

Abstract
The pathogenic model of Alzheimer's disease (AD) posits that aggregates of amyloid β, a product of amyloid precursor protein (APP) processing, cause dementia. However, alterations of normal APP functions could contribute to AD pathogenesis, and it is therefore important to understand the role of APP. APP is a member of a gene family that shows functional redundancy as documented by the evidence that single knock-out mice are viable, whereas mice with combined deletions of APP family genes die shortly after birth. A residue in the APP intracellular region, Y682, is indispensable for these essential functions of APP. It is therefore important to identify pathways that regulate phosphorylation of Y682 as well as the role of Y682 in vivo. TrkA is associated with both phosphorylation of APP-Y682 and alteration of APP processing, suggesting that tyrosine phosphorylation of APP links APP processing and neurotrophic signaling to intracellular pathways associated with cellular differentiation and survival. Here we have tested whether the NGF/TrkA signaling pathway is a physiological regulator of APP phosphorylation. We find that NGF induces tyrosine phosphorylation of APP, and that APP interacts with TrkA and this interaction requires Y682. Unpredictably, we also uncover that APP, and specifically Y682, regulates activation of the NGF/TrkA signaling pathway in vivo, the subcellular distribution of TrkA and the sensitivity of neurons to the trophic action of NGF. This evidence suggests that these two membrane protein's functions are strictly interconnected and that the NGF/TrkA signaling pathway is involved in AD pathogenesis and can be used as a therapeutic target.
Introduction
Genetic evidence points to a key role for amyloid precursor protein (APP) processing in Alzheimer's disease (AD) pathogenesis (Hardy and Selkoe, 2002; Selkoe and Podlisny, 2002). APP is a cell membrane protein that is sequentially cleaved by several enzymes. One of the APP-derived metabolites, amyloid β, is believed to play a key role in AD pathogenesis; however, so far all attempts to validate this hypothesis clinically have failed. There are many possible reasons for these failures, including that other APP-derived metabolites cause the disease. Indeed, several other APP fragments have been implicated in triggering neurodegenerative processes; an NH2-terminal APP fragment interacts with DR6 (death-receptor 6) to trigger axon pruning and neuron death (Nikolaev et al., 2009); and peptides derived from the APP intracellular domain can promote cell death both in vitro and in vivo (Lu et al., 2000; Passer et al., 2000; Bertrand et al., 2001; Madeira et al., 2005; Giliberto et al., 2008).
The intracellular region of APP, and specifically the tyrosine residue at position 682 (Y682, using the numbering of the APP695 isoform), has a critical role for developmental functions of APP (Li et al., 2010; Barbagallo et al., 2011) and in regulating APP processing (Barbagallo et al., 2010). Phosphorylation of Y682 has profound effects on APP protein–protein interactions. Some proteins interact with APP only when Y682 is phosphorylated, others only when this tyrosine is not phosphorylated (Russo et al., 2002; Tarr et al., 2002a; Zhou et al., 2004, 2009; Tamayev et al., 2009). Given this crucial role of Y682, it is conceivable that phosphorylation of this residue has fundamental biological and pathological consequences. Therefore, it is significant to identify the mechanisms and pathways that regulate phosphorylation of Y682 in physiological conditions. We showed that overexpression of the nerve growth factor receptor TrkA produced phosphorylation of APP on Y682 and altered APP processing, suggesting that tyrosine phosphorylation of APP may functionally link APP processing and neurotrophic signaling to intracellular pathways associated with cellular differentiation and survival (Tarr et al., 2002b). The finding that interruption of NGF signaling in hippocampal neurons rapidly activates APP processing and causes neuronal apoptotic death further supported this link (Matrone et al., 2008, 2009). In this study, we asked whether the NGF/TrkA signaling pathway is a physiological regulator of APP phosphorylation. We find that NGF induces tyrosine phosphorylation of APP and, surprisingly enough, unveils a vital role for APP in the activation and localization of the NGF/TrkA signaling pathway.
Materials and Methods
Cell culture.
Hippocampal and medial septal neurons were prepared from embryonic day 17/18 (E17/E18) mice, as previously reported (Matrone et al., 2008). The hippocampus and septum were dissected in HBSS buffered with HEPES and dissociated via trypsin/EDTA treatment. A total of 106 cells was plated on 3.5 cm dishes precoated with poly-l-lysine. After 2 d of culturing in neurobasal medium with B-27 supplement and glutamax, cytosine arabinofuranoside was added to reduce glial proliferation. Neurons were exposed to NGF (50 ng/ml) for 48 h at 3–4 d after plating.
Dorsal root ganglia (DRGs) and sympathetic cervical ganglia (SCGs) were collected from either wild-type (WT) or APPYG/YG mice [postnatal day 0 (P0) to P3]. Cells (30 × 103) were plated on slides, and neurons were cultured for 3 d in NGF-containing DMEM plus serum. The antimitotic agent cytosine arabinose (3 μg/ml) was added 12 h after plating. To determine the response to NGF, the number of neurons, stained by a specific neuronal marker (NeuN, Millipore Bioscience Research Reagents), was assessed.
Mice and ethics statement.
Mice of either sex were on a C57BL/6 background. Mice were handled according to the Ethical Guidelines for Treatment of Laboratory Animals of Albert Einstein College of Medicine. APPYG/YG generation and genotyping have been described (Barbagallo et al., 2010). Genotyping for the APP−/− allele was performed as described on the Jackson Laboratory website.
Hippocampal slices.
Transverse hippocampal slices (400 μm) were transferred to a recording chamber where they were maintained at 29°C and perfused with artificial CSF (ACSF) continuously bubbled with 95% O2 and 5% CO2. The ACSF composition (Stock solution 10×) was as follows (in mm): 124 NaCl, 4.4 KCl, 1 Na2HPO4, 25 NaHCO3, 2 CaCl2, 2 MgSO4, and 10 glucose.
Western blotting.
Equal amounts (10–30 μg) of proteins were separated on 4–12% Bis-Tris SDS-PAGE gels or 4–20% Tris-Gly gels (Invitrogen), blotted onto PVDF membranes (Millipore) and incubated overnight with the appropriate primary antibody. The antibodies used were as follows: mouse monoclonal α-APP (22C11) from Millipore Bioscience Research Reagents; α-αtubulin antibody from Sigma; α-pTrKA (Rajagopal et al., 2004) and α-TrkA, which detects basal levels of Trk receptors, α-pAKT, and α-AKT were from Cell Signaling Technology.
Immuofluorescence.
Hippocampal neurons were fixed for 20 min in PBS containing 4% formaldehyde and 4% sucrose, permeabilized with 0.1% Triton X-100 (5–10 min, 20°C), and processed for single labeling with monoclonal α-AchE (Millipore Bioscience Research Reagents). Secondary antibodies coupled to Alexa dyes (488 or 594) were from Invitrogen. Nuclei were visualized by staining with Hoechst dye 33258 (1 μg/ml) (Sigma). Digital images were obtained with an Olympus BX51 microscope (100× oil and 60× oil objectives) equipped with a Spot Diagnostic Instruments camera and were collected with Spot image analysis software. Controls were performed either by omitting the primary antibody or by preincubating the primary antibody with the corresponding peptide.
For DRG and SCG double immunofluorescence for APP (Y188) and TrkA (clone B3), primary antibodies were from Abcam and Santa Cruz Biotechnology Biotec, respectively. Secondary antibodies coupled to Alexa dyes (488 and 594) were from Invitrogen. Slides were examined with a confocal laser scanning microscope (SP5, Leica Microsystems) equipped with four laser lines: violet diode emitting at 405 nm; argon emitting at 488 nm; helium/neon emitting at 543 nm; and helium/neon emitting at 633 nm. Confocal acquisition modality was multitrack, and all the settings were maintained constant across different cases. For the production of figures, brightness and contrast of images were adjusted. Cells (derived from four independent experiments) from SCG and DRG of both WT and APPYG/YG mice were first visually studied at the fluorescence microscope. Only cells displaying a healthy appearance, characterized by a regular cellular and nuclear pattern (the latter examined in the Hoechst channel), and a medium-high immunofluorescence intensity of both TrkA and APP were selected for confocal acquisition. For each cell, particular care was taken to sample the equatorial plane of the cell, indentified through fast scanning along the z-axis and by selecting the focal plane where the nucleus displayed the larger diameter.
Immunoprecipitation.
For immunoprecipitation, protein samples were added to Dynabeads-ProteinG, according to the procedure described by the manufacturer (Invitrogen), and were eluted with 0.1 m citrate buffer, pH 2.3. pH was adjusted by adding Tris/HCl 2 m. Immunoprecipitation with α-pTyrosine antibody was performed with pY99 or pY20 antibodies (30 μg/100 μl of Dynabeads-ProteinG) from Sigma. For TrkA immunoprecipitation, we used α-TrkA antibody (clone B3) from Santa Cruz Biotechnology.
Statistical analysis.
Values are expressed as the mean ± SE. Statistical analysis was performed with ANOVA, followed by the Newman–Keuls test. Statistical significance was accepted at the 95% confidence level (p < 0.05).
Results
NGF induces Tyr phosphorylation of APP
To test whether NGF triggers phosphorylation of APP on Tyr residues in physiological conditions, we treated primary hippocampal neurons with 50 ng/ml NGF for the indicated time periods (Fig. 1A). Lysates from treated neurons were immunoprecipitated with an α-APP antibody, and precipitants were analyzed by Western blot analysis with an α-pTyr (α-pY) antibody. As shown in Figure 1A, we detected a protein of molecular mass similar to mature APP that is immunoprecipitated by the α-APP antibody and is phosphorylated on Tyr residues. This phosphorylation is detectable 10 min after NGF exposure and persists for at least 24 h (Fig. 1A). In reciprocal experiments, we found that APP is immunoprecipitated by the α-pY antibody from samples treated with NGF (Fig. 1B). The evidence that the TrkA receptor is phosphorylated upon NGF treatment indicates that the NGF/TrkA signaling pathways is properly activated in this experimental setting (Fig. 1B). In addition, the phosphorylation was very rapid and long lasting, indicating a direct and sustained NGF effect and further supporting the specificity of the NGF/TrkA signaling involvement. These results strongly suggest that NGF treatment induces APP phosphorylation on tyrosines in primary neuronal cells.
Figure 1.

NGF triggers phosphorylation of APP via the TrkA receptor. A, Primary cortical neurons from WT mice were treated with 50 ng/ml NGF for the indicated time periods. Lysates from treated neurons were immunoprecipitated with an α-APP antibody (IP-α-APP). Immunoprecipitants and whole lysates were analyzed by Western blot with an α-phospho Tyr antibody (α-pY, pY99). B, In a reciprocal experiment, lysates from treated or untreated neurons were immunoprecipitated with an α-pY antibody. Immunoprecipitants and whole lysates were analyzed by Western blot with an α-APP and an α-pTrkA antibody. C, Immunofluorescence analysis performed with anti-acetylcholinesterase antibody (anti-AchE, red) of cultures exposed or not to NGF for 48 h. Nuclei are marked in blue. Cholinergic neurons account for ∼20–25% of the overall population. NGF exposure does not modify the number of cholinergic neurons. D, Hippocampal slices were treated for 10 min with NGF and treated as in B. E, Two distinct TrKA inhibitors, CEP-2563 (CEP) and K-252a (Matrone et al., 2009), prevented APP phosphorylation. F, Quantization of triplicate experiments as shown in E shows that this inhibition is statistically significant (N = 4, Newman–Keuls test).
It is likely that the NGF signaling in primary hippocampal neuronal cultures (Fig. 1A,B) is due to the cholinergic afferent population, which is considered one of the most responsive NGF populations (Siegel et al., 1999) and accounted for ∼20–25% of the overall population in our experimental conditions (Fig. 1C). We speculated whether such NGF-dependent APP phosphorylation may occur also in hippocampal slices, where most of the neuronal connections are preserved, and in septal nuclei slices (data not shown), which are considered to be the major cholinergic output of the CNS (Hartikka and Hefti, 1988). Ten minutes of NGF exposure induced APP phosphorylation on Tyr residues in hippocampal slices as well as in septum slices. Again, a band corresponding to mature APP is immunoprecipitated by the α-pY antibody in samples treated with NGF, and this phosphorylation is synchronous with the activation of TrkA (Fig. 1D). Activation of TrkA is necessary to mediate Tyr phosphorylation of APP, since the TrkA inhibitors CEP-2563 and K-252a prevented phosphorylation of APP (Fig. 1E,F). Thus, NGF/TrkA signaling mediates Tyr phosphorylation of APP in two anatomical regions known to be involved in Alzheimer pathogenesis. Altogether, the results indicate that activation of TrkA by NGF produces phosphorylation of APP on one or more tyrosine residues under physiological conditions. Although it is conceivable that TrkA, a receptor with tyrosine kinase activity, directly phosphorylates APP, phosphorylation of APP may also be mediated by other kinases that are activated by TrkA.
APP regulates NGF-mediated TrkA activation and the sensitivity of neurons to the trophic action of NGF
APP contains three tyrosine residues in the cytoplasmic tail (Y653, Y682, and Y687 of the APP695 isoform). The latter two are included in the APP-Y682ENPTY687 sequence, which is instrumental for the association with the phosphotyrosine-binding domains of the APP interacting proteins identified to date (King and Scott Turner, 2004). We, and others, have previously shown that overexpression of TrkA as well as the constitutively active form of the tyrosine kinase Abl induces phosphorylation of APP on Y682 (Zambrano et al., 2001; Tarr et al., 2002b), suggesting that this tyrosine is the phosphorylation target of the NGF/TrkA signaling pathway. To test this hypothesis, we have taken advantage of an APP Y682G knock-in (KI) mouse, in which Y682 has been mutated into glycine, to understand the physiological functions of Y682 in vivo (Barbagallo et al., 2010). Hippocampal slices prepared from either APPYG/YG mice or WT littermates were treated with NGF. The APPYG/YG mutant is not phosphorylated on Tyr, 10 min after NGF treatment (Fig. 2A), suggesting that Y682 is either the phosphorylation target of the NGF/TrkA signaling pathway or that it is necessary for phosphorylation of other Tyr residues of APP by TrkA. However, and surprisingly, we found that TrkA phosphorylation induced by NGF was absent in APPYG/YG mice (Fig. 2A, bottom). Lack of TrkA phosphorylation did not depend on reduced TrkA expression by APPYG/YG mice (Fig. 2B) and suggests that TrkA signaling is impaired by the mutation at Y682. This prediction was confirmed by the finding that NGF fails to activate downstream signaling molecules, such as Akt, in APPYG/YG mice (Fig. 2B).
Figure 2.

APP and Y682 are necessary for activation of TrkA by NGF. A, Hippocampal slices from WT and APPYG/YG mice were treated with NGF for 10 min (+NGF). Lysates from treated or untreated slices were immunoprecipitated with an α-pY antibody. Immunoprecipitants and whole lysates were analyzed by Western blot with an α-APP and an α-pTrkA antibody. B, C, Lysates from WT, APPYG/YG, or APP−/− (KO) hippocampal slices treated with NGF were analyzed using antibodies against pTrkA, TrkA, pAKT, AKT, APP, and α-tubulin (α-tub.).
Next, we asked whether APPYG/YG mutation abolishes NGF/TrkA signaling by either a loss or a gain of function mechanism. We answered this question by analyzing APP-null mice. Of note, the Y682G mutation acts like a null allele when the essential function of APP in development are analyzed in vivo (Li et al., 2010; Barbagallo et al., 2011). Consistent with this, we found that NGF-dependent TrkA activation is also impaired in APP−/− hippocampal slices, indicating that APP plays an important role in TrkA signaling and that this function requires Y682 (Fig. 2C). Moreover, no significant differences were observed in p75 expression and processing (data not shown).
We then tested whether the role of APP in TrkA signaling had functional consequences. NGF exerts a trophic activity on DRG and SCG neurons, which express high levels of TrkA (Table 1). Notably, DRG and SCG neurons isolated from APPYG/YG mice were insensitive to this trophic function of NGF (+NGF) and an extensive neuronal loss was assessed when compared with the corresponding +NGF samples from WT mice (table 1).
Table 1.
Neuronal nuclei assessment in APP WT and APPYG/YG mice
| DRGs |
SCGs |
|||
|---|---|---|---|---|
| Ctrl | +NGF | Ctrl | +NGF | |
| APP WT | 14,13 ± 4.52 | 52.97 ± 12.1 | 24.5 ± 3.39 | 165 ± 33.29 |
| APPyg/yg | 12.9 ± 1.48 | 11.3 ± 1.65 | 26.47 ± 9.53 | 33.3 ± 13.6 |
Data are given as the mean ± SEM of four slides per condition of four independent experiments. Ctrl, Neurons plated in absence of NGF; +NGF, neurons exposed to NGF for 3 d.
APP interacts with TrkA, and both modulate their cellular distribution
To dissect the molecular and biochemical mechanism by which APP regulates TrkA signaling, we tested whether APP and TrkA physically interact in vivo, under physiological condition. To this end, we prepared protein samples from septum of WT, APPYG/YG, or APP−/− mice. Samples were immunoprecipitated with an α-TrkA antibody and analyzed by Western blot with an α-APP antibody. As shown in Figure 3A, a fraction of endogenous TrkA is complexed to endogenous APP in mouse brain. Interestingly, the APPYG/YG mutant does not interact with TrkA, although APPYG/YG mice express normal amounts of APP, suggesting an essential role for Y682 in the APP/TrkA interaction. However, the lack of any selective α-TrkA antibody fails to rule out the possibility that TrkB also may be involved in these events.
Figure 3.
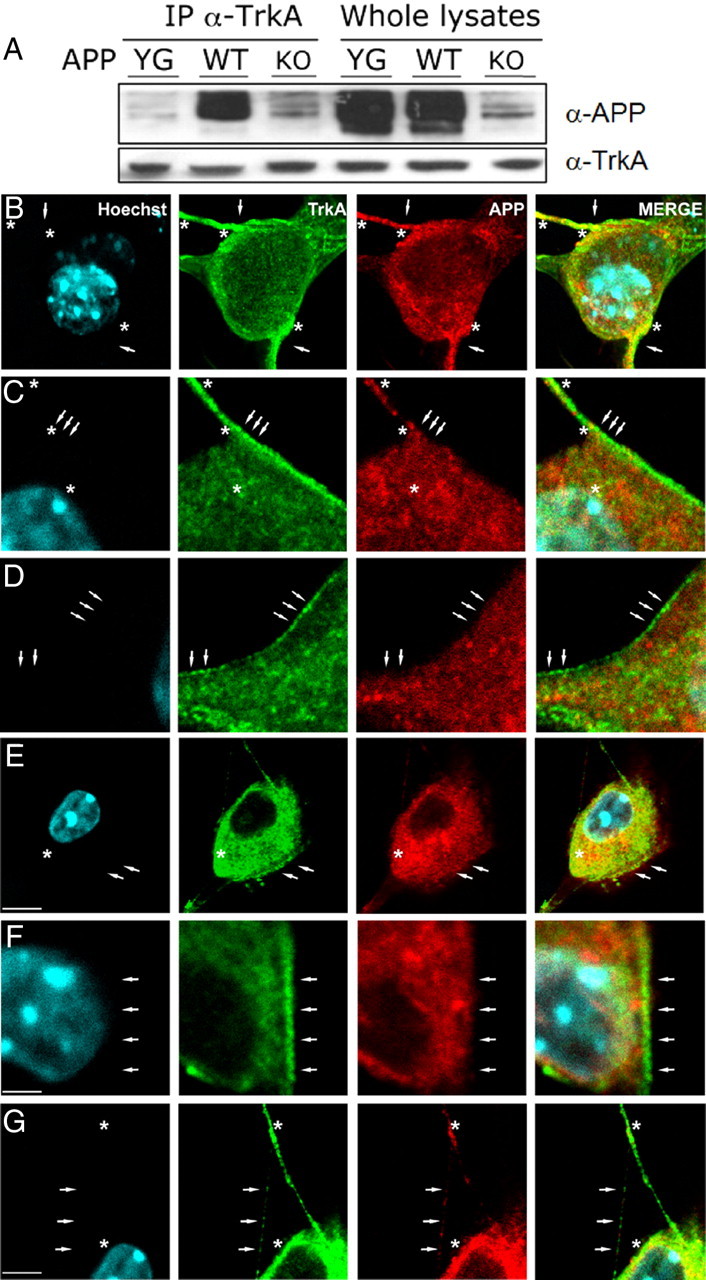
APP interacts with TrkA and regulates TrkA cellular distribution. A, Protein samples from septum of WT, APPYG/YG, or APP−/− mice were immunoprecipitated with an α-TrkA antibody and analyzed by Western blot with an α-APP antibody. B–G, Confocal microscopy of a double immunofluorescence for TrkA (green channel) and APP (red channel) in DRGs of WT (B–D) and APPYG/YG (E–G) mice. In WT mice (B–D), TrkA and APP immunofluorescence appeared homogeneously distributed on the cellular membrane particularly enriching the proximity and the neuritic domains (B, arrows). TrkA-immunostained vesicles (C, D, arrows) were clearly visible on the cellular surface and neuritic domains. Colocalization between TrkA and APP was restricted to the proximity of and along the neuritic domain (A, B, asterisks). E–G, Note in APPYG/YG mice the intracytoplasmatic and perinuclear accumulation of TrkA and APP immunofluorescence and the decrease of immunoreactivity in neuritic structures (G, arrows). Some TrkA-positive vesicles are still present on the cellular membrane (E, F, arrows). APP and TrkA colocalization appeared to increase in the intracytoplasmic domain (E, F, asterisks) and to decrease in neuritic structures (G, asterisks). Scale bars: B, E, 5 μm; C, D, F, 2 μm; G, 3 μm.
Next, we determined whether APP regulates the subcellular distribution of TrkA. For this, we prepared from both WT and APPYG/YG mice primary neuronal cells derived from the DRGs (Fig. 3) and SCGs (data not shown). In WT mice, at low magnification, TrkA immunofluorescence was distributed on neuronal cell bodies and at high intensities along neurites. At high magnification, TrkA immunofluorescence, localized in vesicles of small and homogeneous size, showed a preferential distribution on the cellular membrane or immediately beneath (Fig. 3B,D, arrows). Of particular interest, TrkA staining was increased in proximity to and along the neuritic domains, and neurites displayed a grainier or beaded appearance with the tendency to organize in clusters (Fig. 3B, arrows). These clusters filled the neurites, and sometimes it was possible to observe a certain number of TrkA-positive vesicles organized in a row, suggesting an anterograde transport through cytoskeletric structures (Fig. 3C,D). The distribution pattern of APP was slightly different than that of TrkA. APP-immunopositive vesicles appeared less structurally defined and of more variable size (Fig. 3B–D). As for TrkA, APP also appeared to increase in immunoreactivity in proximity and along the neuritic domains, and showed a preferential membrane distribution (Fig. 3B–D, arrows). The increase and accumulation of both APP and TrkA immunoreactivity in proximity and along neurites (Fig. 3B) suggest the presence of an intense trafficking toward the cellular peripheral structures (axonal terminals, spines), where both proteins may exert their primary functions. Colocalization between TrkA and APP appeared to be selectively confined to the proximity of and along neuritic domains (Fig. 3B–D, asterisks).
In APPYG/YG mice, at low magnification, TrkA immunofluorescence resembled the distribution pattern observed in WT mice. However, at high magnification several marked differences were evident. The TrkA prevalent membrane distribution shown in WT mice was lost in APPYG/YG mice in favor of a more intracytoplasmic, clearly perinuclear, distribution pattern (Fig. 3E). The loss of an organized distribution pattern was also clearly evident when analyzing the neuritic domains where a marked reduction of TrkA immunofluorescence can be observed. Very often, only few labeled grains, distantly interspaced, and organized in a row, could be followed (Fig. 3G), suggesting the disruption of the TrkA cytoskeletric transport. As for TrkA, APP immunoreactivity was highly decreased from the cell membrane and neuritic domains, while it appeared to increase in the intracellular and perinuclear regions (Fig. 3E–G). the colocalization pattern between TrkA and APP appeared to be switched from the proximity of and along neuritic domains (Fig. 3B–D, asterisks) in WT mice to the intracellular and perinuclear regions (Fig. 3E–G, asterisks) in APPYG/YG mice. Altogether, these results suggest that in APPYG/YG mice both TrkA–APP interaction and cellular distribution are affected.
Discussion
This study was initiated to determine whether NGF/TrkA signaling promotes phosphorylation on APP on tyrosine residues under physiological conditions, and to determine whether Y682 is the primary target. We answered the first question conclusively by showing that NGF treatment triggers APP Tyr phosphorylation via activation of TrkA. To address the second question, we studied the APP-KI mutant APPYG/YG, which bears a mutation in Y682. However, we could not determine whether Y682 is the TrkA phosphorylation target since NGF/TrkA signaling is strongly reduced in APPYG/YG hippocampal samples. This observation, and the finding that TrkA signaling is also compromised in APP-null hippocampal samples, unveiled an unexpected role of APP in NGF/TrkA signaling. The evidence that neuronal cells from APPYG/YG mice are insensitive to the trophic function of NGF underlined the functional relevance of this role. TrkA and APP interact in brain, and this association is dependent on Y682. Immunofluorescence studies suggest that APP regulates peripheral export of TrkA and that this function is impaired in APPYG/YG mutant mice. Indeed, localization of both TrkA and APP is altered in APPYG/YG neurons. This evidence further underlines the functional relationship between APP and TrkA, although it does not clarify whether the localization of these proteins is changed because the interaction is abolished or whether the interaction is abolished because the localization of APP is altered by the YG mutation. Since plasma membrane TrkA is accessible to NGF, the data suggest that APP regulates NGF/TrkA signaling by regulating the surface levels of TrkA.
This evidence, together with previous findings involving the NGF/TrkA signaling axis in the regulation of APP processing (Tarr et al., 2002b; Matrone et al., 2008, 2009), suggest that the functions of these two membrane proteins are strictly interconnected, and that the NGF/TrkA signaling pathway may be involved in the deregulation of APP processing that causes AD and be used as a therapeutic target to correct such a deregulation. It would also be important to test whether the APP mutation causing familial Alzheimer disease alters the NGF/TrkA signaling pathway and whether such alterations have a pathogenic role in AD.
Footnotes
This research was supported by the European Molecular Biology Organization (Grant ASTF264-2010) to C.M.; FIRB (Grant 2010–2012) from the Italian Ministry of Higher Education and Scientific Research, IT to P.C.; Alzheimer Disease Research Grants A2003-076 to L.D. and NIRG-10-173876 to A.P.M.B.; NIH (Grant AG033007 to L.D, AG025970 NS21072 to M.V.C.); and a grant from the Thome Foundation (to L.D.). The funders had no role in study design, data collection, and analysis, decision to publish, or preparation of this manuscript. We thank Dr. Jhon Sutachan and Rosalia Castro Perez (from Skirball Institute) for their support and technical assistance in neuronal cell culture preparation. We greatly appreciate administrative support of P. Papa.
The authors declare no competing financial interests.
References
- Barbagallo AP, Weldon R, Tamayev R, Zhou D, Giliberto L, Foreman O, D'Adamio L. Tyr682 in the intracellular domain of APP regulates amyloidogenic APP processing in vivo. PLoS One. 2010;5:e15503. doi: 10.1371/journal.pone.0015503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbagallo AP, Wang Z, Zheng H, D'Adamio L. A single tyrosine residue in the amyloid precursor protein intracellular domain is essential for developmental function. J Biol Chem. 2011;286:8717–8721. doi: 10.1074/jbc.C111.219873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand E, Brouillet E, Caillé I, Bouillot C, Cole GM, Prochiantz A, Allinquant B. A short cytoplasmic domain of the amyloid precursor protein induces apoptosis in vitro and in vivo. Mol Cell Neurosci. 2001;18:503–511. doi: 10.1006/mcne.2001.1030. [DOI] [PubMed] [Google Scholar]
- Giliberto L, Zhou D, Weldon R, Tamagno E, De Luca P, Tabaton M, D'Adamio L. Evidence that the amyloid beta precursor protein-intracellular domain lowers the stress threshold of neurons and has a “regulated” transcriptional role. Mol Neurodegener. 2008;3:12. doi: 10.1186/1750-1326-3-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hartikka J, Hefti F. Comparison of nerve growth factor's effects on development of septum, striatum, and nucleus basalis cholinergic neurons in vitro. J Neurosci Res. 1988;21:352–364. doi: 10.1002/jnr.490210227. [DOI] [PubMed] [Google Scholar]
- King GD, Scott Turner R. Adaptor protein interactions: modulators of amyloid precursor protein metabolism and Alzheimer's disease risk? Exp Neurol. 2004;185:208–219. doi: 10.1016/j.expneurol.2003.10.011. [DOI] [PubMed] [Google Scholar]
- Li H, Wang Z, Wang B, Guo Q, Dolios G, Tabuchi K, Hammer RE, Südhof TC, Wang R, Zheng H. Genetic dissection of the amyloid precursor protein in developmental function and amyloid pathogenesis. J Biol Chem. 2010;285:30598–30605. doi: 10.1074/jbc.M110.137729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu DC, Rabizadeh S, Chandra S, Shayya RF, Ellerby LM, Ye X, Salvesen GS, Koo EH, Bredesen DE. A second cytotoxic proteolytic peptide derived from amyloid beta-protein precursor. Nat Med. 2000;6:397–404. doi: 10.1038/74656. [DOI] [PubMed] [Google Scholar]
- Madeira A, Pommet JM, Prochiantz A, Allinquant B. SET protein (TAF1beta, I2PP2A) is involved in neuronal apoptosis induced by an amyloid precursor protein cytoplasmic subdomain. FASEB J. 2005;19:1905–1907. doi: 10.1096/fj.05-3839fje. [DOI] [PubMed] [Google Scholar]
- Matrone C, Ciotti MT, Mercanti D, Marolda R, Calissano P. NGF and BDNF signaling control amyloidogenic route and Abeta production in hippocampal neurons. Proc Natl Acad Sci U S A. 2008;105:13139–13144. doi: 10.1073/pnas.0806133105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matrone C, Marolda R, Ciafrè S, Ciotti MT, Mercanti D, Calissano P. Tyrosine kinase nerve growth factor receptor switches from prosurvival to proapoptotic activity via Abeta-mediated phosphorylation. Proc Natl Acad Sci U S A. 2009;106:11358–11363. doi: 10.1073/pnas.0904998106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaev A, McLaughlin T, O'Leary DD, Tessier-Lavigne M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature. 2009;457:981–989. doi: 10.1038/nature07767. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Passer B, Pellegrini L, Russo C, Siegel RM, Lenardo MJ, Schettini G, Bachmann M, Tabaton M, D'Adamio L. Generation of an apoptotic intracellular peptide by gamma-secretase cleavage of Alzheimer's amyloid beta protein precursor. J Alzheimers Dis. 2000;2:289–301. doi: 10.3233/jad-2000-23-408. [DOI] [PubMed] [Google Scholar]
- Rajagopal R, Chen Z-Y, Lee FS, Chao MV. Transactivation of Trk neurotrophin receptors by G-protein-coupled receptor ligands occurs on intracellular membranes. J Neuroscience. 2004;24:6650–6658. doi: 10.1523/JNEUROSCI.0010-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo C, Dolcini V, Salis S, Venezia V, Zambrano N, Russo T, Schettini G. Signal transduction through tyrosine-phosphorylated C-terminal fragments of amyloid precursor protein via an enhanced interaction with Shc/Grb2 adaptor proteins in reactive astrocytes of Alzheimer's disease brain. J Biol Chem. 2002;277:35282–35288. doi: 10.1074/jbc.M110785200. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, Podlisny MB. Deciphering the genetic basis of Alzheimer's disease. Annu Rev Genomics Hum Genet. 2002;3:67–99. doi: 10.1146/annurev.genom.3.022502.103022. [DOI] [PubMed] [Google Scholar]
- Siegel GJ, Agranoff BW, Albers RW, Fisher SK, Uhler MD. Basic neurochemistry: molecular, cellular and medical aspects. 6th Ed. Philadelphia: Lippincott Williams & Wilkins; 1999. [Google Scholar]
- Tamayev R, Zhou D, D'Adamio L. The interactome of the Amyloid betaeta Precursor Protein family members is shaped by phosphorylation of their intracellular domains. Mol Neurodegener. 2009;4:28. doi: 10.1186/1750-1326-4-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarr PE, Roncarati R, Pelicci G, Pelicci PG, D'Adamio L. Tyrosine phosphorylation of the beta-amyloid precursor protein cytoplasmic tail promotes interaction with Shc. J Biol Chem. 2002a;277:16798–16804. doi: 10.1074/jbc.M110286200. [DOI] [PubMed] [Google Scholar]
- Tarr PE, Contursi C, Roncarati R, Noviello C, Ghersi E, Scheinfeld MH, Zambrano N, Russo T, D'Adamio L. Evidence for a role of the nerve growth factor receptor TrkA in tyrosine phosphorylation and processing of beta-APP. Biochem Biophys Res Commun. 2002b;295:324–329. doi: 10.1016/s0006-291x(02)00678-2. [DOI] [PubMed] [Google Scholar]
- Zambrano N, Bruni P, Minopoli G, Mosca R, Molino D, Russo C, Schettini G, Sudol M, Russo T. The beta-amyloid precursor protein APP is tyrosine-phosphorylated in cells expressing a constitutively active form of the Abl protoncogene. J Biol Chem. 2001;276:19787–19792. doi: 10.1074/jbc.M100792200. [DOI] [PubMed] [Google Scholar]
- Zhou D, Noviello C, D'Ambrosio C, Scaloni A, D'Adamio L. Growth factor receptor-bound protein 2 interaction with the tyrosine-phosphorylated tail of amyloid beta precursor protein is mediated by its Src homology 2 domain. J Biol Chem. 2004;279:25374–25380. doi: 10.1074/jbc.M400488200. [DOI] [PubMed] [Google Scholar]
- Zhou D, Zambrano N, Russo T, D'Adamio L. Phosphorylation of a tyrosine in the amyloid-beta protein precursor intracellular domain inhibits Fe65 binding and signaling. J Alzheimers Dis. 2009;16:301–307. doi: 10.3233/JAD-2009-0970. [DOI] [PubMed] [Google Scholar]