

The challenge of atropisomer-selective synthesis is often manifest in drug discovery projects,[1] and also in the synthesis of materials with interesting optical properties.[2] With respect to the former, numerous small molecule ligands for proteins and enzyme inhibitors exist as conformational racemates, with low barriers to atropisomerization (Figure 1). For example, terphenyl compounds (e.g., 1, disruptors of protein-protein interactions,)[3] heteroarene- and heteroatom-substituted biphenyls (e.g., kinase inhibitors; 2),[4] and other biologically active scaffolds (e.g., biphenyl tetrazole 3)[5] all exhibit the possibility for stereochemically unique atropisomeric conformations. Nonetheless, binding of the small molecule to the biological target often occurs with enantiospecificity, as the inherent chirality of the receptor effects in situ dynamic kinetic resolution of the ligand, provided the barrier to atropisomerization is low enough. Thus, the preparation of single atropisomer scaffolds could lead to increases in potency for small molecules, through an increase in the effective concentration of the biologically active atropisomer, with the exclusion of a less active, or alternatively active form.
Figure 1.

Representative bioactive polyaryl compounds.
We recently reported an approach to the catalytic enantioselective synthesis of atropisomerically enriched biaryl compounds.[6] Our strategy is predicated on a peptide-catalyzed electrophilic aromatic substitution reaction, wherein compounds such as 4 are converted to tribrominated compounds (e.g. 5), through the action of catalyst 6 (eq 1). The utility of this process may be expanded through a variety of strategies, including the expansion of the types of catalysts and substrates that may be employed. As important, if compounds such as 5 may be differentially, and sequentially functionalized, then the scaffold diversity of atropisomerically pure compounds ultimately derived from catalyst 6 is similarly expanded.
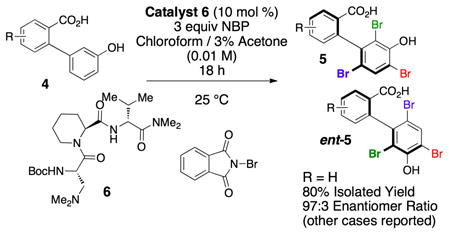 |
(1) |
Thus, a goal in our laboratory has been the development of sequential, regioselective cross-coupling reactions of the tribromides delivered through the dynamic kinetic resolutions of substrates like 4. For example, the ability to convert 7 to 8 (eq 2), in a general sense, would greatly increase the number of easily accessible atropisomerically pure biaryl compounds.[7] While much study has been directed torward the development of enantio- and atropisomer selective[8] cross-coupling reactions, regioselective couplings have received less attention, with most efforts directed toward poly-halogenated heterocycles, and dihalogenated benzene derivatives.[9] Thus, our ambition was to develop sequential cross-couplings at each Br-position, such that one could essentially fashion “A-B-C” differentially functionalized scaffolds with total control (eq 2).
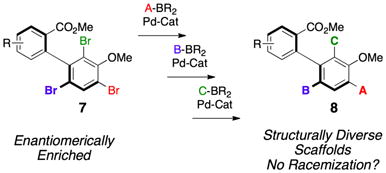 |
(2) |
As noted in eq 2, a critical aspect of the study involves the assurance that there is no racemization of the starting materials during any of the cross-coupling steps. While the barriers to atropisomerization of trisubstituted biaryls are known to be quite high (>30 kcal/mol),[10] one could imagine that racemization might occur under the often-elevated temperatures associated with many Pd-catalyzed cross-coupling protocols. Moreover, the processes of oxidative addition, transmetalation and reductive elimination would all involve the formation of, and cleavage of rather long arene-Pd bonds, that in principle, could lead to lower barriers to atropisomerization/racemization. There are indeed pioneering examples in the literature that demonstrate that such cross-couplings may occur without racemization.[11] Yet, the earliest of the reports considered aryl triflates that undergo cross-coupling under different conditions, and several of these are highly substituted with bulky groups. Such compounds could present higher intrinsic barriers to atropisomerization than the tribromides we projected to explore. Moreover, subsequent reports focus mostly on binaphthyl-based systems, which also generally possess intrinsically high barriers.[12] Thus, for the cases we wished to study, close literature precedent seemed scant. Thus, our initial experiments sought to verify that all three Br-atoms of 7 could be substituted to form enantiomerically pure penta-aryls.
As shown in Table 1, we subjected atropisomerically enriched tribromide 9 to standard Suzuki cross-coupling conditions (5 mol % Pd(PPh3)4, 5 equiv aryl boronic acid; refluxing THF: 2M Na2CO3(aq),[13] which delivered penta-arene 11 in excellent isolated yield (95%; entry 1a). Critically, no degradation of enantiomeric ratio (er) was observed, as determined by chiral HPLC analysis. We also determined that implementing analogous conditions under microwave irradiation[14] for 1 h at 100 °C yielded the desired product; once again no racemization had occurred under these conditions. Thus, compound 11 was once again obtained in 95% yield, with no loss of er (entry 1b). So too, penta-arene 12 was obtained in 95% isolated yield with total preservation of er (entry 2). Tribromide 10 was also converted to its derived penta-arene 13 with no racemization (67% yield; entry 3).
Table 1.
Homo-functionalization of tribromobiaryls.
 | ||||
|---|---|---|---|---|
| entry | substrate | product | SM er Pdt era | % yieldb |
| 1a |
 9 |
 11 |
95.0:5.0 | 95c |
| 1b | 95.0:5.0 | 95d | ||
| 2 |
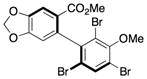 9 |
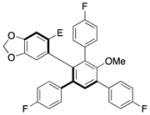 12 |
95.0:5.0 | 95d |
| 96.0:4.0 | ||||
| 3 |
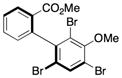 10 |
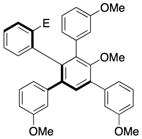 13 |
97.0:3.0< | 67d |
| 97.0:3.0 | ||||
Enantiomer ratios were determined by chiral HPLC.
Average isolated yield of three experimental runs.
Reaction conducted in refluxing THF/H2O.
Reaction conducted at 100 °C under microwave conditions, See supporting information for tables.
With these encouraging observations in hand, we then set out to assess the possibility of regioselective cross-couplings, presumably at the “A-position” first. Our initial attempts of Suzuki coupling with boronic acids resulted in low yields and selectivities.[15] On the other hand, application of the MIDA (N-methyliminodiacetic acid) boronates developed by Burke and coworkers led to improved results.[16] Notably, this approach resulted in good yields, and high regioselectivities, as determined by NMR, for the cross-coupling of aryl groups, heterocyclic functions, and alkenes to the tribromides. In no case was a significant degree of racemization observed. Moreover, regioselectivity, as assigned by HMBC NMR anaylsis (and further supported by changes in 1H NMR shifts due to anisotropy; see Supporting Information), was apparent. For example, as shown in Table 2, the m-nitrophenyl substituent could be introduced into the A-position with 11:1:1 regioselectivity, with 14a isolated in 70% yield (entry 1). The reactions tend to be quite clean, and in cases of incomplete conversion, unreacted tribromide can be recovered (e.g., 20% of 9 recovered, as noted in Table 2). Similarly, the m-methoxy-phenyl moiety may be introduced at the A-position to give 14b with 17:1:1 regioselectivity, allowing isolation of 48% of pure product (entry 2). Heterocyclic coupling partners are also successfully employed in these reactions. The N-tosyl-pyrrolo moiety may be introduced to deliver 14c in 55% isolated yield (entry 3). A 3-pyridyl substituent may be introduced at the A-position to give 14d in 52% yield (entry 4). Formal Heck couplings are also possible, as the α-styryl substituent is introduced to give 14e in 49% yield (entry 5). Comparable results are achieved when tribromide 10 is employed as the starting material, as noted in entries 6–8 (to give 15a–c), reflecting the diversity of atropisomerically pure “A-coupled” products that may be formed. Any undesired regioisomers could be separated using semi-preparative HPLC. The demonstrated ability to substitute the A-position of 9 and 10 may represent one of the first examples of a general asymmetric synthesis of axially chiral p-terphenyls, often studied as mimetics of α-helices.[3]
Table 2.
Regioselective cross-couplings of tribrominated biaryls.
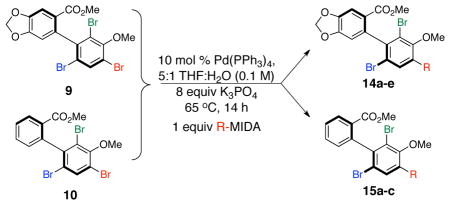 | |||||
|---|---|---|---|---|---|
| entry | product R= | substrate | % yielda (% rec. SM) | regioselectivityb,c,d (A:B:C) | SM er Pdt ere,f |
| 1 |
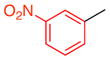 14a |
9 | 70 (20) | 11.0:1.0:1.0 | 95.0:5.0 |
| 97.0:3.0 | |||||
| 2 |
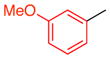 14b |
9 | 48 (20) | 17.0:1.0:1.0 | 95.0:5.0 |
| 98.0:2.0 | |||||
| 3 |
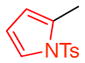 14c |
9 | 55 (20) | >30.0:1.0:1.0 | 95.0:5.0 |
| 97.5:2.5 | |||||
| 4 |
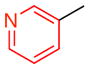 14d |
9 | 52 (23) | 24.0:2.0:1.0 | 95.0:5.0 |
| 94.5:4.5 | |||||
| 5 |
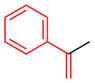 14e |
9 | 49 | 5.0:1.0:1.0 | 95.0:5.0 |
| 95.0:5.0 | |||||
| 6 |
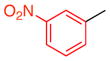 15a |
10 | 45 (19) | 10.0:1.0:1.0 | 95.0:5.0 |
| 97.0:3.0 | |||||
| 7 |
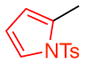 15b |
10 | 45 (30) | 10.0:1.0:-- | 95.0:5.0 |
| 95.5:4.5 | |||||
| 8 |
 15c |
10 | 52 (23) | 14.0:1.0:1.0 | 95.0:5.0 |
| 96.0:4.0 | |||||
Average isolated yields of three experimental runs.
Assigned by NMR analysis.
Ratio determined by 1H NMR.
Averaged over three experimental runs.
Enantiomer ratios were determined by HPLC analysis.
Averaged over three runs.
We also wished to establish whether or not regioselectivity could be achieved in heteroatom-based cross-couplings of enantiomerically enriched tribromides.[17] Indeed, these too were found to be regioselective processes. For example, 9 may be subjected to Pd-catalyzed amination[18] to give highly substituted aniline 14f in 60% yield, with high regioselectivity (>20:1:1) and with no detectable racemization (Table 3, entry 1). Pd-catalyzed etherification[19] proceeds with similarly high regioselectivity to give biaryl ether 14g in 55% yield, once again without perceptible loss of optical activity (entry 2). Quite strikingly, we were also able to replace the remote Br-atom of 9 (i.e., the “A-position”) with an H-atom, employing NaBH4 as the hydride source.[20] While selectivity for the formation of 14h was somewhat lower (4:1), purification was straightforward allowing isolation of 14h in 56% yield (entry 3). Substitution of the “A-position” with an amine, ether or H-atom functionality significantly expands the scope of accessible atropisomerically enriched biaryl compounds that may be obtained under this protocol.
Table 3.
Regioselective heteroatom cross-couplings.
 | |||||
|---|---|---|---|---|---|
| entry | R= | substrate | % yielda | regioselectivityb,c,d | SM er Pdt ere,f |
| 1 |
 14fg |
9 | 60 | >20.0:1.0:1.0 | 95.0:5.0 |
| 95.0:5.0 | |||||
| 2 |
 14gh |
9 | 55h | >20.0:1.0:1.0 | 95.0:5.0 |
| 95.0:5.0 | |||||
| 3 |
14hi |
9 | 56i | 4:1:-- | 95.0:5.0 |
| 97.0:3.0 | |||||
Average Isolated yields of three experimental runs.
Assigned by NMR analysis.
Ratio determined by 1H NMR.
Averaged over three experimental runs.
Enantiomer ratios were determined by HPLC analysis
Averaged over three runs.
10% Pd(OAc)2, 20% rac BINAP, 100 °C, 0.1 M in Toluene.
10% Pd(OAc)2, 13% t-Bu-XPhos, 2 equiv K3PO4, 100 °C 0.3 M in Toluene,
NaBH4, 5 mol% Pd(OAc)2, 5.5% BINAP, 1.5 equiv TMEDA, 50 °C, 0.25 M in THF. The 56% yield refers to the isolated regioisomerically pure product.
With robust methods for synthesis of gram-scale[21] quantities of these “A-coupled” p-terphenyls, we then turned our attention to their further functionalization to yield “A-B” coupled tetra-aryl compounds. Initially, we observed that the standard conditions implemented in the “A-couplings” gave moderate yields and little to no selectivity (low yields, 1.2:1 regioselectivities) in various attempted couplings (e.g., in the formation of 16, Table 4). However, use of the sterically more demanding Pd source (Pd(II)dppf) resulted in modest improvements (~3:1 regioselectivity), as did Pd2dba3/(R)-BINAP system (~3:1). Of particular note, the enantiomeric Pd2dba3/(S)-BINAP system resulted in higher yields and selectivity (~5.5:1 regioselectivity). Regioselectivity was further improved to 10.0:1 with a 52% isolated yield upon lowering the temperature from 100 °C to 65 °C (Table 4, entry 1). These results are striking as they suggest a moderate level of double diastereodiffentiation in the cross-coupling events.[22] Interestingly, application of these reaction conditions to racemic 14b allowed us to observe a modest kinetic resolution with a krel of approximately 4, reflecting the differential behavior of the enantiomeric catalysts with homochiral 14b. In any case, from a pragmatic perspective, these conditions and the Pd2dba3/(S)-BINAP catalyst system proved consistently effective when a range of aryl MIDA boronates were employed for coupling at the B-position.[23]
Table 4.
Regioselective cross-couplings of dibrominated p-terphenyls.
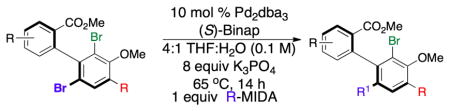 | |||||
|---|---|---|---|---|---|
| entry | substrate | product | % yield (% rec. SM)b | regioselectivityc,d,e | SM er Pdt era |
| 1 | 14b |
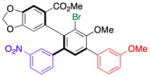 16 |
52 (20) | 10.0:1.0 | 98.0:2.0 |
| 99.0:1.0 | |||||
| 2 | 14b |
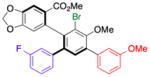 17 |
65 (10) | 5.0:1.0 | 98.0:2.0 |
| 99.0:1.0 | |||||
| 3 | 15c |
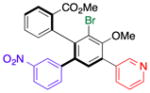 18 |
41 (24) | 7.0:1.0 | 96.0:4.0 |
| 95.0:5.0 | |||||
| 4 | 15c |
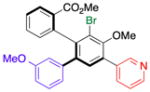 19 |
52 (18) | 6.0:1.0 | 96.0:4.0 |
| 95.0:5.0 | |||||
| 5 | 14h |
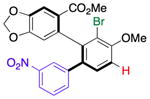 20 |
37 (36) | 2.5:1.0f | 96.0:4.0 |
| 95.0:5.0 | |||||
Enantiomer ratios were determined by chiral HPLC.
Average isolated yield of three experimental runs.
Assigned by NMR analysis.
Ratio determined by 1H NMR.
Averaged over three experimental runs.
Isolated as single regioisomer in 25 % overall yield
Thus, we observed that in addition to formation of 16, m-fluorophenyl-substitution can be achieved such that 17 is obtained with 5:1 regioselectivity and 65% isolated yield (entry 2). Heteroarene-substituted compounds such as 15c perform comparably, with 18 and 19 each formed with 6-7:1 regioselectivity (entries 3 and 4). Isolated yields for these compounds are somewhat lower (41%–52%), but the reactions are clean, with products readily purified, and 15c may be recovered to varying extents. Moreover, even the H-substituted biphenyl 14h is converted to 20 with 2.5:1 regisolectivity (37% yield; entry 5). While the yield of 20 is modest, it is easily purified. Once again, no racemization is detected during any of the cross-coupling reactions of Table 4.
With efficient access to atropisomerically enriched “A-B-coupled” compounds, we then turned our attention to cross-couplings of the final “C-position,” targeting differentially substituted, enantiomerically enriched penta-aryl compounds. While regioselectivity is no longer an issue, this process involves metal insertion into a hindered ortho-ortho’ disubstituted bromide.[24] While standard Suzuki cross-coupling conditions (5 mol% Pd(PPh3)4, excess aryl boronic acid, refluxing 2:1 THF/2M Na2CO3(aq)) resulted in incomplete conversions after 18 h, we found that the microwave-based conditions described in Table 1 resulted in full conversion, and good isolated yields (Table 5). Thus, Table 5 presents a series of atropisomerically defined, optically active penta-arenes. Entry 1 reveals a sequentially cross-coupled product in which three differentially substituted arene moieties have been introduced with control. The final arene is introduced in 77% isolated yield to give 21 with no detectable racemization (entry 1). Entry 2 shows the synthesis of an atropisomerically defined “pseudo-enantiomer” of compound 21,[25] wherein the order of cross-coupling has been swapped. Thus, penta-arene 22 is produced with a final cross-coupling reaction that proceeds in 80% yield. Heteroarenes may be introduced in the final cross-coupling event as well, as compound 23 is obtained in 80% yield (entry 3). A closely related, atropisomerically defined structure 24 may also be prepared, simply by choosing the order of the cross-coupling reactions, such that 24 may be isolated in 70% yield. In all cases, Table 5 presents compounds that were prepared with verified preservation of the enantiomeric ratios.
Table 5.
Atropisomer enriched differentially trifunctionalized biaryls.
 | ||||
|---|---|---|---|---|
| entry | substrate | product | % yieldb | SM er Pdt era |
| 1 | 16 |
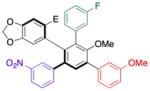 21 |
77 | 99.0:1.0 |
| 99.0:1.0 | ||||
| 2 | 17 |
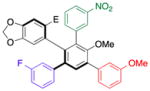 22 |
80 | 99.0:1.0 |
| 99.0:1.0 | ||||
| 3 | 16 |
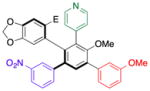 23c |
80 | 99.0:1.0 |
| 99.0:1.0 | ||||
| 4 | 19 |
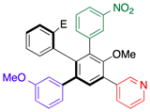 24 |
70 | 96.0:4.0 |
| 96.0:4.0 | ||||
Enantiomer ratios were determined by chiral HPLC.
Isolated yields, averaged over 2 experimental runs.
Used 3 equiv of boronic acid.
In summary, we have reported an iterative atropisomer selective, asymmetric bromination/cross-coupling strategy for the preparation of enantiomerically enriched, complex poly-aryl compounds. Thus far, arenes, heterocycles, and heteroatoms have been introduced with regioselectivity into a tribrominated biaryl scaffold, employing readily available catalysts and straightforward conditions. To the extent that enantiomerically enriched, atropisomerically defined compounds may be of interest in drug discovery or materials science, we are hopeful this work will be of broad interest.
Supplementary Material
Acknowledgments
We are grateful to the National Institutes of General Medical Sciences of the National Institute of Health Foundation (GM-068649) for support.
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.Clayden J, Moran WJ, Edwards PJ, LaPlante SR. Angew Chem Int Ed. 2009;48:6398–6401. doi: 10.1002/anie.200901719. [DOI] [PubMed] [Google Scholar]; b) LaPlante SR, Edwards PJ, Fader LD, Jakalian A, Hucke O. ChemMedChem. 2011;6:505–513. doi: 10.1002/cmdc.201000485. [DOI] [PubMed] [Google Scholar]
- 2.a) Pu L. Chem Rev. 1998;98:2405–2494. doi: 10.1021/cr970463w. [DOI] [PubMed] [Google Scholar]; b) Cornelis D, Franz E, Asselberghs I, Clays K, Verbiest T, Koeckelberghs G. J Am Chem Soc. 2011;132:1317–1327. doi: 10.1021/ja104978t. [DOI] [PubMed] [Google Scholar]
- 3.a) Cummings CG, Hamilton AD. Curr Opin Chem Biol. 2010;14:341–346. doi: 10.1016/j.cbpa.2010.04.001. [DOI] [PubMed] [Google Scholar]; b) Yin H, Lee G-I, Sedey KA, Kutzki O, Park HS, Orner BP, Ernst JT, Wang H-G, Sebti SM, Hamilton AD. J Am Chem Soc. 2005;127:10191–10196. doi: 10.1021/ja050122x. [DOI] [PubMed] [Google Scholar]
- 4.Noble MEM, Endicott JA, Johnson LN. Science. 2004;303:1800–1805. doi: 10.1126/science.1095920. [DOI] [PubMed] [Google Scholar]; b) Da,janov N, Jauffman RS, Scpencer-Green GT. Arthritis & Rheumatism. 2009;60:1232–1241. doi: 10.1002/art.24485. [DOI] [PubMed] [Google Scholar]
- 5.Welsch ME, Snyder SA, Stockwell BR. Curr Opin Chem Biol. 2010;14:347–361. doi: 10.1016/j.cbpa.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gustafson JL, Lim D, Miller SJ. Science. 2010;328:1251–1255. doi: 10.1126/science.1188403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.For complementary approaches to atropisomerically pure biaryl compounds, see Bringmann G, Mortimer AJP, Keller PA, Gresser MJ, Garner J, Breuning M. Angew Chem, Int Ed. 2005;44:5384–5427. doi: 10.1002/anie.200462661.Bringmann G, Menche D. Acc Chem Res. 2001;34:615–624. doi: 10.1021/ar000106z.
- 8.a) Shen X, Jones GO, Watson DA, Bhayana B, Buchwald SL. J Am Chem Soc. 2010;132:11278–11287. doi: 10.1021/ja104297g. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hayashi T, Hayashizaki K, Kiyoi T, Ito Y. J Am Chem Soc. 1988;110:8153–8156. [Google Scholar]; c) Li X, Hewgley JB, Mulrooney CA, Yang J, Kozlowski MC. J Org Chem. 2003;68:5500–5511. doi: 10.1021/jo0340206. [DOI] [PubMed] [Google Scholar]; d) Kakiuchi F, Le Gendre P, Yamada A, Ohtaki H, Murai S. Tetrahedron Asymmetry. 2000;11:2647–2650. [Google Scholar]
- 9.For recent reviews see Schroter S, Stock C, Bach T. Tetrahedron. 2005;61:2245–2267.Fairlamb IJS. Chem Soc Rev. 2007;36:1036–1045. doi: 10.1039/b611177g.Wang JR, Manabe K. Synthesis. 2009:1405–1427.
- 10.a) Eliel EL, Wilen SH. Stereochemistry of Organic Compounds. John Wiley and Sons; New York: 1994. [Google Scholar]; b) Bott G, Field LD, Sternhell S. J, Am Chem Soc. 1980;102:5618–5626. [Google Scholar]
- 11.Hayashi T, Niizuma S, Kamikawa T, Suzuki N, Uozumi Y. J Am Chem Soc. 1995;117:9101–9102. [Google Scholar]
- 12.a) Uozumi Y, Suzuki N, Ogiwara A, Hayashi T. Tetrahedron. 1994;50:4293–4302. [Google Scholar]; b) Berkessel A, Guixa M, Schmidt F, Neudorfl JM, Lex J. Chem Eur J. 2007;13:4483–4498. doi: 10.1002/chem.200600993. [DOI] [PubMed] [Google Scholar]; c) Cho YH, Kina A, Shimada T, Hayashi T. J Org Chem. 2004;69:3811–3823. doi: 10.1021/jo035880p. [DOI] [PubMed] [Google Scholar]; d) Hayashi T. Acc Chem Res. 2000;33:354–362. doi: 10.1021/ar990080f. [DOI] [PubMed] [Google Scholar]
- 13.a) Suzuki A. J Organometallic Chem. 1999;576:147–168. [Google Scholar]; b) Kotha S, Lahiri K, Kashinath D. Tetrahedron. 2002:9633–9695. [Google Scholar]
- 14.Kappe CO. Angew Chem Int Ed. 2004;43:6250–6284. doi: 10.1002/anie.200400655. [DOI] [PubMed] [Google Scholar]
- 15.See supporting information.
- 16.Knapp DM, Gillis EP, Burke MD. J Am Chem Soc. 2009;131:6961–6963. doi: 10.1021/ja901416p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hartwig JF. Nature. 2008;455:314–322. doi: 10.1038/nature07369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schlummer B, Scholz U. Adv Synth Cat. 2004;346:1599–1626. [Google Scholar]
- 19.Burgos CH, Barder TE, Huang X, Buchwald SL. J Am Chem Soc. 2006;45:4321–4326. doi: 10.1002/anie.200601253. [DOI] [PubMed] [Google Scholar]
- 20.Chae J, Buchwald SL. J Org Chem. 2004;69:3336–3339. doi: 10.1021/jo035819k. [DOI] [PubMed] [Google Scholar]
- 21.The reactions perform equally well on a variety of reaction scales. Typical runs were performed with several hundred mg of substrate (1–2 mmol). Reactions on the gram-scale proceed with minimal variation in results.
- 22.Masamune S, Choy W, Petersen JS, Sita LR. Angew Chem Int Ed. 1985;24:1–30. [Google Scholar]
- 23.See the Supporting Information for the regiochemical assignment.
- 24.For representative examples see Altenhoff G, Goddard R, Lehman CW, Glorius F. J Am Chem Soc. 2004;126:15195–15201. doi: 10.1021/ja045349r.Barder TE, Walker SD, Martonelli JR, Buchwald SL. J Am Chem Soc. 2005;127:4685–4696. doi: 10.1021/ja042491j.
- 25.We note that compounds 21 and 22 are nearly enantiomeric, differing as such by the position of the methoxy group. Yet, 21 and 22 exhibit the same sign of [α]D (see SI).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.