

Abstract
Background:
Carney complex (CNC) is a multiple endocrine neoplasia syndrome due to inactivating mutations in the PRKAR1A gene that codes for type Iα regulatory (RIα) subunit of protein kinase A. Most PRKAR1A mutations are subject to nonsense mRNA decay (NMD) and, thus, lead to haploinsufficiency.
Methods and Setting:
Patient phenotyping for CNC features and DNA, RNA, protein, and transfection studies were carried out at a research center.
Results:
We describe in unrelated kindreds with CNC four naturally occurring PRKAR1A mutations (1055del4, 1067del4ins5, 1076delTTins13, and 1142del4) that are predicted to escape NMD because they are located in the last coding exon of the gene. The phenotype of CNC was not different from that in other patients with the condition, although the number of patients was small. Each of the mutations caused a frameshift that led to a new stop codon into the 3′ untranslated open reading frame, predicting an elongated protein that, however, was absent in patient-derived cells. After site-directed mutagenesis, in vitro transcription, and cell-free translation experiments, the expected size mutant proteins were present. However, when the mutant constructs were transfected in adrenal (NCI-295), testicular (N-TERA), and embryonic (HEK293) cells and despite the presence of the mutant mRNA, Western blot analysis indicated that there were no longer proteins. The subsequent application of proteasome inhibitors to cells transfected with the mutant constructs led to the detection of the aberrant proteins, although a compound that affects protein folding had no effect. The wild-type protein was also decreased in both patient-derived cells and/or tissues as well as in the in vitro systems used in this study.
Conclusions:
This was the first demonstration of proteasomal degradation of RIα protein variants leading to PRKAR1A haploinsufficiency and CNC, adding protein surveillance to NMD in the cellular mechanisms overseeing RIα synthesis. In agreement with the molecular data, CNC patients bearing PRKAR1A defects that extend the open reading frame did not have a different phenotype, although this has to be confirmed in a larger number of patients.
Carney complex (CNC) (MIM 160980) is an autosomal dominant multiple endocrine neoplasia syndrome characterized by spotty skin pigmentation, cardiac and other myxomas, and different types of endocrine tumors, including primary pigmented nodular adrenocortical disease, GH-secreting pituitary tumors, and gonadal and thyroid tumors (1, 2). Malignancies are rare, and they include psammomatous melanotic schwannoma and thyroid, ovarian, liver, and pancreatic cancer (1–3). Inactivating mutations in the PRKAR1A gene coding for the type 1A regulatory (R) subunit of protein kinase A (PKA) or cAMP-dependent protein kinase cause the disease in most patients (4–6).
The PKA enzyme plays a major role in eukaryotic cell signaling. In its inactive state, the holoenzyme consists of a tetramer of two homo- or heterodimers of R subunits (RIα, RIβ, RIIα, and RIIβ) and a homodimer of two catalytic (C) subunits (from a choice of four molecules: Cα, Cβ, Cγ, and PRKX) (7). Activation of PKA occurs upon the binding of two molecules of cAMP to each R subunit, followed by the dissociation of the holoenzyme and the release of the active C subunit, which in turn phosphorylates a series of downstream cellular targets (7, 8). Thus, haploinsufficiency for RIα leads to increased and/or dysregulated C subunit activity and abnormal growth and proliferation in cAMP-sensitive tissues causing the characteristic phenotype of CNC (4, 5). To date, more than 100 different PRKAR1A pathogenic mutations have been described; most lead to RIα haploinsufficiency because they result in premature stop codons and subsequent degradation of the mutant transcript by nonsense-mediated mRNA decay (NMD) (5, 6). Rare mutations that escape NMD and lead to expression of a mutant PRKAR1A protein have also been reported (8–10). The mechanism by which these mutations lead to CNC is through impairment of the ability of PRKAR1A to respond normally to cAMP and/or bind the C subunit (8, 9).
In the present study, we report four novel naturally occurring mutations (1055del4, 1067del4ins5, 1076delTTins13, and 1142del4), which evade NMD because of their location in the last coding exon of the PRKAR1A gene. However, they also lead to RIα haploinsufficiency because they result in longer PRKAR1A protein variants that are degraded at the proteasomal level. This is a novel mechanism of RIα haploinsufficiency, and its description adds not only to our understanding of CNC but also to the relatively scarce literature on pathogenic frameshift mutations affecting the last coding exons of genes.
Patients and Methods
Patients
The institutional review boards of the Eunice Kennedy Shriver National Institute of Child Health and Human Development approved the contact of all patients and their families and their participation in protocol 95-CH-0059 after giving informed consent. The manifestations of CNC in the seven patients that were carriers of the novel PRKAR1A mutations are summarized in Table 1; five male and two female patients from four unrelated families were identified.
Table 1.
Clinical manifestations of CNC in patients that carried PRKAR1A mutations abolishing the normal stop codon
| Patient ID | Age (yr) | Gender | Manifestations (age of first diagnosis) |
Manifestations (age of first diagnosis) |
Mutation |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cardiac myxomas | Breast myxomas | Lentigenosis | Other skin lesions | Acromegaly | Cushing's syndrome | LCCSCT | Thyroid tumors | Ovarian tumors | Schwannomas | cDNA | Protein | |||
| CAR15.02 | 50 | F | + (27) | + (27) | + | + | − | + (32) | + (32) | + (32) | − | c.1076_77delTTins13 | p.Leu359fsPLUS62 | |
| CAR15.04 | 14 | M | − | − | + | + | − | − | + (4) | + (4) | − | c.1076_77delTTins13 | p.Leu359fsPLUS62 | |
| CAR66.03 | 19 | F | + | + | + | + | − | + (13) | − | − | + (19) | c.1055_1058delGACC | p.Arg352fsPLUS57 | |
| CAR540.01 | 63 | M | + (33) | + (33) | + | + | + (41) | − | + (25) | − | − | c1067_1070delAACGins5 | p.Glu356fsPLUS45 | |
| CAR540.11 | 27 | M | − | − | + | + | − | − | + (24) | − | − | c1067_1070delAACGins5 | p.Glu356fsPLUS45 | |
| CAR541.01 | 6 | M | − | − | + | − | − | + | − | − | − | c1142_1145delTCTG | p.Val381fsPLUS58 | |
| CAR541.05 | 26 | M | − | − | + | − | − | − | + | − | − | c1142_1145delTCTG | p.Val381fsPLUS58 | |
The age of diagnosis of the respective tumor is indicated in parentheses (when this information was available). Patients CAR15.02 and CAR66.03 had multiple cardiac myxomas and schwannomas, respectively. F, Female; LCCSCT, large-cell calcifying Sertoli cell tumor; M, male.
Methods
Sequencing of the PRKAR1A gene in DNA extracted from tissues and blood samples by standard methods was accomplished as previously published (4–6).
A PCR-cloning method was used to generate both the wild-type and mutant expression constructs. The wild-type RIα (RIα-WT) was created as previously described (10). The elongated mutations were then introduced into the RIα-WT construct using overlapping PCR with mutagenic primers (sequences available upon request). PCR was performed using AcuPfx DNA polymerase (Invitrogen, Carlsbad, CA) and the RIα-WT as the DNA template under the following conditions: 94 C for 2 min; 94 C for 30 sec, annealing temperature (Table 2) for 30 sec, and 72 C for 1 min for 25 cycles; and 72 C for 3 min. The PCR products were gel purified, digested with KpnI and EcoRI (New England Biolabs, Beverly, MA) and ligated in the vector replacing the corresponding wild-type sequence. The construct DNA were amplified via transformation in One Shot TOP10 Escherichia coli competent cells (Invitrogen). The DNA was extracted and purified from the E. coli bacterium using the Wizard Plus SV Minipreps (Promega, Madison, WI) and HiSpeed Plasmid Midi Kit (QIAGEN, Valencia, CA) following the manufacturer's instructions. All constructs were repeatedly sequenced before use in the experiments to ensure the proper constructs.
To determine protein expression and the size of the RIα elongated mutant proteins, we used the cell-free TNT quick-coupled transcription/translation systems (Promega) according to the manufacturer's instructions. Briefly, 2 μg of each expression vector was added to an aliquot of the master mix and incubated in a 50-μl reaction volume for 90 min at 30 C. Synthesized proteins were then analyzed by Western analysis.
For transfections, adrenocortical (NCI-H295) and testicular cancer (N-TERA), and embryonic kidney (HEK293) cells were cultured in DMEM supplemented by 10% fetal bovine serum and 1% antibiotic-antimycotic. The cells were plated in 10-cm dishes and 6 μg of each plasmid containing the mutant construct was transfected using Lipofectamine 2000 (Invitrogen) when plated cells reached approximately 60% confluence. Cells were harvested 24 h later and were used for DNA, RNA, and protein extraction. RNA was extracted from harvested transfected cells or patient sample lymphocytes using the RNeasy Kit (QIAGEN). RNA was reverse transcribed using the SuperScript III first-strand cDNA synthesis for RT-PCR (Invitrogen).
Extracted protein lysates were resolved by electrophoresis on a Novex 10% Tris-glycine (Invitrogen) and then transferred to a 0.2-μm nitrocellulose membrane. Western blotting was performed with the primary mouse antibody against RIα (1:250; BD Biosciences, San Jose, CA) and primary rabbit antibody against β-actin (1:5000; Abcam, Cambridge, MA). Secondary antibodies conjugated with horseradish peroxidase against mouse (1:1000; EMD Biosciences, Darmstadt, Germany) or rabbit IgG (1:10000; Abcam). Bands were detected by enhanced chemiluminescence reagent and densitometer scanning (Molecular Dynamics, Sunnyvale, CA). Protein images were quantified with ImageQuant software and normalized against the expression of β-actin.
To determine whether NMD is involved in the translational repression of the mutant proteins, we treated the transfected cells with cycloheximide, as previously described (4, 5). Transfected cells were also treated overnight with the proteasome inhibitors clasto-lactacystin β-lactone (CLL) at 5 μm, MG132 at 1 μm, and the synthetic chaperone N-nonyl-deoxynojirimycin (NN-DNJ) also at 1 μm and harvested the next day in three separate experiments and in one where both MG132 and NN-DNJ were used in combination, as previously described (11–13).
Statistics
Patient phenotypes (i.e. number of manifestations) were compared by χ2 with Fisher correction, as needed. All in vitro experiments were conducted in triplicate; for quantification, means were calculated and t tests were used for the analysis. For all comparisons (clinical, nonparametric; and in vitro, parametric), a P value <0.05 was considered significant.
Results
Clinical manifestations
The manifestations (Table 1) varied from spotty skin pigmentation and only one characteristic CNC tumor (in CAR541) to skin lesions in association with multiple tumors, such as breast and cardiac myxomas, primary pigmented nodular adrenocortical disease, and thyroid and ovarian cysts (in CAR15). When compared with CNC patients with mutations that are subject to NMD (5, 6, 14), there were no statistically significant differences in the distribution or severity of the lesions (data not shown).
However, within the group of patients described in Table 1, the most severe phenotypes were associated with mutations p.Leu359fsPLUS62 (CAR15), p.Arg352fsPLUS57 (CAR66), and p.Glu356fsPLUS45 (CAR540), in that order. These patients had collectively more tumors than the two patients in family CAR541 with the frameshift starting at the last coding exon (p.Val381fsPLUS58); CAR541 had the mildest phenotype (P < 0.05). Thus, it appeared that a mutant PRKAR1A protein, in which a larger part of the sequence coded by the last exon was replaced by abnormal amino acids, was associated with more CNC lesions, although the number of patients was small, and the finding could be spurious.
Molecular studies
The four mutations, 1055del4, 1067del4ins5, 1076delTTins13, and 1142del4, were small insertions, deletions, or combined rearrangements in the last PRKAR1A exon, all leading to a frameshift and predicted termination codon further downstream from the wild-type TGA codon of the PRKAR1A open reading frame (ORF). As a result, the PRKAR1A protein was predicted to extend by 57, 45, 62, and 58 amino acids, respectively (Supplemental Fig. 1, published on The Endocrine Society's Journals Online web site at http://jcem.endojournals.org). Expression in a cell-free system demonstrated that these proteins could be produced and detected by the commercially available PRKAR1A antibody (Fig. 1); the four mutant isoforms were slightly longer compared with the wild-type protein, and their molecular size corresponded to the expected amino acid additions. However, after transfection of the adrenocortical (NCI-H295), testicular cancer (N-TERA), and embryonic kidney (HEK293) cells (with the same constructs bearing the mutations that were used in the cell-free system), immunoblotting showed the presence of only the wild-type PRKAR1A protein; there were no longer RIα isoforms (Fig. 2). Interestingly, cells transfected with the wild-type constructs showed a significantly higher amount of wild-type PRKAR1A protein, compared with the cells transfected with the mutant constructs and the empty vector (mock transfection), where only the endogenous PRKAR1A was detected. These results indicated successful transcription and translation from the wild-type expressing vector, in addition to the endogenous PRKAR1A expression, that was seen in all transfected cell lines. The efficiency of the transfection was proven by the presence of green fluorescent protein that was tagged to the wild-type expression vector as previously reported (8) (data not shown).
Fig. 1.

Demonstration of longer PRKAR1A protein products in a cell-free system; these are detected by a commercially available PRKAR1A antibody. The second lane is that of a construct leading to a shorter protein (see Ref. 8).
Fig. 2.
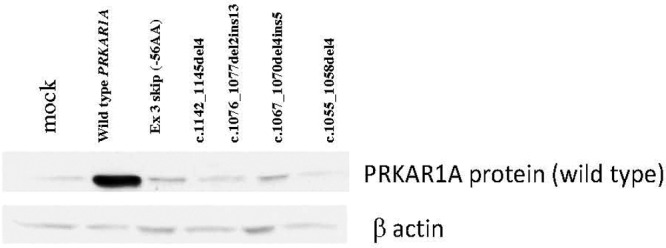
Transfection of HEK293 cells with the same constructs bearing the mutations that were used in the cell-free system (Fig. 1) led to the presence of only the wild-type PRKAR1A protein, without any longer variants.
To determine whether the expression of the mutant proteins was inhibited at the mRNA or protein level, we analyzed the cDNA from all transfected cell lines before and after transfection with the mutant constructs. Sequencing of the cDNA after transfection showed the presence of all four mutations (Supplemental Fig. 2), indicating that the mutant mRNA was there (all samples had been treated by deoxyribonuclease, and sequencing confirmed that this was not genomic DNA contamination). We also treated all transfected HEK293 cells with cycloheximide, which inhibits NMD and allows nonsense RNA expression as we have previously described (4, 5); cycloheximide is also a well-known protein synthesis inhibitor. Again, Western blot analysis detected only the protein corresponding to the wild-type PRKAR1A size in all transfected cell lines; no other protein isoforms were detected. A lymphocytic cell line generated from a patient with CNC (CAR507.01) who was a carrier of a premature stop codon mutation (c.920C→G/p.Ser307X) was used in parallel experiments (data not shown). As expected, the cycloheximide prevented NMD in the CAR507.01 cell line and enabled the expression of the mutant mRNA species, as we have demonstrated elsewhere (4, 5), thus proving the efficiency of the NMD treatment in the experiments described above.
To analyze PRKAR1A expression in vivo in these patients, we extracted protein from lymphocytes of three individuals from one of the affected families (CAR15). Unfortunately, cell lines or tissue were not available from the other three kindreds. Two of the individuals (CAR15.02 and CAR15.04) had CNC and were carriers of the c.1076_77delTTins13 mutation; the third individual (CAR15.03) was an unaffected family member negative for any PRKAR1A defects, whose cells served as a control so that we tested for the protein isoforms in the same genetic background (Fig. 3). Whereas the mutant cDNA was present (data not shown), Western blot analysis detected the wild-type RIα protein at 49 kDa in all three samples, consistent with expression of the wild-type allele only. Instead of the longer protein isoform, in the affected family members (CAR15.02 and CAR15.04), an additional band at 35 kDa was present; this band was not seen in the individual negative for PRKAR1A mutations (CAR15.03). We then studied PRKAR1A cDNA and protein expression in an atrial myxoma tissue of patient CAR15.02. The mutant cDNA was there (data not shown). There was loss of heterozygosity of the normal PRKAR1A allele (data not shown), and consistent with our previous data, we did not detect any protein of the wild-type size (Fig. 3). However, in addition to the shorter 35-kDa band already seen in the lymphocytes, yet another band, corresponding to a 17-kDa protein, was detected in the atrial myxoma tumor tissue. In our previous studies of tumor tissues from patients with CNC (4–6), we had never seen similar protein products in place of the wild-type PRKAR1A protein.
Fig. 3.
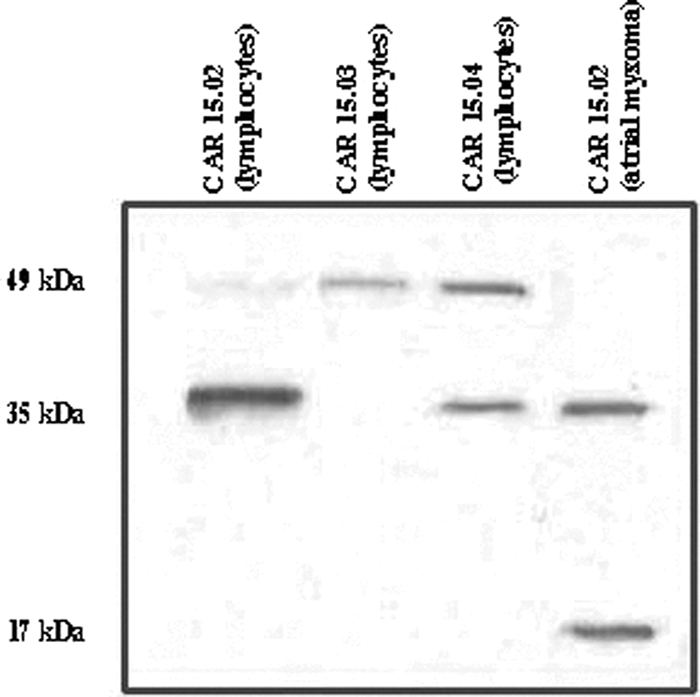
PRKAR1A protein expression in lymphocytes and tissue from an atrial myxoma from patient CAR15.02; no protein of the wild-type size was present. In addition to the shorter 35-kDa band already seen in the lymphocytes, another band, corresponding to a 17-kDa protein, was detected, most likely the product of proteolysis.
We interpreted these data as consistent with proteolytic degradation of the mutant PRKAR1A isoform produced by the c.1076_77delTTins13 PRKAR1A mutation. Thus, we then treated all transfected cell lines with the proteasome and proteolysis inhibitor CLL. Like in the cell-free experiments (Fig. 1), this time, the transfected cells contained the four PRKAR1A elongated proteins; the wild-type size was also present. The data from the HEK293 cells are shown in Fig. 4, but they were identical in the adrenal NCI-H295 and testicular N-TERA cells. As expected, the mock (empty pcDNA 3.1 expression vector) and the wild-type PRKAR1A transfected cell lines showed only the wild-type PRKAR1A protein.
Fig. 4.

HEK293 cells transfected with all four mutant constructs with (+) and without (−) the proteasome and proteolysis inhibitor CLL. When the inhibitor was used, the transfected cells contained the four PRKAR1A elongated variants along with the wild-type protein; in the absence of the inhibitor, only the wild-type PRKAR1A protein was present.
CLL targets the 20S proteasome by an irreversible modification of the amino-terminal threonine of β-subunits; however, it appears to be a nonspecific inhibitor of other cellular proteases (15). Thus, we then used the synthetic molecule MG132, a potent and selective inhibitor of the chymotrypsin-like activity of the proteasome (12, 13, 15). We proceeded in testing all transfected cells with the MG132; the data were identical to those with CLL. Figure 5 demonstrates the data, this time in the adrenortical NCI-H295 cells.
Fig. 5.
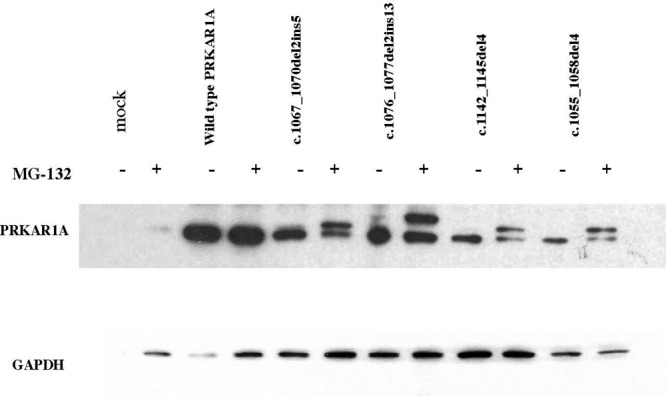
NCI-H295R cells transfected with all four mutant constructs with (+) and without (−) the specific proteasome inhibitor MG132. As in the previous experiment (Fig. 4), when the inhibitor was used, the transfected cells contained the four PRKAR1A elongated variants along with the wild-type protein; in the absence of the inhibitor, only the wild-type PRKAR1A protein was present.
The most likely mechanism for triggering proteasomal degradation of the longer PRKAR1A variants is abnormal protein folding (11). We used NN-DNJ, a synthetic chaperone that allows for the stabilization of misfolded proteins, alone and in combination with MG132 in all transfected cells. We did not get any longer protein product when NN-DNJ was used alone (data not shown) but did get it when MG132 was used, as previously (Fig. 5). NN-DNJ as a pharmacological chaperone binds to misfolded proteins inhibiting their degradation, mostly in the endoplasmic reticulum (ER) (12).
Discussion
CNC is mostly caused by RIα haploinsufficiency due to NMD of mRNA produced by PRKAR1A alleles bearing mutations that lead to a predicted premature stop codon. There are rare exceptions to this rule: a few mutations (8, 9) or larger genomic rearrangements (10) at the 17q22–24 PRKAR1A locus are not associated with RIα haploinsufficiency but instead lead to expression of a defective, shorter protein (14). The mechanism of disease relates to an inappropriate response to cAMP (9) or deficient control of the PKA C subunits (8). In this study, we report yet another mechanism of disease in CNC: when frameshift mutations occur in the last coding exon of the PRKAR1A gene and cause extension of the protein, these mutations escape NMD but still lead to RIα haploinsufficiency due to proteasomal degradation of the abnormally long mutant protein.
Our studies of four novel such mutations showed that in a cell-free system containing the ribosomal subunits, necessary chaperones, and all the other proteins needed to successfully translate a longer PRKAR1A ORF, the corresponding protein products were present. This cell-free system lacked a nucleus and a proteasome, both necessary for the naturally occurring surveillance and destruction of aberrant and malfunctioning protein isoforms. Although the elongated PRKAR1A proteins were not present after transfection in a cell-based system, cDNA sequencing showed the presence of the mutant mRNA, and treatment with cycloheximide (which inhibits NMD) did not change the findings. Instead, treatment of the transfected cells with CLL and MG132, a natural and synthetic, respectively, proteasomal inhibitor (15), led to the detection of the longer RIα isoforms (Figs. 4 and 5). Thus, PRKAR1A should be added to the list of proteins that are surveyed by the proteasome, such as collagen type 1 α1 (11). We also tested the possibility that some of this degradation takes place in the ER; NN-DNJ, a pharmacological chaperone that binds to misfolded proteins inhibiting their degradation in the ER had no effect.
The fact that frameshift mutations involving the last exon of PRKAR1A escape NMD (16) was somewhat unexpected. In several genes, mRNA containing last coding exon mutations are in fact degraded, and longer ORF are generally not present (16, 17). Analysis of the expressed PRKAR1A species in the lymphocytes and affected tumor tissues in one of the families (CAR15, carrying the c.1076_77delTTins13 mutation) showed that in vivo, the situation may in fact be even more complicated; the wild-type size protein (49 kDA) was detected in all examined cell lysates, except in those derived from a heart tumor with loss of heterozygosity of the normal 17q22–24 PRKAR1A allele. However, in all cells, including those from the heat tumor, shorter rather than longer isoforms were detected by the RIα-specific antibody. One band, at 35 kDa, was present in all samples; in the heart tumor, an additional band with molecular mass of approximately 17 kDa was detected. Indeed, two sites of proteolytic cleavage have been described in the PRKAR1A protein (18); the first one contains the sequence Lys-Arg-Arg-Gly-Ala-Ile-Ser-Ala and is recognized by trypsin and thermolysin. These enzymes cut the protein into an approximately 35-kDa fragment containing the carboxy (COOH) terminus of the protein and a shorter fragment containing the amino (NH2) terminus with a molecular mass of approximately 12 kDa. The sequence at the second site is Val-Arg-Arg-Val-Ile-Ala and is recognized by chymotrypsin only, which cuts the PRKAR1A protein into two fragments of approximately 31 and 17 kDa, containing the COOH and NH2 terminus, respectively. Thus, in our samples, we see fragments corresponding to cleavage at both proteolytic sites, 35 and 17 kDa. The lack of 31- and 12-kDa protein products may be due to either further nonspecific degradation or decreased affinity of our antibody to bind shorter PRKAR1A species. It is interesting to speculate a tumor-related increase in activity of one or the other type of cleavage (or different rates of proteolysis of the resulting fragments), given the difference between the PRKAR1A fragments in the heart tumor cells vs. the peripheral lymphocytes of patients with the c.1076_77delTTins13 mutation (Fig. 3). Because the mutant mRNA was present in the tumor cells, another possibility is the expression of alternate protein products, in vivo.
As stated earlier, the mutations in this study caused the stop codon to move further downstream when compared with the wild type. This elongation of the protein could play a role in the inhibition of its expression. Several studies have reported a significant role of the 3′-UTR sequence and the spatial distance between the termination codon and the 3′-end of the mRNA for efficient expression (17–19). In the current study, we demonstrated that both 3′-UTR sequence and the spatial distance between the termination codon and the 3′-mRNA end are disrupted in the mutant proteins, thus possibly inhibiting normal translation.
In summary, we report herein for the first time naturally occurring PRKAR1A mutations that result in elongated, compared with the wild-type, encoded protein sequences. The mutant mRNA messages are expressed and are not subject to NMD. However, the protein expression is inhibited by proteolytic degradation. Thus, defective PRKAR1A is degraded not only at the mRNA but also at the protein level.
Supplementary Material
Acknowledgments
We thank all patients and their referring physicians for their participation in this study.
This work was supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development, Intramural National Institutes of Health Project Z01-HD-000642-04 to C.A.S.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- C
- Catalytic subunit
- CLL
- clasto-lactacystin β-lactone
- CNC
- Carney complex
- ER
- endoplasmic reticulum
- NMD
- nonsense-mediated mRNA decay
- NN-PNJ
- N-nonyl-deoxynojirimycin
- ORF
- open reading frame
- PKA
- protein kinase A
- PRKAR1A
- type 1A regulatory subunit of PKA
- R
- regulatory
- RIα-WT
- wild-type RIα.
References
- 1. Carney JA, Hruska LS, Beauchamp GD, Gordon H. 1986. Dominant inheritance of the complex of myxomas, spotty pigmentation, and endocrine overactivity. Mayo Clin Proc 61:165–172 [DOI] [PubMed] [Google Scholar]
- 2. Stratakis CA, Kirschner LS, Carney JA. 2001. Clinical and molecular features of the Carney complex: diagnostic criteria and recommendations for patient evaluation. J Clin Endocrinol Metab 86:4041–4046 [DOI] [PubMed] [Google Scholar]
- 3. Gennari M, Stratakis CA, Hovarth A, Pirazzoli P, Cicognani A. 2008. A novel PRKAR1A mutation associated with hepatocellular carcinoma in a young patient and a variable Carney complex phenotype in affected subjects in older generations. Clin Endocrinol (Oxf) 69:751–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kirschner LS, Carney JA, Pack SD, Taymans SE, Giatzakis C, Cho YS, Cho-Chung YS, Stratakis CA. 2000. Mutations of the gene encoding the protein kinase A type I-α regulatory subunit in patients with the Carney complex. Nat Genet 26:89–92 [DOI] [PubMed] [Google Scholar]
- 5. Kirschner LS, Sandrini F, Monbo J, Lin JP, Carney JA, Stratakis CA. 2000. Genetic heterogeneity and spectrum of mutations of the PRKAR1A gene in patients with the Carney Complex. Hum Mol Genet 9:3037–3046 [DOI] [PubMed] [Google Scholar]
- 6. Horvath A, Bertherat J, Groussin L, Guillaud-Bataille M, Tsang K, Cazabat L, Libé R, Remmers E, René-Corail F, Faucz FR, Clauser E, Calender A, Bertagna X, Carney JA, Stratakis CA. 2010. Mutations and polymorphisms in the gene encoding regulatory subunit type 1-α of protein kinase A (PRKAR1A): an update. Hum Mutat 31:369–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bossis I, Stratakis CA. 2004. PRKAR1A: normal and abnormal functions. Endocrinology 145:5452–5458 [DOI] [PubMed] [Google Scholar]
- 8. Meoli E, Bossis I, Cazabat L, Mavrakis M, Horvath A, Stergiopoulos S, Shiferaw ML, Fumey G, Perlemoine K, Muchow M, Robinson-White A, Weinberg F, Nesterova M, Patronas Y, Groussin L, Bertherat J, Stratakis CA. 2008. Protein kinase A effects of an expressed PRKAR1A mutation associated with aggressive tumors. Cancer Res 68:3133–3141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Greene EL, Horvath AD, Nesterova M, Giatzakis C, Bossis I, Stratakis CA. 2008. In vitro functional studies of naturally occurring pathogenic PRKAR1A mutations that are not subject to nonsense mRNA decay. Hum Mutat 29:633–639 [DOI] [PubMed] [Google Scholar]
- 10. Horvath A, Bossis I, Giatzakis C, Levine E, Weinberg F, Meoli E, Robinson-White A, Siegel J, Soni P, Groussin L, Matyakhina L, Verma S, Remmers E, Nesterova M, Carney JA, Bertherat J, Stratakis CA. 2008. Large deletions of the PRKAR1A gene in Carney complex. Clin Cancer Res 14:388–395 [DOI] [PubMed] [Google Scholar]
- 11. Fitzgerald J, Lamandé SR, Bateman JF. 1999. Proteasomal degradation of unassembled mutant type I collagen pro-α (I) chains. J Biol Chem 274:27392–27398 [DOI] [PubMed] [Google Scholar]
- 12. Mu TW, Ong DS, Wang YJ, Balch WE, Yates JR, 3rd, Segatori L, Kelly JW. 2008. Chemical and biological approaches synergize to ameliorate protein-folding diseases. Cell 134:769–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Brumshtein B, Greenblatt HM, Butters TD, Shaaltiel Y, Aviezer D, Silman I, Futerman AH, Sussman JL. 2007. Crystal structures of complexes of N-butyl- and N-nonyl-deoxynojirimycin bound to acid beta-glucosidase: insights into the mechanism of chemical chaperone action in Gaucher disease. J Biol Chem 282:29052–29058 [DOI] [PubMed] [Google Scholar]
- 14. Bertherat J, Horvath A, Groussin L, Grabar S, Boikos S, Cazabat L, Libe R, René-Corail F, Stergiopoulos S, Bourdeau I, Bei T, Clauser E, Calender A, Kirschner LS, Bertagna X, Carney JA, Stratakis CA. 2009. Mutations in regulatory subunit type 1A of cyclic adenosine 5′-monophosphate-dependent protein kinase (PRKAR1A): phenotype analysis in 353 patients and 80 different genotypes. J Clin Endocrinol Metab 94:2085–2091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Myung J, Kim KB, Crews CM. 2001. The ubiquitin-proteasome pathway and proteasome inhibitors. Med Res Rev 21:245–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Muhlrad D, Parker R. 1999. Aberrant mRNAs with extended 3′ UTRs are substrates for rapid degradation by mRNA surveillance. RNA 5:1299–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tan JT, Kremer F, Freddi S, Bell KM, Baker NL, Lamandé SR, Bateman JF. 2008. Competency for nonsense-mediated reduction in collagen X mRNA is specified by the 3′ UTR and corresponds to the position of mutations in Schmid metaphyseal chondrodysplasia. Am J Hum Genet 82:786–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Potter RL, Taylor SS. 1980. The structural domains of cAMP-dependent protein kinase I. Characterization of two sites of proteolytic cleavage and homologies to cAMP-dependent protein kinase II. J Biol Chem 255:9706–9712 [PubMed] [Google Scholar]
- 19. Inada T, Aiba H. 2005. Translation of aberrant mRNAs lacking a termination codon or with a shortened 3′ UTR is repressed after initiation in yeast. EMBO J 24:1584–1595 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.