

Abstract
Context:
The most common cause of resistance to thyroid hormone (RTH) is heterozygous thyroid hormone receptor β (THRB) gene mutations. Homozygous mutations in the THRB gene are a rare event.
Objective:
In this study, the clinical findings of three new patients (belonging to two families) homozygous for mutations in the THRB gene are compared to three other families in which affected individuals lack a normal TRβ.
Methods:
We conducted clinical studies and genetic analyses.
Results:
The clinical presentation in all three homozygous subjects was unusually severe; their phenotype was characterized by compromised intellectual development, tachycardia, goiter, growth retardation, and hearing loss. This was comparable with one other reported patient homozygous for mutant TRβ, but not in RTH due to THRB gene deletions.
Conclusion:
We report three new subjects, from two families, in whom RTH was associated with homozygous mutations in the THRB gene. They represent an important addition to the single known patient homozygous for a mutant TRβ. The clinical and laboratory abnormalities indicate a strong dominant-negative effect and are in agreement with data obtained from mice expressing a mutant Thrb in both alleles. This report strengthens the concept that the mutated TRβ interferes with the function of the TRα1 in humans.
The syndrome of resistance to thyroid hormone (RTH) is caused by decreased tissue responsiveness to thyroid hormone (TH) (1, 2). The biochemical hallmark of RTH is abnormally elevated serum free TH levels together with nonsuppressed TSH secretion. The clinical presentation is highly variable, ranging from isolated biochemical abnormalities to a mixture of hypo-hyperthyroid signs and symptoms (1–3). The cause is mostly mutations in TH receptor β (THRB) gene (1, 2).
With the exception of a single family with recessively inherited RTH due to a deletion of the THRB gene (4), inheritance of RTH is autosomal dominant (1). Clinical and biochemical manifestations are attributed to the interference of mutant TRβ with the function of the normal receptor, a phenomenon termed “dominant-negative effect” (DNE) (5).
Two homozygous cases have been reported so far: the complete absence of both THRB alleles, described by Takeda et al. (4, 6), and the homozygous case, caused by an amino acid deletion, reported by Usala et al. (7). The apparent homozygosity of a third case, described by Frank-Raue et al. (8), could have been the result of a deletion of the paternal allele, added to the missense mutation inherited from the mother, producing compound heterozygosity.
We now report two families with RTH in which three subjects are homozygous for two different point mutations in the THRB gene. The clinical manifestations of these three individuals are more severe not only than all heterozygotes reported but also those homozygotes for gene deletion, indicating a deleterious effect of occupancy of TH response elements (TRE) by unliganded receptor and possibly the interference with TRα function.
Patients and Methods
Cases reports
Family Mbyd
The proposita (subject IV-I; Fig. 1A), with XX karyotype, was born to a 17-yr-old single primigravida of mixed Australian Aborigine and White European ancestry. The mother of the proposita (III-1) revealed that her maternal uncle (II-1) was the father of the proposita. The weight at birth was 2470 g. The neonate was not frankly dysmorphic but had a prominent nasal bridge, backward-sloping forehead, and overriding fourth toes and was later noted to have an intermittent squint. No goiter was appreciated on physical examination. Because of very high serum TSH levels, a 99mpertechnetate scan was obtained, which showed a thyroid gland of normal size, contour, and position with increased uptake. Treatment with a β-blocker for tachycardia was started at 1 wk of age.
Fig. 1.

Pedigrees showing the genotype and TFT results. Results are aligned with each symbol. Values represent the mean of two determinations on separate samples collected several months apart. In two instances (A, II-1 and IV-1), when samples were obtained almost 4 yr apart, both values are given. Ages represent those at the time of blood sampling. Tx, Thyroidectomy.
At 1 month of age, brainstem-evoked response audiometry showed a moderate mixed hearing loss bilaterally: air and bone conduction at 2000 Hz was 50 and 35, respectively, for both ears [sensorineural component considered to be mild to moderate at 35 decibels (dB)]. The proposita was fitted with hearing aids at age 9 months. Thyroid function tests (TFT) are shown in Fig. 1A. At that time, there was no palpable goiter.
At 1 yr and 8 months of age, the major issue was failure to thrive, which improved with increased β-blockers (propranolol and then atenolol) and increased calorie intake. The weight curve has subsequently followed the 1st centile. There was delay in verbal development, and IQ score (Griffith Mental Development Scale) was 68. A patent ductus arteriosus and foramen ovale were also noted. A urinary tract infection led to renal ultrasound, which demonstrated unilateral urinary reflux, but morphologically normal kidney. Recurrent episodes of tonsillitis and glue ear, associated with significant snoring and breath holding, were treated by tonsilloadenoidectomy and grommets.
At 3 yr and 2 months of age, the proband underwent an extensive developmental and neuropsychological evaluation. A mild delay in her gross motor development was noted. She was able to walk on tiptoes, to ascend stairs with one foot per step and one held hand, and descend with two feet per step.
She had decreased visual attention while performing motor tasks and tended not to look at the task she was performing. She required help with expressive speech and chewing. She produced long utterances using appropriate sentence-like intonation with unintelligible words. Her receptive language abilities were appropriate when compared with her peers with hearing impairment, but the rate of progress indicated that she was at risk of developing receptive language delay. Overall, her speech skills were delayed, and she was not participating in conversations with others. IQ score [Wechsler Preschool and Primary Scale of Intelligence (WPPSI) III test] was 72. Bone age at 3 yr was 9 months.
No facial dysmorphism was present (Fig. 2A), but a diffuse visible goiter was by now evident (Fig. 2B). TFT at that time are also shown in Fig. 1A.
Fig. 2.

A and B, Proposita of family Mbyd. A, Facial features; B, visible goiter. C–H, Subjects from family Mozv, facial features and appearance of the thyroid gland. C and D, Subject IV-2; E and F, subject IV-3. Both show the dysmorphic features of birdlike facies with prominent nasal bridge. Affected subject IV-2 had thyroidectomy, whereas subject IV-3 has a visible goiter. G and H, Subject IV-1, the normal sister without the facial features of her affected siblings. Thyroid gland is visible due to autoimmune thyroid disease.
At 4 yr and 8 months, her IQ score (WPPSI III test) was 61. Her height was 92.2 cm (<3rd centile), weight was 13 kg (<3rd centile), and the head circumference is 47.2 cm (3rd centile). Bone age at 4 yr remained at 9 months. Testing with Interacoustics Diagnostic Audiometer (AD28) indicated a hearing loss of 45 dB at 500 Hz, sloping to 55 dB at 4000 Hz on the right and left ears.
Family members.
The mother (III-1) had a normal serum TSH but high T4 and T3 levels (Fig. 1A). Her height, weight, and body mass index were 159.6 cm, 80.2 kg, and 31.5 kg/m2, respectively. She did not have tachycardia, but she had a small goiter and a mild hearing deficit. She completed secondary school but for 3 yr was placed in a support class.
The proposita's designated father had a hearing deficit of unknown etiology and was on levothyroxine (l-T4) treatment for an undetermined thyroid problem. He declined any form of investigation.
Family members I-1, II-2, and III-2 had elevated serum iodothyronine concentrations and normal TSH as shown in Fig. 1A, and all had hearing impairment.
Family Mozv
The proposita (IV-2; Fig. 1B), with XX karyotype, was the second child born to consanguineous Turkish parents. She had dysmorphic features consisting of birdlike facies with prominent nasal bridge (Fig. 2, C and D) and hearing impairment (evoked responses, 60 dB normalized hearing level right ear, and at 65 dB left ear) requiring hearing aids.
At 3 yr and 3 months of age, she was started on propranolol (20 mg/d) because of sinus tachycardia. She had TFT compatible with RTH, and she was noted to have a goiter.
At 7 yr of age, serum concentration of free T4 (FT4) was greater than 6.0 ng/dl (normal range, 0.7–1.9 mU/liter), free T3 (FT3) was greater than 30 pg/ml (normal range, 1.8–4.2 mU/liter), and TSH was 14.4 mU/liter. To lower the TSH, 0.26 mg triiodothyroacetic acid (TRIAC) was given in three divided doses, and 1 yr later, the dose was doubled.
At 8.75 yr of age, because of a further increase in goiter size and the presence of macronodules, she underwent thyroidectomy and was given l-T4 75 μg/d. Pathological diagnosis was diffuse adenomatous hyperplasia. Her growth was delayed, with height and weight below the 3rd centile. Bone age was also greatly delayed with a catch-up at 11 yr (Fig. 3). She entered puberty at 9 yr but has no menarche at 11.6 yr. TFT results are shown in Fig. 1B.
Fig. 3.
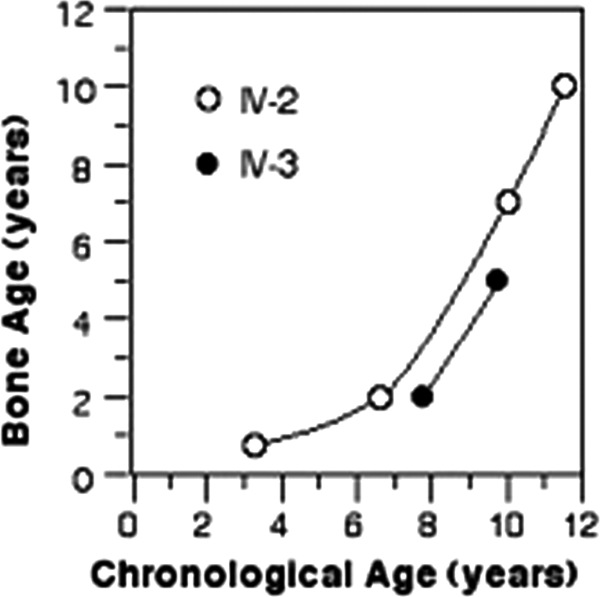
Bone age of affected patients in family Mozv. Note the severe delay in both siblings.
At 11 yr and 7 months of age, her verbal, performance, and total IQ scores (Wechsler Intelligence Scale for Children-Revised intelligence test) were 49, 55, and 50, respectively, categorized as being mildly mentally retarded, with significant speech defect. She was also hyperactive, with improvement in the last 3–4 yr. Frequent upper respiratory tract infections occurred throughout childhood.
The brother of the proposita (IV-3; Fig. 1B), with XY karyotype, had similar dysmorphic features as his sister, IV-2 (Fig. 2, E and F). He had a mild hearing impairment (evoked responses 60 dB left and 80 dB normalized hearing level right ear) requiring hearing aid. There was no tachycardia.
At the age of 5 yr, because of a large goiter, he was started on TRIAC (0.26 mg/d) given in three divided doses, with an increase to 0.525 mg/d at age 8 yr. He grew poorly, height being at the 10th centile through age 6, then at the 3rd centile at 8 yr. Weight has followed the 3rd centile throughout. Bone age was delayed to a similar degree as his sister (Fig. 3).
At the age of 9 yr and 6 months, his verbal, performance, and total IQ scores were 49, 50, and 46, respectively, categorized as having a moderate mental retardation, and he was unable to speak. He was also hyperactive from age 5 yr, with improvement after age 8.5 yr. His TFT are shown in Fig. 1B. He had fewer episodes of upper respiratory tract infections than his sister.
Family members.
Subject IV-1, the eldest sister (Fig. 1B), was 150 cm in height and weighed 48 kg at age 15 yr. She had no facial dysmorphism, no goiter (Fig. 2, G and H), and no tachycardia. Her serum TSH level was mildly elevated, accompanied by positive thyroid peroxidase (TPO) antibodies, consistent with subclinical hypothyroidism due to autoimmune thyroid disease (Fig. 1B).
The mother of the proposita (III-3; Fig. 1B) had neither goiter nor tachycardia. She had high serum total T4 and FT4 concentrations but also had positive TPO antibodies.
The father of the proposita (III-2; Fig. 1B) underwent thyroidectomy for goiter at the age of 28 yr. On 100 μg l-T4/d, his serum TSH level was elevated at 34 mU/liter despite a FT4 index (FT4I) above the upper limit of normal. He did not have tachycardia.
The paternal grandfather (II-1; Fig. 1B) and the maternal uncle (III-4; Fig. 1B) had elevated serum FT4I with a normal TSH concentration. They had no hearing impairment, goiter, or tachycardia.
Studies were approved by the review boards of the University of Chicago, and written consents were obtained from all subjects studied and guardians of minors.
Thyroid function tests
Blood was collected locally and shipped at room temperature for analyses in Chicago, IL. Total T4, total T3, and TSH were measured by chemiluminescence immunometric assays using the Elecsys Automated System (Roche Molecular Biochemicals GmbH and Hitachi, Ltd., Indianapolis, IN). Total rT3 was measured by RIA (Adaltis Italia S.p.A, Bologna, Italy), and thyroglobulin was measured by an in-house RIA. Serum FT4I and FT3 index were calculated as the product of their total serum concentrations of each iodothyronine and the normalized resin T4 uptake ratio. Antibodies to thyroglobulin and TPO were measured by passive hemagglutination (Fujirebio, Inc., Tokyo, Japan).
DNA isolation and THRB sequencing
DNA was isolated from peripheral blood leukocytes using QIAamp DNA Mini Kit (QIAGEN, Valencia, CA), following the manufacturer's protocol. Exons and adjacent intronic THRB gene sequences were amplified by the PCR and sequenced using automated fluorescence-based sequencing (3730XL 96-capillary; Applied Biosystems, Carlsbad, CA). The primers used for amplification and sequencing are available upon request. Nucleotide and amino acid numbering is according to established consensus (9).
Results
Thyroid function tests
In family Mbyd, the proposita's mother (III-1), grandmother (II-1), great-grandmother (I-1), and uncle (III-2) had TFT consistent with RTH (Fig. 1A). In family Mozv, the proposita's parents (III-2 and III-3) had TFT suggestive of RTH, taking into consideration that the father underwent thyroid surgery and was inadequately replaced with l-T4. The eldest sister and mother had autoimmune thyroid disease likely responsible for the sister's subclinical hypothyroidism. Patients II-1 and III-4, who are heterozygous carriers of the THRB gene mutation, have a slightly elevated FT4I, but the remaining TFT were in the normal range. For unknown reasons, subject II-2, who does not carry the mutant allele, also has an elevated value of FT4I.
Mutation identification and genotyping of family members
Family Mbyd
The proposita (IV-1) was found to have a single nucleotide substitution in the THRB gene. The normal guanosine 1325 was replaced with an adenosine, resulting in the substitution of the normal amino acid glycine 347 (GGG) with a glutamic acid (GAG) (G347E). The proposita (IV-1) has only the mutant allele, whereas other affected members of the family (III-1, III-2, II-2, I-1) are heterozygous.
Family Mozv
The proposita (IV-2) and her brother (IV-3) were found to have the normal cytosine 1231 of the THRB gene replaced with a thymidine. This nucleotide substitution is responsible for the replacement of the normal arginine 316 (CGC) with a cysteine (TGC) (R316C). Both are homozygous for the mutation, whereas their parents (III-2 and III-3) and the paternal grandfather (II-1) and maternal uncle (III-4) are heterozygous for the mutation.
The electropherograms of heterozygous and mutant homozygous sequences are showed in Supplemental Fig. 1 (published on The Endocrine Society's Journals Online web site at http://jcem.endojournals.org).
Discussion
In this study, we described three new patients with RTH carrying two different homozygous mutations in the THRB gene. The mutation G347E, found in family Mbyd, was previously reported in heterozygotes of one family (10), but it has not undergone in vitro characterization. The mutation R316C, found in patients from family Mozv, has been recently described in a heterozygous subject with RTH associated to lingual thyroid (11). Functional studies showed that R316C TRβ had a decreased T3-binding and caused impaired transcription of genes both positively and negatively regulated by T3.
So far, individuals from only three families have been shown to have only a mutant allele. In the first family reported to have RTH (family G) (12), the three affected children were homozygous for THRB gene deletion (4). They had stippled epiphyses, dysmorphic features (bird-like facies, pigeon breast, winged scapulae), deaf-mutism, and color blindness (12, 13). Although growth was delayed, they reached an adult height above the parental mean. Furthermore, their IQ were normal compared with the average for hearing-impaired individuals (Table 1). All heterozygous family members, including both consanguineous parents and some of the siblings, were phenotypically normal (12, 14).
Table 1.
Clinical features of subjects devoid of TRβ or expressing only mutant TRβ
| Family ID | G | S | F | Mbyd | Mozv |
|---|---|---|---|---|---|
| Mutation | THRB deletion | T337del | I280S | G347E | R316C |
| Dysmorphic features | Yes | Yes | Not reported | Yes | Yes |
| Associated abnormalities | None | Cardiaca | Unable to walk | Cardiacb | None |
| Recurrent URTI | Yes | Yes | ?c | Yes | Yes |
| Tachycardia | + | +++ | +++ | ++ | ++ (1 of 2) |
| Goiter | + | ++ | +++ | ++ | +++ |
| Failure to thrive | No | Yes | Yes | Yes | No |
| Developmental delayd | + | ++++ | ++++ | +++ | ++ |
| Growth delay | Yes + + | Yes + + + + | Yes + | Yes + + | Yes + + + |
| Bone age retardation | No | Yes | Yes | Yes | Yese |
| Mental retardation | + | ++++ | +++ | ++ | +++ |
| Total IQ score | Nlf | (<60) | (68, 71, 61) | (50, 46) | |
| Hyperactivity | Yes | Yes | Yes | No | Yes |
| Hearing loss (Db) | ++++ | +++ | ++ (30/40) | ++ | ++ (60–65) |
| Speech | Absent | Absent | Absent | Significant defect | Significant defect |
| Vision | Color blind | Myopiag | Severe myopia (9 diopters) | Squint, decreased acuity | Nl |
Nl, Normal; URTI, upper respiratory tract infection; ?, unknown; +, mild; ++, moderate; +++, severe; ++++, very severe.
Patent ductus arteriosus, tricuspid insufficiency, inguinal hernia, noncommunicating, hydrocephalus requiring ventriculoperitoneal shunt, seizures, hydrocoele, recurrent diarrhea.
Patent ductus arteriosus, patent foramen ovale.
Enlarged tonsils (adenoidectomy at age 4 yr), chronic otitis media.
Graded according to the degree in achieving milestones, such as sitting, standing, feeding, walking, talking.
Catching up in adolescence.
Compared to deaf mute children of variable etiology.
B. B. Bercu, personal communication.
The proband of family S, a boy born to consanguineous parents, had threonine-337 deleted from both alleles (15). This mutant TRβ has virtually no ability to bind T3 (7). At birth he presented with respiratory distress, hyperbilirubinemia, hypofibrinogenemia, polycythemia, and very high T4 and TSH (Table 2). He was later found to have a goiter and heart malformation (Table 1). His linear growth and bone age were severely delayed (at 2¾ yr, bone age was 4 months), and he had profound mental retardation, complicated by seizures, and a noncommunicating hydrocephalus. The patient had bilateral sensorineural hearing impairment and myopia (B. B. Bercu, personal communication). Treatments with β-blockers, propylthiouracil, and d-T4 caused little or no improvement. This patient remained quite tachycardic and had great difficulty gaining weight, even with rigorous attempts at alimentation. The child died at 8 yr of age from cardiogenic shock complicating staphylococcal pneumonia (B. B. Bercu, personal communication). The heterozygous family members had goiter, tachycardia, and variable elevation of the FT4 with nonsuppressed TSH, typical for RTH (7, 15, 16).
Table 2.
Other characteristics of the probands
| Family ID | G | S | F | Mbyd | Mozv |
|---|---|---|---|---|---|
| Mutation | THRB deletion | T337del | I280S | G347E | R316C |
| Ethnic origin | Mexican | Italian/Irish | German | Australian Aborigine/White | Turkish |
| Age at recognition | 6 yr | 3 d | 1.2 yr | 3 d | 3 yr |
| Age at genetic diagnosis | 32 yr | 3.5 yr | 2 yr | 9 wk | 9 yr |
| Reason for thyroid investigation | Stippled epiphyses | Tachycardia, goiter, high TSH | Tachycardia, goiter, sweating | High TSH at neonatal screen | Speech delay |
| Birth weight (g) | 2268 | 1480a | 3000 | 2470 | 2000 |
| TFT at birth | High T4b | High T4 and TSH | Nl T4 and TSH | High T4, T3, TSH | Not measured |
| Postnatal problems | None | Respiratory distress, hyperbilirubinemia, hypofibrinogenemia, polycythemia | Respiratory distress | Temperature instability, hyperbilirubinemia, vomiting | None |
| Treatment | None | β-blockers, d-T4 | Antithyroid drugs, Tx at age 9, 13, and 16 yr, l-T4 | β-blockers | Tx at 8 and 9.5 yr, TRIAC, l-T4 |
Nl, Normal; Tx, thyroidectomy.
Premature birth at 35 wk.
Third born tested because of known thyroid defect in sibling.
The proband of family F was born to nonconsanguineous parents but was found to harbor only the mutant I280V THRB allele inherited from the mother, who was heterozygous for this mutation. It is unclear whether the father, who was not available, had a deletion that included the THRB gene (8). In contrast to all other patients with absent wild-type (WT) THRB gene, the patient had normal serum T4 and TSH levels at birth. However, within 3 months, goiter; greatly increased T4, T3, and TSH levels; sweating; and tachycardia were noted (Table 2). She had myopia, a mixed sensorineural and conductive hearing defect. Development was severely retarded; she was unable to walk at age 3 yr; and at 25 yr of age, intellectual development was severely impaired. Until 3 yr of age, her height and weight were in the 3rd centile; at the age of 7 yr, in the 10th centile; and at the age of 10 and beyond, in the 25th centile. Because of a large goiter, she underwent three thyroidectomies (Table 2). Her mother had thyroidectomy because of goiter with tracheal compression but maintained a normal FT4 and TSH with 100 μg/d l-T4 replacement (8).
The cases reported herein presented with the classic findings of RTH, including abnormal TFT, tachycardia, and goiter. However, as with the previously reported case homozygous for THRB gene mutations, the severity of symptoms and magnitude of TFT abnormalities are greater than those found in subjects with heterozygous mutations. This is attributed to the DNE of the mutant receptor combined with the absence of a WT TRβ. In fact, with the exception of deaf-mutism, the manifestations of RTH and the magnitude of TFT abnormalities of homozygotes in family G were the least severe (Tables 1 and 3) and comparable to many subjects carrying heterozygous mutations.
Table 3.
Mean values of FT4, FT3, and TSH in homozygous (Homo) and heterozygous (Hetero) individuals belonging to five families with different THRB gene mutations
| Mutation (family ID) | FT4 (% ULN) |
FT3 (% ULN) |
TSH (mU/liter) |
No. of subjects |
Refs. | ||||
|---|---|---|---|---|---|---|---|---|---|
| Homo | Hetero | Homo | Hetero | Homo | Hetero | Homo | Hetero | ||
| THRBdel (G) | 214 ± 23 | 80 ± 2 | 228 ± 84 | 59 ± 8 | 4.3 ± 2.3 | 2.5 ± 0.9 | 3 | 5 | 5, 7, 8, 9, and PDSA |
| T337del (S)a | 587b | 132 ± 29 | 774 | 117 ± 16 | 239b | 2.5 ± 1.2 | 1 | 8 | 10, 11, 21 |
| I280S (F) | 205c | 86c | 130c | 71c | 24c | 1.5c | 1 | 1 | 12 |
| G347E (Mbyd) | 488 | 173 ± 6 | 1028 | 103 ± 22 | 490 | 4.8 ± 3.6 | 1 | 4 | This report |
| R316C (Mozv) | 299 | 113 ± 9 | >714 | 85 ± 15 | 35 | 2.6 ± 1.1 | 2 | 4 | This report |
Values for individual homozygotes were derived from an average of two to more than 10 determinations off treatment, except for members of family F for whom data before thyroidectomy were not available. When more than one subject, values are expressed as mean ± sd. ULN, Upper limit of normal; PDAS, personal data from the senior author.
Originally reported as T33, prior to the identification of an error in the initial report of the human TRβ sequence (33) which added five amino acids to the amino terminus of the molecule (9).
Values ranged from 440 to 680% ULN for FT4 and from 155 to 395 mU/liter for TSH.
Thyroidectomy, both on 100 μg l-T4/d.
We used the thyrotroph T4 resistance index (TT4RI), the product of FT4I and TSH (17), to estimate the relative resistance of the hypothalamo-pituitary axis to TH (calculated using data from Table 3). This allows the inclusion of individuals on l-T4 replacement. In increasing order of resistance, subjects without WT TRβ from the five families ranked as follows: G<F<Mozv<S<Mbyd, with scores of 920, 4,920, 10,456, 140,290, and 239,120 (normal, 136 ± 73), respectively. It is of interest that heterozygous relatives ranked in almost similar order F<G<Mozv<S<Mbyd, with scores of 129, 200, 293, 330, and 830, respectively. Note that the TT4RI of heterozygotes of families G and F are well within the normal range.
Ranking the effect of the various mutations on the global manifestations is more difficult. However, taking into account the severity of manifestations as listed in Table 1, ranking is similar to that of the TT4RI, with the exception of the proband of family F, who had more severe mental and physical incapacity. This may be due to the effect of a major gene deletion on one of the two chromosomes 3. Furthermore, in the homozygous member of family S, the additional hydrocephalus and cardiac abnormalities, possibly not directly related to the THRB gene defect, may have contributed to the severity of the phenotype and undoubtedly to his early demise.
It is noteworthy that findings less commonly seen in RTH with heterozygous mutations, such as effects on growth, bone, cognitive development, and hearing loss, were found in all subjects of the four families expressing only mutant TRβ molecules.
The severity of RTH is dependent on the magnitude of impaired ligand-binding of the mutant TRβ and, in its heterozygous form, on the degree of DNE. The mutant TRβ may interfere with the WT TRβ function in several ways: by occupying TRE on target genes or by engaging the WT TRβ in homodimerization or heterodimerization with retinoid X receptor and possibly with TRα (18). Some mutant TRβ have increased affinity to corepressors (19) or reduced association with coactivators (20). Thus, mutant TRβ with complete inability to bind T3 can produce minimal dysfunction if their association to corepressors is also reduced (21). Therefore, several mechanisms may account for the variability of clinical manifestation in individuals carrying a mutant THRB genes in one or both alleles.
Some clinical and laboratory findings in patients with RTH can be explained in light of the information derived from animal models for Thrb gene mutations (22–28). Deletion of the Thrb gene in the mouse (Thrb−/−) reproduces a phenotype similar to that observed in subjects of family G. Heterozygous mice are normal, and homozygotes have abnormal TFTs, goiter, sensorineural deafness, and monochromatic vision but normal growth and normal learning abilities (22, 23, 27). However, as in humans, the magnitude of TFT abnormalities in the absence of TRβ is not as pronounced as in mice homozygous for point mutations in the Thrb gene (Thrbmut/mut) (25–27). Such mice, as do the subjects of families S, Mbyd, and Mozv that express only a mutant TRβ in both alleles, show goiter, growth delay, and learning disabilities. An accurate mouse model for family S, with deletion of threonine-337, has been generated in Wondisford's laboratory, and the effect of this deletion on the heart has been reported in the heterozygous animals (29). The homozygotes were described as small, frail, and weak, similar to the homozygote of family S known to the same investigator (F. E. Wondisford, personal communication).
We can speculate that the severe phenotype observed in the Thrbmut/mut mouse compared with the Thrb−/− mouse and, similarly, that of patients S, Mbyd, and Mozv compared with those of G, are the result of several effects of the mutant TRβ in addition to the complete loss of TRβ function. First was the maintenance of aporeceptor-mediated repression or stimulation of genes regulated positively or negatively, respectively, by TH. This is achieved by the occupation of TRE by the mutant TRβ. Second was the interference with TRα1 function through formation of inactive TRα/TRβmut heterodimers and/or direct binding of the mutant TRβ to TREs normally occupied by TRα1 molecules (30).
The amount of the mutant TRβ, acting as aporeceptor, undoubtedly has an effect on the severity of the phenotype, even in the absence of a WT TRβ. Thus, the proband of family F, expressing a single mutant TRβ allele, manifests a pituitary resistance intermediate to that of homozygotes of the G family with no TRβ and all other families with two copies of a mutant TRβ, the TT4RI values being 920, 4,920, and at least 10,456 for G, F, and all others.
TRα1 is essential for heart function, including heart rate up-regulation, for bone maturation, and for the function of specific regions of the brain (cerebellum and hippocampus) and intestine. All patients expressing only the mutant TRβ had marked growth retardation with different degrees of neurological impairment. Using the above reasoning, it is expected that interference of the mutant TRβ with the function of TRα1 should, as in the Thrb−/−/Thra10/0 mouse (31), reduce the tachycardia by blocking TRα1-mediated TH action. This is not the case, probably because of the effect of the astronomically high TH levels of these animals on the WT TRα1, overriding the effect of the mutant TRβ expressed at a low level in the heart (32, 33).
In conclusion, we report two new families of RTH associated to homozygous mutations in the THRB gene. They represent an important addition to the single patient homozygous for a mutant TRβ with DNE. The clinical and laboratory abnormalities are in agreement with data obtained from mouse models and strengthen the concept of the interference of the mutated TRβ on the function of the TRα1.
Acknowledgments
This work was supported in part by Grants R37DK15070 and P60DK20595 from the National Institute of Diabetes and Digestive and Kidney Diseases and National Institutes of Health Grant 5M01RR04999. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Diabetes and Digestive and Kidney Diseases or the National Institutes of Health.
Current address for K.O.: Department of Pediatrics, Shimane University Faculty of Medicine, Izumo 693-8501, Japan.
Disclosure Summary: A.M.F., K.O., O.E., H.W., and R.E.W. have nothing to declare. S.R. is academic associate for Quest Diagnostic Laboratories and, therefore, is considered as a consultant.
Footnotes
- dB
- Decibel
- DNE
- dominant-negative effect
- FT3
- free T3
- FT4
- free T4
- FT4I
- FT4 index
- l-T4
- levothyroxine
- RTH
- resistance to TH
- TFT
- thyroid function test
- TH
- thyroid hormone
- THRB
- TH receptor β
- TPO
- thyroid peroxidase
- TRα
- TH receptor α
- TRE
- TH response element
- TRIAC
- triiodothyroacetic acid
- TT4RI
- thyrotroph T4 resistance index
- WT
- wild-type.
References
- 1. Refetoff S, Weiss RE, Usala SJ. 1993. The syndromes of resistance to thyroid hormone. Endocr Rev 14:348–399 [DOI] [PubMed] [Google Scholar]
- 2. Refetoff S, Dumitrescu AM. 2007. Syndromes of reduced sensitivity to thyroid hormone: genetic defects in hormone receptors, cell transporters and deiodination. Best Pract Res Clin Endocrinol Metab 21:277–305 [DOI] [PubMed] [Google Scholar]
- 3. Brucker-Davis F, Skarulis MC, Grace MB, Benichou J, Hauser P, Wiggs E, Weintraub BD. 1995. Genetic and clinical features of 42 kindreds with resistance to thyroid hormone. The National Institutes of Health Prospective Study. Ann Intern Med 123:572–583 [DOI] [PubMed] [Google Scholar]
- 4. Takeda K, Sakurai A, DeGroot LJ, Refetoff S. 1992. Recessive inheritance of thyroid hormone resistance caused by complete deletion of the protein-coding region of the thyroid hormone receptor-β gene. J Clin Endocrinol Metab 74:49–55 [DOI] [PubMed] [Google Scholar]
- 5. Jameson JL. 1994. Mechanisms by which thyroid hormone receptor mutations cause clinical syndromes of resistance to thyroid hormone. Thyroid 4:485–492 [DOI] [PubMed] [Google Scholar]
- 6. Takeda K, Balzano S, Sakurai A, DeGroot LJ, Refetoff S. 1991. Screening of nineteen unrelated families with generalized resistance to thyroid hormone for known point mutations in the thyroid hormone receptor β gene and the detection of a new mutation. J Clin Invest 87:496–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Usala SJ, Menke JB, Watson TL, Wondisford FE, Weintraub BD, Bérard J, Bradley WE, Ono S, Mueller OT, Bercu BB. 1991. A homozygous deletion in the c-erbA β thyroid hormone receptor gene in a patient with generalized thyroid hormone resistance: isolation and characterization of the mutant receptor. Mol Endocrinol 5:327–335 [DOI] [PubMed] [Google Scholar]
- 8. Frank-Raue K, Lorenz A, Haag C, Höppner W, Boll HU, Knorr D, Hentze S, Raue F. 2004. Severe form of thyroid hormone resistance in a patient with homozygous/hemizygous mutation of T3 receptor gene. Eur J Endocrinol 150:819–823 [DOI] [PubMed] [Google Scholar]
- 9. Beck-Peccoz P, Chatterjee VK, Chin WW, DeGroot LJ, Jameson JL, Nakamura H, Refetoff S, Usala SJ, Weintraub BD. 1994. Nomenclature of thyroid hormone receptor β-gene mutations in resistance to thyroid hormone: consensus statement from the first workshop on thyroid hormone resistance, July 10–11, 1993, Cambridge, United Kingdom. J Clin Endocrinol Metab 78:990–993 [DOI] [PubMed] [Google Scholar]
- 10. Parrilla R, Mixson AJ, McPherson JA, McClaskey JH, Weintraub BD. 1991. Characterization of seven novel mutations of the c-erbA β gene in unrelated kindreds with generalized thyroid hormone resistance. Evidence for two “hot spot” regions of the ligand binding domain. J Clin Invest 88:2123–2130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nakajima Y, Yamada M, Horiguchi K, Satoh T, Hashimoto K, Tokuhiro E, Onigata K, Mori M. 2010. Resistance to thyroid hormone due to a novel thyroid hormone receptor mutant in a patient with hypothyroidism secondary to lingual thyroid and functional characterization of the mutant receptor. Thyroid 20:917–926 [DOI] [PubMed] [Google Scholar]
- 12. Refetoff S, DeWind LT, DeGroot LJ. 1967. Familial syndrome combining deaf-mutism, stuppled epiphyses, goiter and abnormally high PBI: possible target organ refractoriness to thyroid hormone. J Clin Endocrinol Metab 27:279–294 [DOI] [PubMed] [Google Scholar]
- 13. Newell FW, Diddie KR. 1977. Typische Monochromasie, angeborene Taubheit und Resistenz gegenüber der intrazellulären Wikung des Thyroideahormons. Klin Mbl Augenheilk 171:731–734 [PubMed] [Google Scholar]
- 14. Refetoff S, DeGroot LJ, Benard B, DeWind LT. 1972. Studies of a sibship with apparent hereditary resistance to the intracellular action of thyroid hormone. Metabolism 21:723–756 [DOI] [PubMed] [Google Scholar]
- 15. Ono S, Schwartz ID, Mueller OT, Root AW, Usala SJ, Bercu BB. 1991. Homozygosity for a dominant negative thyroid hormone receptor gene responsible for generalized resistance to thyroid hormone. J Clin Endocrinol Metab 73:990–994 [DOI] [PubMed] [Google Scholar]
- 16. Schwartz ID, Bercu BB. 1992. Dextrothyroxine in the treatment of generalized thyroid hormone resistance in a boy homozygous for a defect in the T3 receptor. Thyroid 2:15–19 [DOI] [PubMed] [Google Scholar]
- 17. Yagi H, Pohlenz J, Hayashi Y, Sakurai A, Refetoff S. 1997. Resistance to thyroid hormone caused by two mutant thyroid hormone receptors β, R243Q and R243W, with marked impairment of function that cannot be explained by altered in vitro 3,5,3′-triiodothyroinine binding affinity. J Clin Endocrinol Metab 82:1608–1614 [DOI] [PubMed] [Google Scholar]
- 18. Yen PM, Sugawara A, Refetoff S, Chin WW. 1992. New insights on the mechanism(s) of the dominant negative effect of mutant thyroid hormone receptor in generalized resistance to thyroid hormone. J Clin Invest 90:1825–1831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yoh SM, Chatterjee VK, Privalsky ML. 1997. Thyroid hormone resistance syndrome manifests as an aberrant interaction between mutant T3 receptors and transcriptional corepressors. Mol Endocrinol 11:470–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Collingwood TN, Wagner R, Matthews CH, Clifton-Bligh RJ, Gurnell M, Rajanayagam O, Agostini M, Fletterick RJ, Beck-Peccoz P, Reinhardt W, Binder G, Ranke MB, Hermus A, Hesch RD, Lazarus J, Newrick P, Parfitt V, Raggatt P, de Zegher F, Chatterjee VK. 1998. A role for helix 3 of the TRβ ligand-binding domain in coactivator recruitment identified by characterization of a third cluster of mutations in resistance to thyroid hormone. EMBO J 17:4760–4770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Weiss RE, Hayashi Y, Nagaya T, Petty KJ, Murata Y, Tunca H, Seo H, Refetoff S. 1996. Dominant inheritance of resistance to thyroid hormone not linked to defects in the thyroid hormone receptor α or β genes may be due to a defective cofactor. J Clin Endocrinol Metab 81:4196–4203 [DOI] [PubMed] [Google Scholar]
- 22. Forrest D, Hanebuth E, Smeyne RJ, Everds N, Stewart CL, Wehner JM, Curran T. 1996. Recessive resistance to thyroid hormone in mice lacking thyroid hormone receptor β: evidence for tissue-specific modulation of receptor function. EMBO J 15:3006–3015 [PMC free article] [PubMed] [Google Scholar]
- 23. Forrest D, Erway LC, Ng L, Altschuler R, Curran T. 1996. Thyroid hormone receptor β is essential for development of auditory function. Nat Genet 13:354–357 [DOI] [PubMed] [Google Scholar]
- 24. Hashimoto K, Curty FH, Borges PP, Lee CE, Abel ED, Elmquist JK, Cohen RN, Wondisford FE. 2001. An unliganded thyroid hormone receptor causes severe neurological dysfunction. Proc Natl Acad Sci USA 98:3998–4003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kaneshige M, Kaneshige K, Zhu X, Dace A, Garrett L, Carter TA, Kazlauskaite R, Pankratz DG, Wynshaw-Boris A, Refetoff S, Weintraub B, Willingham MC, Barlow C, Cheng S. 2000. Mice with a targeted mutation in the thyroid hormone β receptor gene exhibit impaired growth and resistance to thyroid hormone. Proc Natl Acad Sci USA 97:13209–13214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Suzuki H, Cheng SY. 2003. Compensatory role of thyroid hormone receptor (TR) α 1 in resistance to thyroid hormone: study in mice with a targeted mutation in the TR β gene and deficient in TR α 1. Mol Endocrinol 17:1647–1655 [DOI] [PubMed] [Google Scholar]
- 27. Chassande O, Flamant F, Samarut J. 1999. Thyroid hormone receptor knockouts: their contribution to our understanding of thyroid hormone resistance. Curr Opin Endocrinol Diabet 6:293–300 [Google Scholar]
- 28. Weiss RE, Murata Y, Cua K, Hayashi Y, Seo H, Refetoff S. 1998. Thyroid hormone action on liver, heart, and energy expenditure in thyroid hormone receptor β-deficient mice. Endocrinology 139:4945–4952 [DOI] [PubMed] [Google Scholar]
- 29. Ortiga-Carvalho TM, Hashimoto K, Pazos-Moura CC, Geenen D, Cohen R, Lang RM, Wondisford FE. 2004. Thyroid hormone resistance in the heart: role of the thyroid hormone receptor β isoform. Endocrinology 145:1625–1633 [DOI] [PubMed] [Google Scholar]
- 30. Gloss B, Sayen MR, Trost SU, Bluhm WF, Meyer M, Swanson EA, Usala SJ, Dillmann WH. 1999. Altered cardiac phenotype in transgenic mice carrying the Δ337 threonine thyroid hormone receptor β mutant derived from the S family. Endocrinology 140:897–902 [DOI] [PubMed] [Google Scholar]
- 31. Johansson C, Göthe S, Forrest D, Vennström B, Thorén P. 1999. Cardiovascular phenotype and temperature control in mice lacking thyroid hormone receptor-β or both α1 and β. Am J Physiol 276:H2006–H2012 [DOI] [PubMed] [Google Scholar]
- 32. Dillmann WH. 2002. Cellular action of thyroid hormone on the heart. Thyroid 12:447–452 [DOI] [PubMed] [Google Scholar]
- 33. Swanson EA, Gloss B, Belke DD, Kaneshige M, Cheng SY, Dillmann WH. 2003. Cardiac expression and function of thyroid hormone receptor β and its PV mutant. Endocrinology 144:4820–4825 [DOI] [PubMed] [Google Scholar]