 min before cytokine stimulation. Cells were then stimulated for 15 min without or with 15 nM recombinant mouse growth hormone (mGH). The cells were then lysed, and STAT5 was immunoprecipitated from the PAS-precleared lysate and analyzed via SDS–PAGE and Western immunoblot as described under Material and methods to measure the ratio of phosphorylated STAT5 to total STAT5; these ratios were expressed as the mean (n = 3) ± standard deviation. #P<0.05 relative to mGH-stimulated, nonoxidized quiescent cells.
Discussion
The results presented in Fig. 6 provide the first direct evidence for a cysteine-based redox switch that reversibly inhibits JAK2's autocatalytic activity. This observation provides a mechanistic basis for reports that oxidants associated with “oxidative stress” inhibit Janus kinases and downstream pathway events [19–23,25]. The discovery of the redox switch is an extension of previous work identifying four cysteines in the catalytic domain that cooperatively maintain JAK2's kinase activity [30]. The C-lobe cysteines and the N-lobe cysteines cooperate, as supported by the data in Figs. 4 and 5, and it is now evident that these two sets of cysteines have distinctly different functional roles.
The structure of the catalytic domain of JAK2, like other tyrosine kinases, consists of an N-terminal lobe comprising alternating β-sheets and a C-terminal lobe comprising α-helices [31,44]. The N-lobe cysteines are close to both the ATP-binding site and the invariant lysine, Lys882, which is absolutely essential for catalytic activity. It is conceivable that the reduced thiols of Cys866 and Cys917 contribute directly to the catalytic cycle and that oxidation prevents these thiols from participating in their normal function in the catalytic cycle. The precise mechanism through which this “redox switch” controls catalysis remains undefined.
In JAK2, the redox switch motif can be simplified to a three-point cluster containing the invariant lysine as a reference point and in which the two sulfur centers are approximately 9 Å apart. As shown in Fig. 7B, a nearly identical motif is present in the structure of JAK1 [45]. This is consistent with recent evidence describing the oxidative inhibition of JAK1 in cells exposed to parthenolide [46]. If the oxidation of one cysteine to sulfenic acid is sufficient to regulate catalysis, then this simplifies to a two-point motif, which could be common among protein kinases.
The functional roles of the C-lobe cysteines located at residues 1094 and 1105 have yet to be determined. At this stage, we can state only that they maintain catalytic competency, and they do so in a cooperative fashion with the N-lobe cysteines. Naturally occurring mutations in JAK3 that disrupt or abolish this helix also impair JAIG's activity [47]. One might speculate that they are broadly important because they appear in a highly conserved CysPro(X9)CysTrp motif found in the C lobe of many mammalian protein-tyrosine kinases, commonly in the form of CysPro(X3)Tyr(X5)CysTrp (data not shown).
Cysteine residues serve a variety of important functions due to the chemical versatility of reactive sulfhydryl groups [48–51]. The reduction potential of a generic disulfide/dithiol exchange (− 220 mV) is within the estimated reduction potential of the mammalian cytosol (− 220 to −240 mV [52]), and reversible dithiol/disulfide exchanges are well established as covalent activity switches [53–55]. Sulfenic acids represent another important oxidation state of cysteines, both as stabilized end states and as reactive intermediates [56,57]. Sulfenic acid formation directly regulates enzymes containing reactive-site cysteines, such as protein tyrosine phosphatases [26,29].
The oxidation of cysteines to sulfenic acids could explain why the substitution of serines for cysteines at residues 866 and 917 caused a near-complete loss of activity, whereas the substitution of alanines at these positions allowed relatively high activity levels to be retained. The supposition is that the substitution of alanines for cysteines was less disruptive to the folded conformation of JAK2's catalytic domain than the substitution of serines, based on hydropathy [58], electronegativity [59], and hydrogen bonding [60] considerations. If the introduction of a hydroxyl moiety at this site via site-directed mutagenesis impairs activity by mimicking the hydroxyl moiety resulting from sulfenic acid formation, then the CC866,917SS mutant may represent a functional model of the oxidized state of JAK2.
Oxidation to sulfenic acid would also explain why a single substitution of either serine or alanine for either Cys866 or Cys917 conserves sensitivity to redox regulation (Fig. 3). Whereas o-lBZ treatment is known to cause the reversible formation of disulfide bonds, it has not yet been shown to oxidize cysteines into sulfenic acids. To determine whether this is plausible, the sulfenic acid content of GST/(NΔ661) rJAK2 was assessed after treatment with dimedone, which selectively condenses with sulfenic acid to form a stable thioether product detectable through immunochemical techniques [61]. Pretreatment of GST/ (NΔ661)rJAK2 with o-IBZ substantially increased the amount of sulfenic acid detected in JAK2, and the sulfenic acid content of DTT-pretreated JAK2 was less than half that of the o-lBZ-pretreated enzyme (45.6 ±15.0% SE, n = 4, data not shown). Thus, sulfenic acid formation in the Cys866/Cys917 redox switch is one plausible explanation for our observations. However, the fact that sulfenic acid formation can account for the oxidative inhibition of JAK2 mutants in vitro does not exclude the possibility that a disulfide bond is formed in vivo upon the oxidation of Cys866 and Cys917. Nearby reduced cysteines destabilize cysteine sulfenic acids [62], and cysteine sulfenic acids serve as intermediates in nonenzymatic disulfide bond formation [63]. JAK2 might be alternatively oxidized to either the sulfenic or the disulfide state, resulting in an equilibrium mixture.
Based on this demonstration of a cysteine-based redox switch, obtained via site-directed mutagenesis of the recombinant form of JAK2, one can propose that the redox-mediated activation of this switch provides an important physiological regulatory mechanism, which should apply to native forms of JAK1 and JAK2. We propose that the redox switch is as important to catalysis as the essential invariant lysine [64] and the activation loop [65] and has biological relevance under physiological and pathophysiological conditions [6–8,19–25]. In addition to identifying this redox switch, we also presented data to test the prediction that it modulates JAK2-coupled responses to cytokine stimulation in response to oxidative stress. The data in Figs. 8 and 9 provide preliminary evidence for the hypothesis that this switch may provide insights into the pathogenesis of type II diabetes mellitus, a disease affected by chronic oxidative stress. However, one must recognize that the results shown in Figs. 8 and 9 do not provide direct evidence that Cys866 and Cys917 serve the physiological role of a redox-sensitive regulatory switch. This proposition is a viable, but as yet unconfirmed, hypothesis.
Clearly, the implications of this hypothesis will require further exploration, and because of the complexity of type II diabetes mellitus and other diseases associated with oxidative stress, more sophisticated experimental techniques will be required. Several of our key experiments (e.g., Fig. 6) employed in vitro model chemistry and recombinant proteins to clearly demonstrate a fully reversible “all or none” biochemical phenomenon. Although this approach may be useful to demonstrate a concept, all or none responses are unlikely to occur under physiological circumstances, in which chronic attenuation of signal transduction by a factor of 10% or less may occur. Unfortunately, whereas 50% inhibition of signal transduction can be revealed as a statistically significant change via Western blot assays (e.g., Fig. 9), these assays lack the sensitivity and precision to detect smaller changes that may have biologically significant repercussions. In type II diabetes mellitus or in ischemia–reperfusion injuries, there are further complications introduced by initial compensatory responses, such that the reproducibility of biological experiments will be highly dependent upon the precise timing of an experiment within the sequence of events after the initial oxidative stress. Indeed, because the biology of redox regulation of cytokine signaling is rife with complications, it is important to fully understand the biochemical underpinnings of the direct and indirect mechanisms for redox regulation of key signal transducers. The identification of a redox switch in JAK2 should improve our comprehension of this fascinatingly complex field.
Acknowledgments
This research was made possible by Grant 1-R56-DK082781-01 from the National Institute of Diabetes and Digestive and Kidney Diseases (R.J.D.). Additional support was provided by Grant 5-R01-HL088101-05 from the National Heart, Lung, and Blood Institute (G.W.B.), the Alliance for Graduate Education in Mississippi (J.K.S.), the Summer Undergraduate Research Experience Program, the School of Graduate Studies in the Health Sciences (S.P.), and an Institutional Research Support Grant from the University of Mississippi Medical Center (R.J.D.). The authors gratefully acknowledge the excellent resources provided by Dr. Albert F. Parlow, and many helpful discussions with Dr. Naila M. Mamoon, Dr. Jonathan Hosier, Dr. Stanley V. Smith, and Ms. Loraine C. Duhé.
Abbreviations
- DTT
dithiothreitol
- GST
glutathione S-transferase
- JAK
Janus protein-tyrosine kinase
- o-IBZ
ortho-iodosobenzoate
- rJAK2
rat JAK2
- ROS
reactive oxygen species
- SDS–PAGE
sodium dodecyl sulfate–polyacrylamide gel electrophoresis
- Sf21
Spodoptera frugiperda cell line 21
- STAT
signal transducer and activator of transcription
References
- 1.Sarsour EH, Kumar MG, Chaudhuri L, Kalen AL, Goswami PC. Redox control of the cell cycle in health and disease. Antioxid Redox Signal. 2009;11:2985–3011. doi: 10.1089/ars.2009.2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 3.Faraci FM. Reactive oxygen species: influence on cerebral vascular tone. J Appl Physiol. 2006;100:739–743. doi: 10.1152/japplphysiol.01044.2005. [DOI] [PubMed] [Google Scholar]
- 4.Freedman JE. Oxidative stress and platelets. Arterioscler Thromb Vase Biol. 2008;28:s11–16. doi: 10.1161/ATVBAHA.107.159178. [DOI] [PubMed] [Google Scholar]
- 5.Forman HJ, Torres M. Reactive oxygen species and cell signaling: respiratory burst in macrophage signaling. Am J Respir Crit Care Med. 2002;166:S4–8. doi: 10.1164/rccm.2206007. [DOI] [PubMed] [Google Scholar]
- 6.Nistala R, Whaley-Connell A, Sowers JR. Redox control of renal function and hypertension. Antioxid Redox Signal. 2008;10:2047–2089. doi: 10.1089/ars.2008.2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rahman I, Adcock IM. Oxidative stress and redox regulation of lung inflammation in COPD. Eur Respir J. 2006;28:219–242. doi: 10.1183/09031936.06.00053805. [DOI] [PubMed] [Google Scholar]
- 8.Forbes JM, Coughlan MT, Cooper ME. Oxidative stress as a major culprit in kidney disease in diabetes. Diabetes. 2008;57:1446–1454. doi: 10.2337/db08-0057. [DOI] [PubMed] [Google Scholar]
- 9.Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 10.Droge W. Oxidative stress and ageing: is ageing a cysteine deficiency syndrome? Philos Trans R Soc London B Biol Sci. 2005;360:2355–2372. doi: 10.1098/rstb.2005.1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Radosevic N, Winterstein D, Keller JR, Neubauer H, Pfeffer K, Linnekin D. JAK2 contributes to the intrinsic capacity of primary hematopoietic cells to respond to stem cell factor. Exp Hematol. 2004;32:149–156. doi: 10.1016/j.exphem.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 12.Shillingford JM, Miyoshi K, Robinson GW, Grimm SL, Rosen JM, Neubauer H, Pfeffer K, Hennighausen L. Jak2 is an essential tyrosine kinase involved in pregnancy-mediated development of mammary secretory epithelium. Mol Endocriol. 2002;16:563–570. doi: 10.1210/mend.16.3.0805. [DOI] [PubMed] [Google Scholar]
- 13.Neubauer H, Cumano A, Muller M, Wu H, Huffstadt U, Pfeffer K. Jak2 deficiency defines an essential developmental checkpoint in definitive hematopoiesis. Cell. 1998;93:397–409. doi: 10.1016/s0092-8674(00)81168-x. [DOI] [PubMed] [Google Scholar]
- 14.Parganas E, Wang D, Stravopodis D, Topham DJ, Marine JC, Teglund S, Vanin EF, Bodner S, Colamonici OR, van Deursen JM, Grosveld G, Ihle JN. Jak2 is essential for signaling through a variety of cytokine receptors. Cell. 1998;93:385–395. doi: 10.1016/s0092-8674(00)81167-8. [DOI] [PubMed] [Google Scholar]
- 15.Maziere C, Conte MA, Maziere JC. Activation of JAK2 by the oxidative stress generated with oxidized low-density lipoprotein. Free Radic Biol Med. 2001;31:1334–1340. doi: 10.1016/s0891-5849(01)00649-9. [DOI] [PubMed] [Google Scholar]
- 16.Sandberg EM, Sayeski PP. Jak2 tyrosine kinase mediates oxidative stress-induced apoptosis in vascular smooth muscle cells. J Biol Client. 2004;279:34547–34552. doi: 10.1074/jbc.M405045200. [DOI] [PubMed] [Google Scholar]
- 17.Carballo M, Conde M, El Bekay R, Martin-Nieto J, Camacho MJ, Monteseirin J, Conde J, Bedoya FJ, Sobrino F. Oxidative stress triggers STAT3 tyrosine phosphorylation and nuclear translocation in human lymphocytes. J Biol Client. 1999;274:17580–17586. doi: 10.1074/jbc.274.25.17580. [DOI] [PubMed] [Google Scholar]
- 18.Simon AR, Rai U, Fanburg BL, Cochran BH. Activation of the JAK-STAT pathway by reactive oxygen species. Am J Physiol. 1998;275:C1640–1652. doi: 10.1152/ajpcell.1998.275.6.C1640. [DOI] [PubMed] [Google Scholar]
- 19.Jang EH, Park CS, Lee SK, Pie JE, Kang JH. Excessive nitric oxide attenuates leptin-mediated signal transducer and activator of transcription 3 activation. Life Sci. 2007;80:609–617. doi: 10.1016/j.lfs.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 20.Duhe RJ, Evans GA, Erwin RA, Kirken RA, Cox GW, Farrar WL. Nitric oxide and thiol redox regulation of Janus kinase activity. proc Natl Acad Sci U S A. 1998;95:126–131. doi: 10.1073/pnas.95.1.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bingisser RM, Tilbrook PA, Holt PG, Kees UR. Macrophage-derived nitric oxide regulates T cell activation via reversible disruption of the Jak3/STAT5 signaling pathway. J Immunol. 1998;160:5729–5734. [PubMed] [Google Scholar]
- 22.Mazzoni A, Bronte V, Visintin A, Spitzer JH, Apolloni E, Serafini P, Zano-vello P, Segal DM. Myeloid suppressor lines inhibit T cell responses by an NO-dependent mechanism. J Immunol. 2002;168:689–695. doi: 10.4049/jimmunol.168.2.689. [DOI] [PubMed] [Google Scholar]
- 23.Kaur N, Lu B, Monroe RK, Ward SM, Halvorsen SW. Inducers of oxidative stress block ciliary neurotrophic factor activation of Jak/STAT signaling in neurons. J Neurochem. 2005;92:1521–1530. doi: 10.1111/j.1471-4159.2004.02990.x. [DOI] [PubMed] [Google Scholar]
- 24.Monroe RK, Halvorsen SW. Cadmium blocks receptor-mediated Jak/STAT signaling in neurons by oxidative stress. Free Radic Biol Med. 2006;41:493–502. doi: 10.1016/j.freeradbiomed.2006.04.023. [DOI] [PubMed] [Google Scholar]
- 25.Di Bona D, Cippitelli M, Fionda C, Camma C, Licata A, Santoni A, Craxi A. Oxidative stress inhibits IFN-α-induced antiviral gene expression by blocking the JAK–STAT pathway. J Hepatol. 2006;45:271–279. doi: 10.1016/j.jhep.2006.01.037. [DOI] [PubMed] [Google Scholar]
- 26.Tonks NK. Redox redux: revisiting PTPs and the control of cell signaling. Cell. 2005;121:667–670. doi: 10.1016/j.cell.2005.05.016. [DOI] [PubMed] [Google Scholar]
- 27.Meng TC, Hsu SF, Tonks NK. Development of a modified in-gel assay to identify protein tyrosine phosphatases that are oxidized and inactivated in vivo. Methods. 2005;35:28–36. doi: 10.1016/j.ymeth.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 28.Groen A, Lemeer S, vander Wijk T, Overvoorde J, Heck AJ, Ostman A, Barford D, Slijper M, den Hertog J. Differential oxidation of protein-tyrosine phosphatases. J Biol Chem. 2005;280:10298–10304. doi: 10.1074/jbc.M412424200. [DOI] [PubMed] [Google Scholar]
- 29.van Montfort RL, Congreve M, Tisi D, Carr R, Jhoti H. Oxidation state of the active-site cysteine in protein tyrosine phosphatase 1B. Nature. 2003;423:773–777. doi: 10.1038/nature01681. [DOI] [PubMed] [Google Scholar]
- 30.Mamoon NM, Smith JK, Chatti K, Lee S, Kundrapu K, Duhe RJ. Multiple cysteine residues are implicated in Janus kinase 2-mediated catalysis. Biochemistry. 2007;46:14810–14818. doi: 10.1021/bi701118u. [DOI] [PubMed] [Google Scholar]
- 31.Lucet IS, Fantino E, Styles M, Bamert R, Patel O, Broughton SE, Walter M, Burns CJ, Treutlein H, Wilks AF, Rossjohn J. The structural basis of Janus kinase 2 inhibition by a potent and specific pan-Janus kinase inhibitor. Blood. 2006;107:176–183. doi: 10.1182/blood-2005-06-2413. [DOI] [PubMed] [Google Scholar]
- 32.Foa PP, Galansino G, Costa E. Prolactin and the secretion of insulin and glucagon by the pancreas. Am J Physiol. 1955;182:493–496. doi: 10.1152/ajplegacy.1955.182.3.493. [DOI] [PubMed] [Google Scholar]
- 33.Kinash B, Macdougall I, Evans MA, Bryans FE, Haist RE. Effects of anterior pituitary extracts and of growth hormone preparations on the islets of Langerhans and the pancreas. Diabetes. 1953;2:112–121. doi: 10.2337/diab.2.2.112. [DOI] [PubMed] [Google Scholar]
- 34.Brelje TC, Stout LE, Bhagroo NV, Sorenson RL. Distinctive roles for prolactin and growth hormone in the activation of signal transducer and activator of transcription 5 in pancreatic islets of Langerhans. Endocrinology. 2004;145:4162–4175. doi: 10.1210/en.2004-0201. [DOI] [PubMed] [Google Scholar]
- 35.Freemark M, Avril I, Fleenor D, Fleenor D, Driscoll P, Petro A, Opara E, Kendall W, Oden J, Bridges S, Binart N, Breant B, Kelly PA. Targeted deletion of the PRL receptor: effects on islet development, insulin production, and glucose tolerance. Endocrinology. 2002;143:1378–1385. doi: 10.1210/endo.143.4.8722. [DOI] [PubMed] [Google Scholar]
- 36.Liu JL, Coschigano KT, Robertson K, Lipsett M, Guo Y, Kopchick JJ, Kumar U, Liu YL. Disruption of growth hormone receptor gene causes diminished pancreatic islet size and increased insulin sensitivity in mice. Am J Physiol Endocrinol Metab. 2004;287:E405–413. doi: 10.1152/ajpendo.00423.2003. [DOI] [PubMed] [Google Scholar]
- 37.Jackerott M, Moldrup A, Thams P, Galsgaard ED, Knudsen J, Lee YC, Nielsen JH. STAT5 activity in pancreatic β-cells influences the severity of diabetes in animal models of type 1 and 2 diabetes. Diabetes. 2006;55:2705–2712. doi: 10.2337/db06-0244. [DOI] [PubMed] [Google Scholar]
- 38.Robertson RP, Harmon J, Tran PO, Poitout V. β-Cell glucose toxicity, lipotoxicity, and chronic oxidative stress in type 2 diabetes. Diabetes. 2004;53(Suppl. 1):S119–124. doi: 10.2337/diabetes.53.2007.s119. [DOI] [PubMed] [Google Scholar]
- 39.Chatti K, Farrar WL, Duhe RJ. Tyrosine phosphorylation of the Janus kinase 2 activation loop is essential for a high-activity catalytic state but dispensable for a basal catalytic state. Biochemistry. 2004;43:4272–4283. doi: 10.1021/bi036109b. [DOI] [PubMed] [Google Scholar]
- 40.Duhe RJ, Farrar WL. Characterization of active and inactive forms of theJAK2 protein-tyrosine kinase produced via the baculovirus expression vector system. J Biol Chem. 1995;270:2308–423089. doi: 10.1074/jbc.270.39.23084. [DOI] [PubMed] [Google Scholar]
- 41.Duhe R, Clark EA, Farrar WL. Characterization of the in vitro kinase activity of a partially purified soluble GST/JAK2 fusion protein. Mol Cell Biochem. 2002;236:23–35. doi: 10.1023/a:1016186907376. [DOI] [PubMed] [Google Scholar]
- 42.Poitout V, Stout LE, Armstrong MB, Walseth TF, Sorenson RL, Robertson RP. Morphological and functional characterization of β TC-6 cells—an insulin-secreting cell line derived from transgenic mice. Diabetes. 1995;44:306–313. doi: 10.2337/diab.44.3.306. [DOI] [PubMed] [Google Scholar]
- 43.Robertson RP. Chronic oxidative stress as a central mechanism for glucose toxicity in pancreatic islet β cells in diabetes. J Biol Chem. 2004;279:42351–42354. doi: 10.1074/jbc.R400019200. [DOI] [PubMed] [Google Scholar]
- 44.Taylor SS, Radzio-Andzelm E, Hunter T. How do protein kinases discriminate between serine/threonine and tyrosine? Structural insights from the insulin receptor protein-tyrosine ldnase. FASEB J. 1995;9:1255–1266. doi: 10.1096/fasebj.9.13.7557015. [DOI] [PubMed] [Google Scholar]
- 45.Williams NK, Bamert RS, Patel O, Wang C, Walden PM, Wills AF, Fantino E, Rossjohn J, Lucet IS. Dissecting specificity in the Janus kinases: the structures of JAK-specific inhibitors complexed to the JAK1 and JAK2 protein tyrosine kinase domains. J Mol Biol. 2009;387:219–232. doi: 10.1016/j.jmb.2009.01.041. [DOI] [PubMed] [Google Scholar]
- 46.Kurdi M, Booz GW. Evidence that IL-6-type cytoldne signaling in cardiomyocytes is inhibited by oxidative stress: parthenolide targets JAK1 activation by generating ROS. J Cell Physiol. 2007;212:424–431. doi: 10.1002/jcp.21033. [DOI] [PubMed] [Google Scholar]
- 47.Lai KS, Jin Y, Graham DK, Witthuhn BA, Ihle JN, Liu ET. A kinase-deficient splice variant of the human JAK3 is expressed in hematopoietic and epithelial cancer cells. J Biol Chem. 1995;270:25028–25036. doi: 10.1074/jbc.270.42.25028. [DOI] [PubMed] [Google Scholar]
- 48.Bindoli A, Fukuto JM, Forman HJ. Thiol chemistry in peroxidase catalysis and redox signaling. Antioxid Redox Signal. 2008;10:1549–1564. doi: 10.1089/ars.2008.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brandes N, Schmitt S, Jakob U. Thiol-based redox switches in eukaryotic proteins. Antioxid Redox Signal. 2009;11:997–1014. doi: 10.1089/ars.2008.2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gilbert HF. Thiol/disulfide exchange equilibria and disulfide bond stability. Methods Enzymol. 1995;251:8–28. doi: 10.1016/0076-6879(95)51107-5. [DOI] [PubMed] [Google Scholar]
- 51.Giles NM, Giles GI, Jacob C. Multiple roles of cysteine in biocatalysis. Biochem Biophys Res Commun. 2003;300:1–4. doi: 10.1016/s0006-291x(02)02770-5. [DOI] [PubMed] [Google Scholar]
- 52.Hwang C, Sinskey AJ, Lodish HF. Oxidized redox state of glutathione in the endoplasmic reticulum. Science. 1992;257:1496–1502. doi: 10.1126/science.1523409. [DOI] [PubMed] [Google Scholar]
- 53.Appenzeller-Herzog C, Riemer J, Christensen B, Sorensen ES, Ellgaard LA. novel disulphide switch mechanism in Ero1α balances ER oxidation in human cells. EMBO J. 2008;27:2977–2987. doi: 10.1038/emboj.2008.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barford D. The role of cysteine residues as redox-sensitive regulatory switches. Curr Opin Struct Biol. 2004;14:679–686. doi: 10.1016/j.sbi.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 55.Gilbert HF. Biological disulfides: the third messenger? Modulation of phosphofruc-tokinase activity by thiol/disulfide exchange. J Biol Chem. 1982;257:12086–12091. [PubMed] [Google Scholar]
- 56.Poole LB, Karplus PA, Claiborne A. Protein sulfenic acids in redox signaling. Annu Rev Pharmacol Toxicol. 2004;44:325–347. doi: 10.1146/annurev.pharmtox.44.101802.121735. [DOI] [PubMed] [Google Scholar]
- 57.Saurin AT, Neubert H, Brennan JP, Eaton P. Widespread sulfenic acid formation in tissues in response to hydrogen peroxide. Proc Natl Acad Sci U S A. 2004;101:17982–17987. doi: 10.1073/pnas.0404762101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 59.Pauling L. The Nature of the Chemical Bond and the Structure of Molecules and Crystals: an Introduction to Modern Structural Chemistry. Cornell Univ. Press; Ithaca, NY: p. 1960. [Google Scholar]
- 60.Raso SW, Clark PL, Haase-Pettingell C, King J, Thomas GJ., Jr Distinct cysteine sulfhydryl environments detected by analysis of Raman S-hh markers of Cys→Ser mutant proteins. J Mol Biol. 2001;307:899–911. doi: 10.1006/jmbi.2001.4476. [DOI] [PubMed] [Google Scholar]
- 61.Seo YH, Carroll KS. Profiling protein thiol oxidation in tumor cells using sulfenic acid-specific antibodies. Proc Natl Acad Sci U S A. 2009;106:16163–16168. doi: 10.1073/pnas.0903015106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Claiborne A, Miller H, Parsonage D, Ross RP. Protein-sulfenic acid stabilization and function in enzyme catalysis and gene regulation. FASEB J. 1993;7:1483–1490. doi: 10.1096/fasebj.7.15.8262333. [DOI] [PubMed] [Google Scholar]
- 63.Rehder DS, Borges CR. Cysteine sulfenic acid as an intermediate in disulfide bond formation and nonenzymatic protein folding. Biochemistry. 2010;49:7748–7755. doi: 10.1021/bi1008694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Carrera AC, Alexandrov K, Roberts TM. The conserved lysine of the catalytic domain of protein kinases is actively involved in the phosphotransfer reaction and not required for anchoring ATP. Proc Natl Acad Sci U S A. 1993;90:442–446. doi: 10.1073/pnas.90.2.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Feng J, Witthuhn BA, Matsuda T, Kohlhuber F, Kerr IM, Ihle JN. Activation of Jak2 catalytic activity requires phosphorylation of Y1007 in the kinase activation loop. Mol Cell Biol. 1997;17:2497–2501. doi: 10.1128/mcb.17.5.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]