

Abstract
KCNH1 (EAG1) is a member of the Kv family of voltage-gated potassium channels. However, KCNH1 channels also show some amino-acid sequence similarity to cyclic-nucleotide-regulated channels: they harbor an N-terminal PAS domain, a C-terminal cyclic nucleotide binding homology domain (cNBHD), and N- and C-terminal binding sites for calmodulin. Another notable feature is the channels’ high sensitivity toward oxidative modification. Using human KCNH1 expressed in Xenopus oocytes and HEK 293 cells we investigated how oxidative modification alters channel function. Intracellular application of H2O2 or cysteine-specific modifiers potently inhibited KCNH1 channels in two phases. Our systematic cysteine mutagenesis study showed that the rapid and dominant phase was attributed to a right-shift in the voltage dependence of activation, caused by chemical modification of residues C145 and C214. The slow component depended on the C-terminal residues C532 and C562. The cysteine pairs are situated at structural elements linking the transmembrane S1 segment with the PAS domain (N-linker) and the transmembrane channel gate S6 with the cNBH domain (C-linker), respectively. The functional state of KCNH1 channels is determined by the oxidative status of these linkers that provide an additional dimension of channel regulation.
Keywords: Potassium channel, Reactive oxygen species, Oxidation, Sulfhydryl modification, Ether à go-go, Gating
1. Introduction
The ether à go-go (EAG) gene encodes a voltage-dependent potassium channel and was first cloned from Drosophila [1] during analysis of mutations affecting membrane excitability. Subsequent molecular studies and sequence comparison resulted in the formation of a distinct potassium channel group: the EAG family. The EAG family now consists of 3 subfamilies: EAG, EAG-related gene (ERG), and EAG-like K+ channel (ELK). The mammalian EAG subfamily comprises of two members, termed EAG1 (KCNH1, KV10.1 [2,3]) and EAG2 (KCNH5, KV10.2 [4,5]). The pore-forming α-subunit of KCNH1 channels contains a core domain resembling that of other voltage-dependent K+ channels with six transmembrane segments (S1–S6) including a charged S4 voltage sensor, and functional channels are suggested to be homotetrameric. In addition, the channel contains large cytosolic amino and carboxyl termini. In its N terminus, KCNH1 harbors a Per-ARNT-Sim (PAS) dimerization domain [6] and a calmodulin-binding motif [7]. The C terminus contains a cyclic nucleotide binding homology domain sequence (cNBHD) and two additional calmodulin/S100B binding motifs [8,9].
KCNH1 is widely expressed in the central nervous system, probably participating in neuronal signaling. In situ hybridization studies have detected KCNH1 transcripts predominantly in the hippocampus, cerebral cortex, dopaminergic neurons of basal ganglia, and olfactory bulb and granular layer of the cerebellum of adult rats [4,10–12]. KCNH1 channels may also be involved in sensory functions of auditory transduction and phototransduction. Rat KCNH1 mRNA has been detected in the organ of Corti and in the fibrocytes of the spiral ligament [13]. In situ hybridization of bovine retinal tissue localized two splice variants of the KCNH1 channel transcript to photoreceptors and retinal ganglion cells [14]. In rod photoreceptors, KCNH1 channels probably underlie an outward K+ current, named IKx, which stabilizes the dark resting potential and accelerates the voltage response to small photocurrents [15]. KCNH1 channels are also essential for induction of human myoblast differentiation and fusion [3,16]. Apart from their physiological function, KCNH1 channels were found abnormally up-regulated in many cancer cells while absent from the corresponding healthy tissues. First observations of this aberrant expression included cell lines derived from breast carcinoma (MCF7, EFM-19 and BT-474), cervical carcinoma (HeLa), neuroblastoma (SH-SY5Y), and melanoma (IGR1) [17–19]. Later studies directly confirmed the overexpression of KCNH1 in tumor tissue of a large variety of human cancers [20]. A recent study on Danio rerio KCNH1 channels suggests their involvement in early development [21].
Functional properties of membrane ion channels can be modulated by a plethora of posttranslational modifications, including oxidation of certain amino acid residues such as cysteines [22–25]. The functional activity of human ether à go-go-related gene 1 (hERG1, KCNH2) potassium channels is efficiently diminished when cellular ROS levels increase, e.g. in response to hyperglycemia [26,27]. We recently identified a cysteine residue (C723) in the cytosolic C terminus of KCNH2 as a major determinant of this oxidation sensitivity. Oxidation of this residue by H2O2 or by thiol-specific reagents resulted in channel inhibition and faster deactivation kinetics [28]. Given the vital function of KCNH2 channels in the control of cardiac action potential repolarization [29], this illustrates the potentially profound impact of seemingly mild modifications on channel proteins. Intriguingly, a splice variant of KCNH2 (hERG1b) that shows specifically high expression in the heart [30] was found to be less sensitive to oxidative modification [28].
In this study we ask if KCNH1 channels are also subject to ROS-mediated functional modulation. We show that KCNH1 channel function is tightly controlled by each two cysteine residues situated in the structural elements linking the cytosolic N-terminal (N-linker) and C-terminal (C-linker) domains to the transmembrane domain, which harbors the channel gate and pore.
2. Material and methods
2.1. Chemicals
DTNB (5, 5′-dithio-bis[2-nitrobenzoic acid]), DTNP (2,2′-dithio-bis [5-nitropyridine]), and DTT (dithiothreitol) were obtained from Sigma (Taufkirchen, Germany); MTSET ([2-(trimethylammonium)ethyl] methane thiosulfonate bromide) and MTSES (2-sulfonatoethyl methanethiosulfonate sodium salt) were from Toronto Research Chemicals (North York, ON, Canada). All reagents were freshly prepared in intracellular solution prior to each experiment and stored at 4 °C and used within 20 min.
2.2. Channel constructs and site-directed mutagenesis
The following channel types were used in this study: human EAG1 (hEAG1, KCNH1), rat Kv1.1 (rKv1.1, KCNA1), and human Kv1.5 (hKv1.5, KCNA5).
Within the Xenopus oocyte expression vector KCNH1-pGEMHE, the following point mutants of KCNH1 were generated by overlap-extension methods as described previously [8] and verified by sequencing: C50A, C67A, C128A, C145A, C214A, C303A, C532A, C541A, C562A, C575A, C592A, C631A, C640A, C794-804-817-853A, Cys-less N (C50-67-128-145-214A), Cys-less T (C303A-C357V-C368V), and Cys-less C (C532-541-562-575-592-631-640-794-804-817-853A). Cysteine residues were replaced with alanine except for the residues in the S5 segment: C357V, C368V. In the background of a mutant lacking all cysteines from the cytosolic termini (Cys-less NC), individual cysteine residues (A128C, A145C, A214C, A532C, and A562C) were reintroduced to determine their individual impact.
2.3. Channel expression in Xenopus oocytes and HEK 293 cells
Capped mRNA was synthesized in vitro using the mMessage mMachine kit (Ambion, Austin, TX, USA). Oocytes were surgically removed from the ovarian tissue of Xenopus laevis that had been anesthetized by immersion in ice water tricaine according to the local animal care program. The oocytes were defolliculated, and healthy stage V and VI oocytes were isolated and microinjected with 50 nl of a solution containing channel wild-type or mutant mRNA. The removal of the vitelline membrane was carried out by enzymatic treatment as described by Wang [31]. Electrophysiological measurements were performed 2–4 days after mRNA injection.
KCNH1 in the mammalian cell expression vector pcDNA3 (Invitrogen, Darmstadt, Germany) was transiently expressed in HEK 293 cells using Superfect transfection kit (Qiagen, Hilden, Germany). Dynabeads (Deutsche Dynal GmbH, Hamburg, Germany) were used for visual identification of individual cells, cotransfected with CD8. Electrophysiological recordings were performed 2–3 days after transfection.
2.4. Electrophysiological measurements
Ionic currents were recorded using the inside-out or whole-cell configuration at room temperature using an EPC-9 patch-clamp amplifier operated with PatchMaster software (both HEKA Elektronik, Lambrecht, Germany). Inside-out patch-clamp experiments were carried out with Xenopus oocytes; macroscopic currents were measured using aluminum silicate glass pipettes with resistances of about 1 MΩ. The intracellular solutions contained (in mM): 100 K-aspartate, 10 EGTA, 15 KCl, 10 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (pH 7.2 or 8.0 with KOH). The extracellular solution contained (in mM): 103.6 Na-aspartate (for recordings under symmetrical K+ conditions 103.6 K-aspartate), 11.4 KCl, 1.8 CaCl2, 10 HEPES (pH 7.2 with NaOH). Solution changes in patch-clamp experiments were performed using a multi-channel perfusion system in which the patch was placed directly in the center of streaming solution. Whole-cell voltage-clamp experiments were carried out with transiently transfected HEK 293 cells. Patch pipettes from borosilicate glass with resistances of 0.9–2 MΩ were used. Pipette solution contained (in mM): 130 KCl, 2.56 MgCl2, 10 EGTA, 10 HEPES (pH 7.4, KOH); bath solution: 135 NaCl, 5 KCl, 2 CaCl2, 10 HEPES (pH 7.4, NaOH). Cells with series resistance above 5 MΩ were discarded; the series resistance was compensated for by more than 70%.
Voltage dependence of channel opening was measured by applying 200-ms depolarizations in steps of 10 mV. The resulting maximal current, subjected to a p/6 leak correction procedure, was plotted as a function of test voltage and described with a Hodgkin-Huxley approach involving four activation gates and a linear single-channel current-voltage relationship:
| (Eq. 1) |
with the linear conductance Γ, the reversal potential Erev, the half-maximal activation voltage per gate Vm, and the corresponding slope factor km.
2.5. Non-stationary noise analysis
Non-stationary noise analysis was performed from sets of at least 200 successive current sweeps elicited by depolarizations to −20, 0, 20, 40, and 80 mV. Ensemble variances were compiled from differences of neighboring records [32] and variance-current plots of the activating current phases were simultaneously fit for all voltages (V) according to Starkus et al. [33] to yield the single-channel current i(V), the maximal open probability Po(V), and the number of active channels. Data fits were always performed simultaneously for control and DTNB-modified patches for all voltages available constraining N, the total number of active channels. This procedure particularly ensured reliable estimates of low Po values at low depolarization and upon DTNB application.
Data were analyzed with FitMaster (HEKA Elektronik) and IgorPro (WaveMetrics, Lake Oswego, OR, USA). Data are presented as mean ± SEM (n = number of independent measurements). P values refer to two-sided Student’s t-tests with unequal variances.
3. Results
3.1. KCNH1 channels are particularly sensitive to intracellular cysteine modification
KCNH2 channels are tightly regulated by the redox status of intracellular cysteine residues [28]. We thus tested the sensitivity of the non-inactivating close relative KCNH1 on cysteine-modifying substances. KCNH1 was expressed in HEK 293 cells and currents were recorded in the whole-cell configuration in response to repetitive depolarizations to 40 mV. Extracellular application of the thiol-modifying reagent DTNB (100 μM), a hydrophilic poorly membrane permeable compound that modifies proteins based on the principle of thiol-disulfide exchange reaction, only slightly increased the KCNH1-mediated outward current (Fig. 1A, B). In contrast, extracellular application of 20 μM of DTNP, a lipophilic membrane permeable thiol-modifying reagent, caused rapid and complete inhibition of macroscopic KCNH1 current (Fig. 1A, B). Treatment with the slowly membrane permeable physiological oxidant hydrogen peroxide (H2O2, 1 mM) only induced slow KCNH1 channel inhibition with 30 ± 8% remaining current after exposure of 200 s (n = 6, Fig. 1A, B). These results suggest that, similar to KCNH2, intracellular cysteines are responsible for the profound alteration of the KCNH1 channel activity by DTNP or H2O2.
Fig. 1.

Intracellular cysteines of KCNH1 are susceptible to oxidative modification. (A) Superposition of current traces recorded from KCNH1-transfected HEK 293 cells at 40 mV before (black) and 200 s after (gray) extracellular application of 100 μM DTNB, 20 μM DTNP, and 1 mM H2O2, respectively. (B) Mean time courses of normalized currents at 40 mV for the application of DTNB, DTNP, and H2O2; substances in concentration as in (A) were applied at time zero. (C) Superposition of current traces at 40 mV from inside-out patches of Xenopus oocytes expressing the indicated channel types before (black) and 100 s after (gray) cytosolic application of 100 μM DTNB. (D) Mean time courses of normalized inside-out patch currents at 40 mV for the indicated channel types upon application of 100 μM DTNB at time zero. Relative remaining current after 100 s was 29 ± 1% for KCNH1 (n = 5, gray circles); for rat KCNA1 (open circles) and human KCNA5 (black circles) current amplitude did not significantly decrease (P > 0.05). (E) Superposition of inside-out patch KCNH1 current traces before (black) and 100 s after (gray) application of 300 μM MTSET and 100 μM MTSES, respectively. Straight lines connect the data points in B and D; data are mean ± SEM with n in parentheses. Holding potential was −80 mV in all cases.
Unlike for KCNH2 channels, however, KCNH1 channels are readily investigated in inside-out patches taken from Xenopus oocytes because in the excised patch configuration only a small degree of current loss (rundown) is observed. We therefore compared the effects of 100 μM DTNB when applied to the intracellular side of the membrane. As shown in Fig. 1C and D, KCNH1 currents were reduced by 72 ± 3%, while the voltage-gated potassium channels KCNA1 and KCNA5 were resistant to DTNB treatment. The sensitivity of KCNH1 was not specific for DTNB because also thiol-modifiers MTSET (positively charged) and MTSES (negatively charged) strongly diminished KCNH1 currents (Fig. 1E; remaining current after 160 s: 18 ± 1% for 100 μM MTSES and 10 ± 1% for 300 μM MTSET). Further experiments were performed with DTNB, in particular because DTNB is a relatively small molecule and the effect on the channel could be reversed with the reducing agent DTT (see below, Fig. 2B).
Fig. 2.

Time course of DTNB modification of KCNH1 channel function. (A) Current traces from an inside-out patch with KCNH1 channels at the holding potential of −120 mV, pulsing to the indicated voltages (for 50 ms each) under control conditions (black) and upon application of 20 μM DTNB for the indicated incubation times (gray). (B) Time course of DTNB-mediated (20 μM) current rundown with double-exponential fits superimposed followed by wash with control saline at three voltages (open circles, 100 mV, gray circles, 40 mV, and triangles, −40 mV). Pulse interval was 10 s. (C) Time course of KCNH1 current at 40 mV with application of 20 μM DTNB and wash with saline containing 3 mM DTT. (D & E) Analysis of inhibitory kinetics: time constants (D) and relative fast fraction (E) at three voltages and with two different concentrations (gray, 20 μM and open, 100 μM). Two time constants (τfast, squares; τslow, circles) were obtained by fitting the data with a double-exponential function (n = 5).
3.2. Time course of DTNB effect on KCNH1 is indicative of multiple targets
The time course of channel modification by 20 μM DTNB on the cytosolic side was analyzed in more detail using symmetrical K+ solutions and a pulse protocol assaying for channel activation at −40, 40, and 100 mV (Fig. 2A). At each of the three voltages the peak current elicited was decreased by DTNB application in a biphasic manner (Fig. 2B). Wash with control saline did not recover the current, but application of 3 mM DTT restored the currents almost completely strongly suggesting that DTNB action is mediated by cysteine-specific modification (Fig. 2C).
Analysis of the current decay as a function of time revealed two exponential components, characterized by two time constants (τfast, τslow, Fig. 2D) with the respective amplitudes (afast, aslow, Fig. 2E): for example, at 40 mV τfast = 12.8 ± 2.0 s, τslow = 369 ± 91 s, with a relative fast fraction (afast/(afast+aslow)) of 62 ± 5.9% (n = 5). At higher DTNB concentration (100 μM) the kinetics was about 2–3 fold faster. In addition, the reaction was slower at stronger depolarization (100 mV vs −40 mV; P < 0.01) suggesting that the DTNB effect on hEAG1 channels depends on the voltage and/or the channel conformational change.
3.3. DTNB alters the gating of KCNH1 channels
The observation that DTNB decreases KCNH1 currents could be interpreted to indicate that DTNB is a simple pore blocker. However, several lines of evidence collectively show that DTNB is rather a gating modifier of KCNH1 channels. For example, intracellular application of 20 μM DTNB slowed the activation kinetics at 40 mV (from to 4.9 ± 0.8 ms to 11.5 ± 1.1 ms), and accelerated the deactivation kinetics at −120 mV (from 1.88 ± 0.15 ms to 1.06 ± 0.06 ms) (Fig. 3B). The voltage dependence of channel activation was analyzed by measuring channel opening in symmetrical K+ solutions and the normalized maximal currents were fit with Eq. 1. As shown in Fig. 3C & D, DTNB not only reduced the current, but the remaining current displayed altered voltage dependence: DTNB shifted the half-activation voltage (Vm) from −65.7 ± 3.6 mV to −49.3 ± 0.9 mV and the slope factor (km) from 29.5 ± 1.9 mV to 40.9 ± 5.6 mV (n = 5).
Fig. 3.

Impact of DTNB on KCNH1 channel function. (A) Pulse protocol (top) and superimposed current traces from a representative inside-out patch containing KCNH1 channels before (black) and 200 s after (gray) application of 20 μM DTNB. Current in the presence of DTNB is also shown scaled in amplitude to match the maximal control value (dashed, gray). (B) Kinetics of activation at 40 mV and deactivation at −120 mV were estimated by fitting single-exponential functions to the data traces. Data points show the resulting time constants before (white) and 200 s after application of 20 μM DTNB (gray). (C) Normalized peak conductance – voltage relationship of KCNH1 currents before (open circles) and after application of 20 μM DTNB (gray circles). Smooth curves are results of data fits according to a Hodgkin & Huxley formalism involving four activation gates (Eq. 1). The dashed curve represents data after DTNB application, normalized to the maximal conductance. (D) Fit results characterizing voltage dependence of activation before (open symbols) and after DTNB application (gray symbols). Data shown in A–D were obtained under symmetrical K+ conditions. (E, F) Non-stationary noise analysis at five different potentials was performed to estimate the maximal open probability (E) and the single-channel current amplitude (F) of KCNH1 channels before (open symbols) and after DTNB application (gray symbols). Straight lines connect the data points in E and F for illustrative purposes and have no physical meaning. An average single-channel conductance was estimated to 14.0 ± 0.6 pS before and 12.0 ± 0.6 pS after DTNB application. All data in B–F are means ± SEM with n indicated in parentheses.
Macroscopic KCNH1 current is a function of the number of functional KCNH1 molecules expressed at the cell surface (N), the single-channel current amplitude (i), and the open probability (Popen) of the channel. In principle, a change in any of these three components of the macroscopic current could account for the effect of thiol reagents. To differentiate between changes in single-channel conductance and channel Popen, non-stationary noise analysis was performed in the inside-out patch clamp configuration. As illustrated in Fig. 3E & F, application of 20 μM DTNB to the intracellular side of KCNH1 resulted in a substantial decrease in Popen (e.g. at 0 mV from 45.5 ± 0.6% to 0.6 ± 0.4% and at 80 mV from 64.8 ± 0.6% to 14.3 ± 1.4%) and only a mild reduction of the unitary current size assuming a constant number of active channels. This finding shows that DTNB is not a pore blocker but rather affects channel gating, effectively reducing the probability of channel opening. The effect of DTNB is weaker at stronger depolarization such as if voltage-dependent conformational changes would counteract the action of the thiol modifier.
3.4. Identification of critical cysteines responsible for thiol-dependent alteration of channel function
To gain structural insight into the thiol-dependence of KCNH1 channel function we used site-directed mutagenesis to identify critical cysteine residues. As illustrated in Fig. 4A, a KCNH1 α-subunit harbors 19 cysteine residues, 3 located in the predicted transmembrane segments, 5 in the N-terminal, and 11 in the C-terminal domains; the latter two domains are facing the intracellular side. We thus examined the effects of DTNB on KCNH1 constructs in which one or more cysteines were mutated to alanine except for two that were mutated to valine. When all 19 cysteines were replaced, channels expressed poorly and were difficult to study.
Fig. 4.

Identification of cysteine residues taking part in KCNH1 regulation. (A) Schematic representation of a KCNH1 subunit with the intracellular N terminus, the C terminus, and six transmembrane segments, indicating cysteine residues (orange circles) and regulatory protein domains (CaM, calmodulin binding domains; PAS, Per-ARNT-Sim domain; cNBHD, cyclic nucleotide binding homology domain area). Cysteine residues identified as targets of oxidative channel modification are highlighted in red. Also indicated are the elements connecting the PAS domain and S1 (N-linker) and S6 with the cytosolic C-terminal domain (C-linker). (B) Normalized current for the indicated KCNH1 constructs at 40 mV with DTNB application (20 μM) at time zero. (C) Analysis of the mean remaining current after 200 s DTNB application (I200, left) and up to two time constants of current reduction (right). All data are means ± SEM with n indicated in parentheses. “N.D.” denotes cases in which the corresponding exponential component was not present and, hence, was not determined.
Constructs lacking 3 cysteines in the transmembrane region (Cys-less T), 5 cysteines in the N terminus (Cys-less N), 11 cysteines in the C terminus (Cys-less C), or lacking 16 cysteines in both N and C terminus (Cys-less NC) expressed well and generated macroscopic currents. When cysteine residues within the predicted transmembrane region were mutated, the resulting channels retained their sensitivity towards 20 μM cytosolic DTNB (not shown), while the construct lacking all cysteines in the N- and C-terminal domains (Cyc-less NC) was not affected at all by DTNB (Fig. 4B, C). In Cys-less N, the fast component of the DTNB effect was abolished; the remaining slow block developed with a time constant (τslow) of 404 ± 73 s. In Cys-less C, only a rapid current block with a time constant (τfast) of 20 ± 7 s was observed (Fig. 4B, C). Comparison with the two time constants obtained for the wild type (τfast = 12.8 s, τslow = 369 s) suggests that cysteine residues in the N terminus must be responsible for the rapid effect and cysteine residues in the C terminus for the slow effect of DTNB.
From this observation it appears that N-terminal cysteines play a major role in DTNB-mediated channel inhibition. To assess the functional importance of the five cysteine residues in the N terminus, each was mutated to alanine and assayed for the fractional current remaining after DTNB treatment. Mutants C145A and C214A showed the weakest response to DTNB with ~65% of the current remaining after 200-s application of 100 μM DTNB (not shown), here operationally defined as DTNB sensitivity. From the analysis of half-activation voltage (Vm), mutants C145A and C214A exhibited a smaller shift when exposed to DTNB (11 and 6 mV, respectively), while the shifts in Vm of KCNH1 wild type, C50A, C67A, and C128A were 29, 15, 16, and 19 mV, respectively. Based on this graded response, we generated the double mutant C145-214A; this showed a markedly diminished sensitivity towards DTNB, leaving only a minor slow component (Fig. 5A, B). Interestingly, the slow onset of DTNB-induced inhibition only occurred with a substantial delay, presumably indicative of the involvement of multiple targets in this response. In addition, the voltage-dependence of channel activation, characterized by Vm and km, was resistant to DTNB (Fig. 5C).
Fig. 5.

Identification of cysteine residues taking part in KCNH1 regulation. (A) Normalized current on a logarithmic scale for the indicated KCNH1 constructs at 40 mV with DTNB application (20 μM) at time zero. (B) Analysis of the mean remaining current after 200 s DTNB application (I200, left) and up to two time constants of current reduction (right). (C) Analysis of voltage-dependent activation (Vm, left; km, right) for the indicated mutants before (open circles) and after application of 20 μM DTNB (gray). All data are means ± SEM with n indicated in parentheses. “N.D.” denotes cases in which the corresponding exponential component was not present and, hence, was not determined.
A similar approach was followed for the C-terminal cysteines. Out of all individual C-to-A substitutions, none strongly affected the sensitivity towards DTNB, most likely because of the presence of the sensitive N-terminal residues C145 and C214. Given the conservation of the major cytosolic domains of KCNH1 channels such as C-linker and CNG-binding domain within the EAG family, KCNH1 channels may share some similarity to KCNH2 channels. A study from our group [28] has found that C723 and C740 of KCNH2 are responsible for oxidative modification and mutation of these cysteine residues rendered the KCNH2 channel insensitive to reactive oxygen species. The homologous positions in KCNH1 are C532 and C562. Combination of these mutations (C532-562A) resulted in channels largely resembling the DTNB sensitivity of Cys-less C, namely only displaying a rapid but saturating response to DTNB (Fig. 5A, B). Unlike to mutant C145-214A, the DTNB impact on voltage dependence was not affected by mutation C532-562A, i.e. Vm and km of the resulting channels were shifted like the wild type (Fig. 5C). Combination of both constructions, i.e. C145-214-532-562A, resulted in complete loss of DTNB sensitivity, much as in Cys-less NC (Fig. 5).
Thus the mutagenesis results collectively show that residues C145 and C214 are important for the rapid component of the DTNB effect and, once thiol-modified, result in a shift in the voltage dependence of channel activation. The C-terminal residues C532 and C562, located in the C-linker region, play a minor role, being responsible for a slow, but complete loss of channel function.
Elimination of cysteines and observation of an altered response to a thiol modifier not necessarily mean that a particular residue is a target of thiol modification. We thus applied an additional approach by starting with the DTNB-insensitive Cys-less NC construct and reintroduced individual cysteines. As shown in Fig. 6A, C, introduction of C128 did not induce DTNB sensitivity, serving as a negative control. Introduction of either C145 or C214 reestablished part of the rapid DTNB response. Likewise, reintroduction of C532 and, particularly, C562 resulted in a slow component of DTNB-medicated current reduction (Fig. 6B,C). Both sets of experiments support the aforementioned that, indeed, C145/C214 in the N-terminal domain and C532/C562 in the C-terminal domain are of prime importance of the functional state of KCNH1 channels.
Fig. 6.

Reintroduction of cysteines into DTNB-insensitive channels. (A, B) Normalized current for the indicated KCNH1 constructs at 40 mV with DTNB application (20 μM) at time zero. In (A) N-terminal and in (B) C-terminal cysteines were introduced into the background of the cysteine-less NC construct. (C) Analysis of the mean remaining current after 200 s DTNB application (I200, left) and up to two time constants of current reduction (right). All data are means ± SEM with n indicated in parentheses.
To test if the identified cysteine residues are only of relevance for KNCH1 function when modified with DTNB, we subjected inside-out patches with wild-type channels and mutant C145-214-532-562A to H2O2 and the thiol modifiers MTSET and MTSES. As illustrated in Fig. 7, in all cases modification resulted in a biphasic loss of channel function in the wild type while the mutations eliminated the channel’s sensitivity toward oxidative modification.
Fig. 7.

Effect of various thiol modifiers. Mean currents from inside-out patches in response to 10 mM H2O2 (top), 300 μM MTSES (center), and 100 μM MTSET (bottom) at time zero during repetitive 50-ms pulses to 40 mV. Open symbols denote results from the wild type (wt), gray symbols for mutant C145-214-532-562A. All data are means ± SEM with n indicated in parentheses.
3.5. State dependence of KCNH1 thiol modification
Data shown thus far indicated that the effect of DTNB on KCNH1 channels depends on the membrane voltage. In addition, DTNB-mediated alteration of the gating parameters is associated with modification of the N-terminal cysteines C145/C214. Since such residues apparently have an impact on channel gating, the channel’s conformational state could, in turn, affect the accessibility of such residues. We therefore measured the effect of 20 μM DTNB on KCNH1 channels in inside-out patches during repetitive pulsing to 40 mV at an interval of 20 s; between these pulses the holding voltage was either set to −80 mV or 20 mV, thus keeping the channel most of the time in a deactivated or activated configuration, respectively. As shown in Fig. 8A, DTNB inhibited the channels significantly more strongly when the membrane potential was held at −80 mV indicating that critical cysteines are more accessible and/or reactive when the channel is in a resting conformation. Since the protocol for the “20 mV condition” involved sojourns around resting voltage for about 500 ms to acquire P/n pulses, channels were not kept in an activated state all of the time. We therefore assayed the effect of DTNB on channels, either held at −120 or 20 mV for 100 s, without pulsing. As shown in Fig. 8B, C, current inhibition was substantially greater under resting conditions compared to 20 mV (P > 0.01) illustrating a strong use- or voltage dependence of channel modification.
Fig. 8.
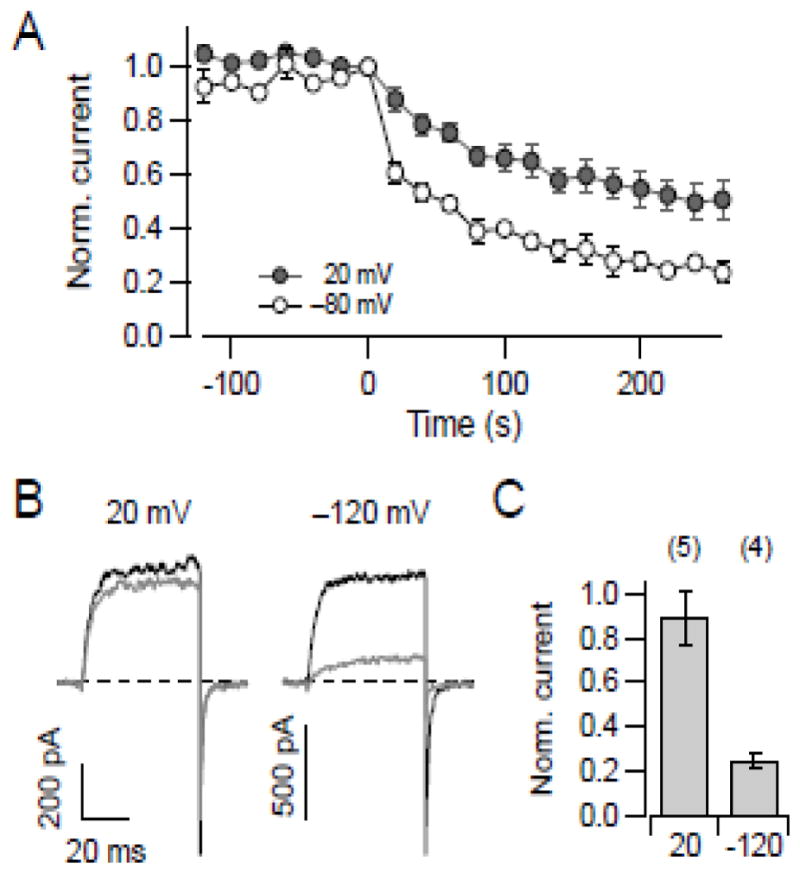
State dependence of DTNB-medicated channel modification. (A) Mean KCNH1 currents from inside-out patches in response to 20 μM DTNB application starting at time zero during repetitively pulsing to 60 mV (for 40 ms, interval of 20 s) with a holding voltage of −80 mV (n = 3; open circles) and 20 mV (n = 4; filled circles). All data are means ± SEM; straight lines connect the data points. (B) Superposition of current traces at 40 mV obtained before (black) and after (gray) exposure to 20 μM DTNB for 100 s. During that exposure the membrane was clamped to the indicated voltage without applying depolarizing pulses. (C) Mean normalized current upon DTNB treatment for the indicated holding voltages. Data are means ± SEM with n indicated in parentheses.
4. Discussion
4.1. Structure-function implications
The identification of four critical cysteine residues not only revealed the major target sites of oxidative modification in KCNH1, but also provides new insights into the dynamic interplay of functional channel domains. The four targets of oxidation fall into two functional groups, located in the N-linker (C145, C214) and in the C-linker (C532, C562), respectively. Notably, the amino acids conferring oxidation sensitivity are not located in any of the defined protein domains, such as the PAS domain, the cyclic nucleotide binding homology domain or the transmembrane helices (Fig. 4A), but in linker regions for which no precise functional roles have been attributed thus far.
Oxidative modification of cysteines in the N-linker is characterized by rapid kinetics and by a resulting right shift in voltage dependence. Thus, both sites must be well exposed to the attack by DTNB, H2O2, and MTS reagents and are most likely in a polar environment. Unlike chemical modification of the C-linker cysteines, modification of the N-terminal oxidation targets did not result in complete current loss, regardless of the oxidant concentration or the time of exposure. The modified channels are still active, but functionally altered. Oxidation of a mutant channel lacking the C-terminal target sites (C532A·C562A) induced a prominent shift inactivation of about 20 mV, while mutagenesis of the N-terminal residues (C145A·C214A) resulted in a similar shift (Fig. 5C). Thus, complete oxidation of the N-linker sites shifts the voltage dependence of activation and therefore results in markedly lower current amplitudes at the test potential of 40 mV without completely impairing channel function.
By contrast, oxidation of critical cysteines in the C-linker was characterized by slower kinetics; this reduced reactivity is indicative of a more buried and hydrophobic localization in the overall structure. Despite this slower kinetics, the effect of oxidation here was more dramatic, ultimately leading to complete loss of function. Thus, the impact of C-linker oxidation either is an extremely large shift in the voltage dependence of activation to the positive direction or it must involve other mechanisms leading to channel dysfunction. The latter is supported by a mild impact of mutagenesis of the critical residues (C532A·C562A) on half-maximal activation voltage (Fig. 5C).
Several molecular mechanisms are conceivable to provide a mechanistic link between cysteine modification and loss of current. (I) Most straightforwardly one could postulate a direct interference of modified residues with the ion permeation pathway. Given the cytoplasmic location of the critical cysteines this is unlikely and non-stationary noise analysis of DTNB-modified KCNH1 channels (Fig. 3E,F) directly ruled out alterations in the single-channel conductance as cause of current loss. (II) Channel exposure to oxidative agents might be suspected to either break or induce disulfide bridges between the critical cysteine residues. However, in this case for each functional pair of cysteines one should expect that mutagenic replacements of either of the sites or of both sites have equal functional effects unless intersubunit disulfide bonds are involved. This is not supported by our data, as cysteine mutagenesis had additive protective effects. (III) More complex scenarios might be summarized as “allosteric mechanisms”, assuming that the modifications influence endogenous interactions within functional channel domains. Such an assumption appears to be plausible from a study by Stevens et al. [34] where they observed a structural and functional interaction between PAS and cNBHD of the KCNH1 channel. DTNB might induce its inhibitory effect by interfering with the cross talk between PAS and cNBH domain. An interaction between the N-terminal and the cNBHD regions of CNGA1 channels has been previously demonstrated by Gordon et al. [35]. In summary, the functional similarities between N-linker cysteines on the one hand and C-linker cysteines on the other hand imply that each pair of cysteines affects a distinct functional aspect of the channel.
Neither for the cytosolic N terminus nor for the C terminus of the KCNH1 protein a clear function has been defined yet, but mutagenesis studies imply that they have principally opposing effects on channel opening. At least the proximal half of the C terminus, including C-linker and cNBHD is essential for the formation of functional channels. A systematic deletion study by Chen and coworkers [36] showed that removal of the distal C terminus by introduced stop codons in the C-linker (Q477X or R521X) or closely behind the cNBHD (N673X or E722X) of the rat KCNH1 protein abolished the formation of electrophysiologically functional channels. If we therefore assume that the cNBHD takes part in the process of channel opening, any allosteric control mechanism most likely involves the C-linker as a coupler between this domain and the cytosolic end of S6 as the channel’s probable gate structure. In this context, it is tempting to speculate that oxidation of cysteines in the C-linker disrupts this essential interaction between C terminus and gate. Interestingly, the critical role of cysteines in the C-linker of KCNH1 parallels the oxidation sensitivity of the C-linker residue C723 in the human KCNH2 channel [28]. Just like in KCNH1, presence of the C-linker and the cNBH domain in KCNH2 channels is required for active channel formation and oxidation of C723 results in current reduction and faster channel deactivation – more rapid channel closure. Likewise, a recent high-resolution structure of a zebrafish KCNH paralog (ELK) demonstrated the importance of the C-linker structure for intersubunit interaction and, hence, presumably the coordination of channel gating events [37].
In contrast to the requirement of the C terminus for the formation of a functional KCNH1 channel, complete deletion of the N terminus (Δ2–190) from KCNH1 still results in functional voltage-gated K+ channels [7,38]. Compared to the wild type, such truncated channels open at very negative voltages, with a shift of the half-activation voltage of about −70 mV [35]. Apparently, the function of the N terminus in KCNH1 is not to promote channel opening, but rather to stabilize a closed state that is only opened upon depolarization. The underlying mechanism is unknown, but a physical interaction between residues in the distal N terminus and the S4–S5 linker was strongly suggested by mutagenesis studies in rat KCNH1 [38]. More recent structural analyses revealed that the S4–S5 linkers in voltage-gated K+ channels [39,40] as well as in voltage-gated Na+ channels [41] form short helices that contact the cytosolic end of the S6 helices and link movements in the voltage sensor to movements in S6 to open or close the gate. If the S4–S5 helix forms a “cuff” that constricts the gate [41], it is conceivable that interaction of the N terminus in KCNH1 with the S4–S5 helix stabilizes the closed conformation of this cuff, shifting the activation voltage to more positive potentials. In this context, the observed right shift of the half-activation voltage upon cysteine oxidation in the N-linker suggests that one function of the N terminus is not disturbed through oxidation, but rather augmented. Cysteine oxidation by DTNB is a relatively dramatic modification adding an aromatic ring structure and a negative charge to the site. Interestingly, a more subtle modification, the mutagenic replacement of alanine for cysteine (C145A, C214A), introduced a very similar right shift in voltage dependence as the oxidation. Quite remarkably, the major component of channel inhibition by DTNB, i.e. a component associated with modification of C145/C214 in the N terminus, was strongly dependent on the channel’s conformational state (Fig. 8). Depolarization-induced channel opening effectively protected these sites from modification arguing that in KCNH1 channels movement of the voltage-sensor elements has a profound impact on the structure of the cytosolic N-terminal domains.
In summary, cysteine oxidation in KCNH1 can induce current reduction by two mechanisms: oxidation in the N terminus augments a right shift in voltage dependence that is physiologically conferred by the N terminus. By contrast, oxidation in the C-linker most likely disturbs an essential mechanical link between the cNBHD and the S6 helix.
4.2. Physiological role of ROS-mediated channel regulation
Elevated levels of physiological ROS generation are known to play a key role in aging and many neuronal disorders such as in Alzheimer’s disease and Parkinson’s disease. Oxidative stress generated during these conditions induces progressive loss of neuronal function, resulting in gradual decrease of motor and non-motor function (see review [42]). One of such ROS-induced changes could possibly be due to modification of K+ channels that are responsible for repolarizing the action potential. In support of this idea, Cai & Sesti [43] showed that in the nematode Caenorhabditis elegans oxidation of K+ channels by ROS is a major mechanism underlying the loss of neuronal function. This observation supports an assertion that K+ channels controlling neuronal excitability and survival might provide a common, functionally important substrate for ROS in aging in mammals. In fact, a recent study by de Oliveira et al. [11] showed that global brain ischemia altered the gene expression of KCNH1 (eag1) and KCNH5 (eag2) in the hippocampus. These observations suggest a significant functional role of EAG channels in synaptic plasticity processes in response to ischemic insult. The localization of KCNH1 channels in the brain may permit selective neuronal modulation by redox reactions, conferred by the presence of specific cysteine residues. In hippocampal neurons, ROS-mediated inhibition of KCNH1 current would shorten the after-hyperpolarization, resulting in hyperexcitability. In physiological levels of ROS, mild oxidative stress as occurring during increased neuronal activity may act through a positive feedback by reducing K+ channel activity and thereby augmenting neuronal firing.
A growing body of evidence suggests that increased generation of ROS and an altered redox status is observed in cancer cells [44]. In addition, the participation of ion channels in cancer and apoptosis is increasingly documented [45]. However, to date, no studies have provided information regarding the influence of ROS on ion channels in cancer cells. KCNH1 channels are ectopically expressed in many human cancer cell lines and its expression induces their progression and proliferation. The specific blockade of KCNH1 by monoclonal antibodies inhibited tumor cell growth both in vitro and in vivo [46]. Thus, cysteine oxidation of KCNH1 by ROS in cancer cells is expected to inhibit cancer cell growth. Further studies are needed to advance our knowledge of redox regulation of these ion channels in cancer cells in situ.
Highlights.
KCNH1 potassium channels are exceptionally sensitive to oxidative modification
Thiol-dependent modification of channel gating occurs in two different ways
C145/C214 in the N-linker are responsible for fast modification
C532/C562 in the C-linker are responsible for slow modification
Redox regulation determines information flow from the cytosolic termini to the channel’s gate.
Acknowledgments
This work was supported by ProExcellence Thuringia, PE114-1 (SHH, NS), DFG HE2993/8 (SHH), and NIH (TH).
Abbreviations
- EAG
ether à go-go
- DTNB
5, 5′-dithio-bis[2-nitrobenzoic acid])
- DTNP
2,2′-dithio-bis [5-nitropyridine]
- DTT
dithiothreitol
- MTSET
[2-(trimethylammonium)ethyl] methane thiosulfonate bromide
- MTSES
2-sulfonatoethyl methanethiosulfonate sodium
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Warmke J, Drysdale R, Ganetzky B. A distinct potassium channel polypeptide encoded by the Drosophila eag locus. Science. 1991;252:1560–1562. doi: 10.1126/science.1840699. [DOI] [PubMed] [Google Scholar]
- 2.Warmke JW, Ganetzky B. A family of potassium channel genes related to eag in Drosophila and mammals. Proc Natl Acad Sci USA. 1994;91:3438–3442. doi: 10.1073/pnas.91.8.3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Occhiodoro T, Bernheim L, Liu JH, Bijlenga P, Sinnreich M, Bader CR, Fischer-Lougheed J. Cloning of a human ether-a-go-go potassium channel expressed in myoblasts at the onset of fusion. FEBS Lett. 1998;434:177–182. doi: 10.1016/s0014-5793(98)00973-9. [DOI] [PubMed] [Google Scholar]
- 4.Ludwig J, Weseloh R, Karschin C, Liu Q, Netzer R, Engeland B, Stansfeld C, Pongs O. Cloning and functional expression of rat eag2, a new member of the ether-à-go-go family of potassium channels and comparison of its distribution with that of eag1. Mol Cell Neurosci. 2000;16:59–70. doi: 10.1006/mcne.2000.0851. [DOI] [PubMed] [Google Scholar]
- 5.Ju M, Wray D. Molecular identification and characterisation of the human eag 2 potassium channel. FEBS Lett. 2002;524:204–210. doi: 10.1016/s0014-5793(02)03055-7. [DOI] [PubMed] [Google Scholar]
- 6.Morais Cabral JH, Lee A, Cohen SL, Chait BT, Li M, Mackinnon R. Crystal structure and functional analysis of the HERG potassium channel N terminus: a eukaryotic PAS domain. Cell. 1998;95:649–655. doi: 10.1016/s0092-8674(00)81635-9. [DOI] [PubMed] [Google Scholar]
- 7.Ziechner U, Schönherr R, Born AK, Gavrilova-Ruch O, Glaser R, Malesevic M, Küllertz G, Heinemann SH. Inhibition of human ether à go-go potassium channels by Ca2+/calmodulin binding to the cytosolic N- and C-termini. FEBS J. 2006;273:1074–1086. doi: 10.1111/j.1742-4658.2006.05134.x. [DOI] [PubMed] [Google Scholar]
- 8.Schönherr R, Löber K, Heinemann SH. Inhibition of human ether à go-go potassium channels by Ca2+/calmodulin. EMBO J. 2000;19:3263–3271. doi: 10.1093/emboj/19.13.3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sahoo N, Tröger J, Heinemann SH, Schönherr R. Current inhibition of human EAG1 potassium channel by the Ca2+-binding protein S100B. FEBS Lett. 2010;584:3896–3900. doi: 10.1016/j.febslet.2010.07.063. [DOI] [PubMed] [Google Scholar]
- 10.Saganich MJ, Machado E, Rudy B. Differential expression of genes encoding subthreshold-operating voltage-gated K+ channels in brain. J Neurosci. 2001;21:4609–4624. doi: 10.1523/JNEUROSCI.21-13-04609.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Oliveira RM, Martin S, de Oliveira CL, Milani H, Schiavon AP, Joca S, Pardo LA, Stühmer W, Del Bel EA. Eag1, Eag2, and SK3 potassium channel expression in the rat hippocampus after global transient brain ischemia. J Neurosci Res. 2012;90:632–640. doi: 10.1002/jnr.22772. [DOI] [PubMed] [Google Scholar]
- 12.Ferreira NR, Mitkovski M, Stühmer W, Pardo LA, Del Bel EA. Ether-à-go-go 1 (Eag1) potassium channel expression in dopaminergic neurons of basal ganglia is modulated by 6-hydroxydopamine lesion. Neurotox Res. 2011 doi: 10.1007/s12640-011-9286-3. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 13.Lecain E, Sauvaget E, Crisanti P, Van Den Abbeele T, Huy PT. Potassium channel ether à go-go mRNA expression in the spiral ligament of the rat. Hear Res. 1999;133:133–138. doi: 10.1016/s0378-5955(99)00068-4. [DOI] [PubMed] [Google Scholar]
- 14.Frings S, Brüll N, Dzeja C, Angele A, Hagen V, Kaupp UB, Baumann A. Characterization of ether-à-go-go channels present in photoreceptors reveals similarity to IKx, a K+ current in rod inner segments. J Gen Physiol. 1998;111:583–599. doi: 10.1085/jgp.111.4.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beech DJ, Barnes S. Characterization of a voltage-gated K+ channel that accelerates the rod response to dim light. Neuron. 1989;3:573–581. doi: 10.1016/0896-6273(89)90267-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bijlenga P, Occhiodoro T, Liu JH, Bader CR, Bernheim L, Fischer-Lougheed J. An ether -à-go-go K+ current, Ih-eag, contributes to the hyperpolarization of human fusion-competent myoblasts. J Physiol. 1998;512:317–323. doi: 10.1111/j.1469-7793.1998.317be.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pardo LA, del Camino D, Sánchez A, Alves F, Brüggemann A, Beckh S, Stühmer W. Oncogenic potential of EAG K+ channels. EMBO J. 1999;18:5540–5547. doi: 10.1093/emboj/18.20.5540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meyer R, Heinemann SH. Characterization of an eag-like potassium channel in human neuroblastoma cells. J Physiol. 1998;508:49–56. doi: 10.1111/j.1469-7793.1998.049br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meyer R, Schönherr R, Gavrilova-Ruch O, Wohlrab W, Heinemann SH. Identification of ether à go-go and calcium-activated potassium channels in human melanoma cells. J Membr Biol. 1999;171:107–115. doi: 10.1007/s002329900563. [DOI] [PubMed] [Google Scholar]
- 20.Hemmerlein B, Weseloh RM, Mello de Queiroz F, Knötgen H, Sánchez A, Rubio ME, Martin S, Schliephacke T, Jenke M, Radzun H-J, Stühmer W, Pardo LA. Overexpression of Eag1 potassium channels in clinical tumours. Mol Cancer. 2006;5:41. doi: 10.1186/1476-4598-5-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stengel R, Rivera-Milla E, Sahoo N, Ebert C, Bolling F, Heinemann SH, Schönherr R, Englert C. Kcnh1 voltage-gated potassium channels are essential for zebrafish early development. 2012 doi: 10.1074/jbc.M112.363978. (submitted) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ruppersberg JP, Stocker M, Pongs O, Heinemann SH, Frank R, Koenen M. Regulation of fast inactivation of cloned mammalian IK(A) channels by cysteine oxidation. Nature. 1991;352:711–714. doi: 10.1038/352711a0. [DOI] [PubMed] [Google Scholar]
- 23.Duprat F, Guillemare E, Romey G, Fink M, Lesage F, Lazdunski M, Honore E. Susceptibility of cloned K 1 channels to reactive oxygen species. Proc Natl Acad Sci USA. 1995;92:11796–11800. doi: 10.1073/pnas.92.25.11796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stephens GJ, Owen DG, Robertson B. Cysteine-modifying reagents alter the gating of the rat cloned potassium channel Kv1.4. Pflügers Arch. 1996;431:435–442. doi: 10.1007/BF02207283. [DOI] [PubMed] [Google Scholar]
- 25.Rozanski GJ, Xu Z. Sulfhydryl modulation of K+ channels in rat ventricular myocytes. J Mol Cell Cardiol. 2002;34:1623–1632. doi: 10.1006/jmcc.2002.2112. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Y, Han H, Wang J, Wang H, Yang B, Wang Z. Impairment of human ether-à-go-go-related gene (HERG) K+ channel function by hypoglycemia and hyperglycemia. J Biol Chem. 2003;278:10417–10426. doi: 10.1074/jbc.M211044200. [DOI] [PubMed] [Google Scholar]
- 27.Zhang Y, Xiao J, Wang H, Luo X, Wang J, Villeneuve LR, Zhang H, Bai Y, Yang B, Wang Z. Restoring depressed HERG K+ channel function as a mechanism for insulin treatment of abnormal QT prolongation and associated arrhythmias in diabetic rabbits. Am J Physiol Heart Circ Physiol. 2006;291:H1446–H1455. doi: 10.1152/ajpheart.01356.2005. [DOI] [PubMed] [Google Scholar]
- 28.Kolbe K, Schönherr R, Gessner G, Sahoo N, Hoshi T, Heinemann SH. Cysteine 723 in the C-linker segment confers oxidation inhibition of hERG1 potassium channels. J Physiol. 2010;588:2999–3009. doi: 10.1113/jphysiol.2010.192468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sanguinetti MC, Tristani-Firouzi M. hERG potassium channels and cardiac arrhythmia. Nature. 2006;440:463–469. doi: 10.1038/nature04710. [DOI] [PubMed] [Google Scholar]
- 30.Sale H, Wang J, O’Hara TJ, Tester DJ, Phartiyal P, He JQ, Rudy Y, Ackermann MJ, Robertson GA. Physiological properties of hERG 1a/1b heteromeric currents and a hERG 1b-specific mutation associated with long-QT syndrome. Circ Res. 2008;103:e81–95. doi: 10.1161/CIRCRESAHA.108.185249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang MH. A technical consideration concerning the removal of oocyte vitelline membranes for patch clamp recording. Biochem Biophys Res Commun. 2004;324:971–972. doi: 10.1016/j.bbrc.2004.09.162. [DOI] [PubMed] [Google Scholar]
- 32.Heinemann SH, Conti F. Non-stationary noise analysis and its application to patch clamp recordings. In: Rudy B, Iverson LE, editors. Ion Channels, Methods in Enzymology. Vol. 207. Academic Press; 1992. pp. 131–148. [DOI] [PubMed] [Google Scholar]
- 33.Starkus JG, Varga Z, Schönherr R, Heinemann SH. Mechanisms for the inhibition of Shaker potassium channels by protons. Pflügers Arch. 2003;447:44–54. doi: 10.1007/s00424-003-1121-0. [DOI] [PubMed] [Google Scholar]
- 34.Stevens L, Ju M, Wray D. Roles of surface residues of intracellular domains of heag potassium channels. Eur Biophys J. 2009;38:523–532. doi: 10.1007/s00249-009-0402-8. [DOI] [PubMed] [Google Scholar]
- 35.Gordon SE, Varnum MD, Zagotta WN. Direct interaction between amino and carboxyl-terminal domains of cyclic nucleotide-gated channels. Neuron. 1997;19:431–441. doi: 10.1016/s0896-6273(00)80951-4. [DOI] [PubMed] [Google Scholar]
- 36.Chen IH, Hu JH, Jow GM, Chuang CC, Lee TT, Liu DC, Jeng CJ. Distal end of carboxyl terminus is not essential for the assembly of rat eag1 potassium channels. J Biol Chem. 2011;286:27183–27196. doi: 10.1074/jbc.M111.233825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brelidze TI, Carlson AE, Sankaran B, Zagotta WN. Structure of the carboxy-terminal region of a KCNH channel. Nature. 2012 doi: 10.1038/nature10735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Terlau H, Heinemann SH, Stühmer W, Pongs O, Ludwig J. Amino terminal-dependent gating of the potassium channel rat eag is compensated by a mutation in the S4 segment. J Physiol. 1997;502:537–543. doi: 10.1111/j.1469-7793.1997.537bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Long SB, Campbell EB, Mackinnon R. Voltage sensor of Kv1.2: structural basis of electromechanical coupling. Science. 2005;309:903–908. doi: 10.1126/science.1116270. [DOI] [PubMed] [Google Scholar]
- 40.Long SB, Campbell EB, Mackinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 2005;309:897–903. doi: 10.1126/science.1116269. [DOI] [PubMed] [Google Scholar]
- 41.Payandeh J, Scheuer T, Zheng N, Catterall WA. The crystal structure of a voltage-gated sodium channel. Nature. 2011;475:353–358. doi: 10.1038/nature10238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Uttara B, Singh AV, Zamboni P, Mahajan RT. Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Curr Neuropharmacol. 2009;7:65–74. doi: 10.2174/157015909787602823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cai SQ, Sesti F. Oxidation of a potassium channel causes progressive sensory function loss during aging. Nat Neurosci. 2009;12:611–617. doi: 10.1038/nn.2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schumacker PT. Reactive oxygen species in cancer cells: live by the sword, die by the sword. Cancer Cell. 2006;10:175–176. doi: 10.1016/j.ccr.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 45.Kunzelmann K. Ion channels and cancer. J Membr Biol. 2005;205:159–173. doi: 10.1007/s00232-005-0781-4. [DOI] [PubMed] [Google Scholar]
- 46.Gómez-Varela D, Zwick-Wallasch E, Knötgen H, Sánchez A, Hettmann T, Ossipov D, Weseloh R, Contreras-Jurado C, Rothe M, Stühmer W, Pardo LA. Monoclonal antibody blockade of the human Eag1 potassium channel function exerts antitumor activity. Cancer Res. 2007;15:7343–7349. doi: 10.1158/0008-5472.CAN-07-0107. [DOI] [PubMed] [Google Scholar]