

Abstract
Both sporadic and autosomal dominant forms of Parkinson's disease (PD) have been causally linked to mutations in leucine-rich repeat kinase 2 (LRRK2), a large protein with multiple domains. The kinase domain plays an important role in LRRK2-mediated toxicity. Although a number of investigations have focused on LRRK2 kinase activity, less is known about the GTPase function of LRRK2. The activity of GTPases is regulated by GTPase activating proteins (GAPs) and GTP exchange factors. Here, we identify ArfGAP1 as the first GAP for LRRK2. ArfGAP1 binds LRRK2 predominantly via the WD40 and kinase domain of LRRK2, and it increases LRRK2 GTPase activity and regulates LRRK2 toxicity both in vitro and in vivo in Drosophila melanogaster. Unexpectedly, ArfGAP1 is an LRRK2 kinase substrate whose GAP activity is inhibited by LRRK2, whereas wild-type and G2019S LRRK2 autophosphorylation and kinase activity are significantly reduced in the presence of ArfGAP1. Overexpressed ArfGAP1 exhibits toxicity that is reduced by LRRK2 both in vitro and in vivo. Δ64–ArfGAP1, a dominant-negative ArfGAP1, and shRNA knockdown of ArfGAP1 reduce LRRK2 toxicity. Thus, LRRK2 and ArfGAP1 reciprocally regulate the activity of each other. Our results provide insight into the basic pathobiology of LRRK2 and indicate an important role for the GTPase domain and ArfGAP1 in LRRK2-mediated toxicity. These data suggest that agents targeted toward regulation of LRRK2 GTP hydrolysis might be therapeutic agents for the treatment of PD.
Introduction
Leucine-rich repeat kinase 2 (LRRK2) is a large multifunction protein that contains multiple domains, including an LRRK2-specific repeat region, a kinase domain, a GTPase domain, a C terminal of Ras of complex domain, and multiple leucine-rich repeats (Mata et al., 2006; Cookson, 2010). Mutations in LRRK2 are a common cause of autosomal dominant Parkinson's disease (Paisán-Ruíz et al., 2004; Zimprich et al., 2004). The kinase activity of LRRK2 is important in the loss of dopamine neurons attributable to LRRK2 mutations, because genetic and pharmacologic inactivation of LRRK2 kinase activity is neuroprotective (Smith et al., 2006; Lee et al., 2010; Deng et al., 2011; Liu et al., 2011). The role of the GTPase domain in LRRK2 toxicity is less clear. However, a recent yeast model of LRRK2 toxicity using truncated LRRK2 constructs suggests that enhanced GTP hydrolysis leads to reduced LRRK2 toxicity, whereas impaired GTP hydrolysis leads to enhanced LRRK2 toxicity (Xiong et al., 2010). The role of GTP hydrolysis in the context of full-length LRRK2 is poorly understood.
GTPase activity is regulated by GTPase activating proteins (GAPs) and guanine exchange factors (GEFs) (Barr and Lambright, 2010; Vigil et al., 2010; East and Kahn, 2011). GAPs enhance hydrolysis of GTP by GTPases and GEFs promote the release of GDP, allowing access of GTP to the GTPase (Barr and Lambright, 2010; Vigil et al., 2010; East and Kahn, 2011). GAPs and GEFs tend to have some specificity for their target proteins. Little is known about the GAPs and GEFs that regulate LRRK2, other than ARFGEF7 may be a GEF for LRRK2 (Haebig et al., 2010). In a genome wide genetic screen to identify modifiers of LRRK2 toxicity in yeast we identified GCS1, whose human homolog is ADP ribosylation factor GTPase activating protein 1 (ArfGAP1) (Xiong et al., 2010). Here we show that ArfGAP1 is a GAP for LRRK2 that enhances GTP hydrolysis of LRRK2 and reduces LRRK2 toxicity both in vitro and in vivo. Unexpectedly, LRRK2 phosphorylates ArfGAP1, resulting in inhibition of the GAP activity of ArfGAP1 providing reciprocal regulation of LRRK2 and ArfGAP1.
Materials and Methods
Plasmids and antibodies.
Full-length human ArfGAP1, Arf1 was cloned into the mammalian expression vector pcDNA3.1/nV5-DEST and Escherichia coli expression vector pDEST15 with a GST tag (Invitrogen) from an entry clone carrying ArfGAP1 full-length cDNA (Invitrogen Ultimate ORF Clone IOH57771) or Arf1 (Invitrogen Ultimate ORF Clone IOH13571) by Gateway technology. To generate ArfGAP1 single or multiple mutants, site-directed mutagenesis was performed using the QuickChange site-directed mutagenesis kit (Stratagene). Deletion mutation ArfGAP (1–64 aa) and Δ64–ArfGAP1 was cloned into pcDNA3.1–Myc–His vector with an N-terminal HA tag or into pEGFP–N1 vector with an N-terminal V5 tag. Full-length ArfGAP1 and Δ64–ArfGAP1 were cloned into Drosophila expression vector pUASTattB vector with an N-terminal HA tag. All cDNAs or mutation sites were confirmed by DNA sequencing analysis. Full-length wild-type (WT) and mutant LRRK2 were generated in our laboratory as described previously (West et al., 2007). Truncated mutants for human LRRK2 were cloned into the mammalian expression vector pcDNA3.1 with three flag tags at N terminus as described previously (Smith et al., 2005; Ko et al., 2009).
Mouse monoclonal anti-ArfGAP1 antibodies were obtained from Santa Cruz Biotechnology. Rabbit polyclonal anti-ArfGAP1, rabbit polyclonal anti-GFP, rabbit polyclonal anti-VAMP2, rabbit polyclonal anti-Histone H4, and rabbit polyclonal anti-PGK1 antibodies were obtained from Abcam. Rabbit monoclonal anti-LRRK2 was from the Michael J. Fox Foundation. Mouse monoclonal anti-LRRK2 (clone N138/6) was from University of California, Davis/National Institutes of Health NeuroMab facility. Mouse monoclonal anti-V5 and anti-V5–HRP antibodies were purchased from Invitrogen. Mouse monoclonal anti-flag, anti-flag–HRP, anti-HA–HRP, anti-phosphothreonine, anti-phosphoserine, and anti-tubulin antibodies were obtained from Sigma-Aldrich. Anti-actin antibodies were from Sungene Biotech. Mouse monoclonal anti-Myc and anti-HA were obtained from Roche Applied Science. HRP-linked anti-rabbit or anti-mouse IgG antibodies were obtained from Jackson ImmunoResearch. Alexa Fluor-488 anti-mouse IgG and Alexa Fluor-594 anti-rabbit IgG antibodies were from Invitrogen.
Cell culture, transfections, and coimmunoprecipitation.
HEK 293T cells were cultured in OPTI-MEM media supplemented with 10% fetal bovine serum. Transient transfection with Myc–LRRK2 or V5–ArfGAP1, V5–Δ64–ArfGAP1, and V5–ArfGAP (1–64 aa) was performed using Fugene HD transfection reagent (Roche) as per the instructions of the manufacturer. After 48 h, cells were washed by PBS once and lysed in immunoprecipitation (IP) buffer [1% Triton X-100, 0.5% NP-40, 150 mm NaCl, 10 mm Tris, 1 mm EGTA, pH 7.4, 1× phosphatase inhibitor cocktails 1 and 2 (Sigma-Aldrich), and 1× Complete mini protease inhibitor cocktail (Roche)] by rotation at 4°C for 1 h. Cell lysates were centrifuged at 15,000 rpm for 15 min. Supernatants were incubated with protein-G Dyna beads (Invitrogen) precoated with anti-V5 or anti-Myc antibodies, followed by rotating overnight at 4°C. The Dyna beads were pelleted and stringently washed five times with IP buffer supplemented with 500 mm NaCl. The immunoprecipitated proteins were resolved on SDS-PAGE and subjected to immunoblotting.
Subcellular fractionation.
Subcellular fractionation was performed as described previously (Sharp et al., 1995; Zhang et al., 2011). Briefly, whole mouse brain was homogenized in 9 vol of ice-cold homogenization buffer [320 mm sucrose, 4 mm HEPES, pH 7.4, 1× phosphatase inhibitor cocktails 1 and 2 (Sigma-Aldrich), and 1× Complete mini protease inhibitor cocktail (Roche)]. The homogenates were centrifuged at 1000 × g for 10 min to produce the pellet including nuclei and large debris fraction (P1) and the supernatant (S1). The S1 fractions were further fractionated into the supernatant (S2) and the pellet (P2) by centrifugation at 10,000 × g for 20 min. P2 contains the crude synaptosomal fraction as well as membranous organelles such as Golgi, endoplasmic reticulum, and plasma membrane. The S2 fraction was further fractionated into a light membrane/microsome-enriched part (P3) and the cytosol (S3) by centrifugation at 165,000 × g for 1 h. The P2 fraction was lysed and fractionated into the supernatant (LS1) and a synaptosomal membrane fraction (LP1) by centrifugation at 25,000 × g for 20 min. LS1 was then fractionated into the synaptic vesicle-enriched fraction (LP2) and the supernatant with soluble synaptosomal proteins (LS2) by centrifugation at 165,000 × g for 2 h.
GTPase activity assay.
GTP hydrolysis activity was measured by monitoring the release of free γ-phosphate (Pi) from GTP as described previously (Xiong et al., 2010). Briefly, recombinant proteins GST–ArfGAP1, GST–Δ64–ArfGAP1, and GST–Arf1 were expressed in E. coli BL21 and purified by glutathione beads as per the instructions of the manufacturer. The purity of the GST peptides was assessed with denaturing SDS-PAGE, followed by Coomassie blue staining. The GTPase activity assay was performed by measuring the concentration of free Pi using the colorimetric GTPase assay kit (Innova Biosciences), and LRRK2 recombinant proteins (deletion of the first 970 aa; Invitrogen) and Arf1 were incubated with or without ArfGAP1 together as per the instructions of the manufacturer.
In vitro kinase assay.
An in vitro kinase assay was performed as described previously (West et al., 2007). WT and mutant ArfGAP1 with GST tag were expressed in E. coli BL21 and purified by glutathione beads as per the instructions of the manufacturer. The purity of the GST peptides was assessed with denaturing SDS-PAGE, followed by Coomassie blue staining. Forty nanograms of WT and mutant LRRK2 recombinant proteins (Invitrogen) with or without recombinant ArfGAP1 protein or dephosphorylated myelin basic protein (MBP) (Millipore) were subjected to an LRRK2 kinase reaction in kinase buffer (20 mm HEPES, pH 7.4, 150 mm NaCl, 10 mm, EGTA, pH 8.0, 20 mm β-glycerol phosphate, 10 μm ATP, 0.5 μCi [γ-32P]ATP, and 20 mm MgCl2). The reactions were incubated at 30°C for 20 min, put on ice, and resolved on SDS-PAGE. LRRK2 autophosphorylation, ArfGAP1 phosphorylation, and MBP phosphorylation were imaged using a Typhoon Phosphoimager and quantified using NIH Image J. Input levels of protein were determined by Coomassie blue staining or immunoblotting. The reactions incubated without radioisotope were subjected to mass spectrometric analysis for ArfGAP1 phosphorylation sites by LRRK2.
Mass spectrometric analysis.
Mass spectrometry analysis was performed by the Taplin Biological Mass Spectrometry Facility (Harvard Medical School, Boston, MA).
Primary cortical neuronal cultures and viability assay.
Primary cortical neuronal cultures were prepared from embryonic day 15–16 CD1 fetal mice or LRRK2 WT and KO fetal mice and transiently cotransfected with LRRK2 and GFP–ArfGAP1 constructs as described previously (West et al., 2007; Xiong et al., 2010). Briefly, cortices were dissected and dissociated by TrypLE (Invitrogen). The cells were seeded into 12-well plates precoated with poly-l-ornithine and were maintained in Neurobasal media (Invitrogen) supplemented with B27 supplement and l-glutamine. The glial cells were inhibited by adding 5-fluoro-20-deoxyuridine (30 μm; Sigma) at days in vitro (DIV) 4. Half of the growth medium was replaced every 3–4 d. At DIV 10, neurons represented >90% of total cells in the culture. To assess LRRK2 and ArfGAP1-induced toxicity, neurons at DIV 10 were transiently cotransfected with LRRK2 and GFP, ArfGAP1–GFP, or Δ64–ArfGAP1–GFP constructs at a molar ratio of 10:1, respectively, using Lipofectamine 2000 reagent (Invitrogen) as per the instructions of the manufacturer. At 48 h after transfection (DIV 12), neurons were fixed with 4% paraformaldehyde (PFA). Immunostaining with GFP antibody and TUNEL staining were performed with these neurons. Fluorescent images were collected on a Carl Zeiss Automatic stage microscope with Axiovision 6.0 software. Neurons with no obvious neurite process and positive TUNEL staining were counted as nonviable cells. For each independent experiment, the percentage viability of GFP-positive neurons (n = 200) was determined and normalized to the control neurons transfected with GFP and pcDNA3.1 empty construct. LRRK2 expression was confirmed in >95% of GFP-positive neurons by performing immunocytochemistry with anti-Myc and anti-GFP antibodies and appropriate fluorescent secondary antibodies.
TUNEL staining.
Neurons were fixed in 4% PFA after 48 h transfection. TUNEL staining was performed using the In Situ Cell Death Detection Kit (Roche) as per the instructions of the manufacturer.
Immunocytochemistry.
Neurons were fixed in 4% PFA for 20 min after 48 h transfection before being permeabilized and blocked for 1 h with PBS containing 10% goat serum and 0.3% Triton X-100. After washing with PBS, the neurons were incubated with mouse anti-Myc and rabbit anti-GFP antibodies at 4°C overnight. After washing with PBS, the neurons were incubated with goat anti-mouse IgG conjugated to Alexa Fluor-594 or goat anti-mouse IgG conjugated to Alexa Fluor-405 and goat anti-rabbit IgG conjugated to Alexa Fluor-488 at room temperature for 1 h. The coverslips were mounted onto slides using Vectashield mounting medium (Vector Laboratories). The imaging was conducted on a Carl Zeiss Automatic stage microscope with Axiovision 6.0 software.
ArfGAP1 lentiviral shRNA preparations.
To knock down ArfGAP1, four shRNA pGIPZ clones were purchased from Open Biosystem (catalog #RMM4532-NM_145760). The second-generation lentiviral packaging system was used for high-titer viral preparations for effective transduction in primary neuronal cultures. Briefly, pGIPZ lentiviral vectors were transfected into HEK 293FT cells along with viral packaging plasmids (psPAX2 and pMD2G) using FuGene HD transfection reagent (Roche). After 48 h, the culture media were collected, and viral particles were precipitated by centrifugation at 25,000 × g for 2 h. Viral particles were resuspended into serum-free medium and stored at −80°C.
Drosophila genetics.
Fly lines for the tyrosine hydroxylase–galactosidase-4 (TH–GAL4), Glass Multiple Reporter (GMR)–GAL4, and UAS–GFP were obtained from Bloomington Stock Center. Human ArfGAP1 cDNAs with an HA tag were subcloned into pUASTattB plasmid, sequenced, and microinjected into Drosophila embryos (Bestgene). The transgenic ArfGAP1 flies were crossed with flies carrying either WT LRRK2 or G2019S LRRK2 (Liu et al., 2008). The resulting bigenic flies carrying both ArfGAP1 and WT LRRK2 or G2019S LRRK2 were crossed with TH–GAL4>UAS–GFP or GMR–GAL4 driver flies and therefore induced the coexpression of ArfGAP1 and WT LRRK2 or G2019S LRRK2 in dopamine neurons or fly eyes.
Climbing assay.
The climbing assay was performed as described previously (Ng et al., 2009). Briefly, six groups of 20 flies of either sex from each genotype were subjected to the assay every 2 weeks from 1 to 10 weeks. The tested flies were anesthetized and placed in a vertical plastic column (length, 10 cm; diameter, 1.5 cm). After a 1 h recovery from CO2 exposure, flies were gently tapped to the bottom of the column. The percentage of flies reaching the 6 cm height in 15 s was counted. Each group was repeated three times.
Statistical analysis.
Two-way ANOVA and two-tailed unpaired Student's t test were used for data analysis. Data represent mean ± SEM, and p ≤ 0.05 was considered statistically significant.
Results
ArfGAP1 interacts with LRRK2
To determine whether ArfGAP1 interacts with LRRK2, V5–ArfGAP1, and LRRK2–Myc were transfected into HEK 293 cells. IP with anti-Myc coimmunoprecipitates V5–ArfGAP1 (Fig. 1A). ArfGAP1 and LRRK2 interact in vivo because IP of ArfGAP1 pulls down LRRK2 in WT mouse brain but not LRRK2 knock-out (KO) brain (Fig. 1B), and IP of LRRK2 pulls down ArfGAP1 in WT mouse brain but not LRRK2 KO brain (Fig. 1C). There is no significant difference in binding between ArfGAP1 and WT LRRK2 or the familial mutations R1441C, R1441G, G2019S, or I2020T LRRK2 (Fig. 1D,E). Subcellular fractionation was performed to determine whether LRRK2 and ArfGAP1 reside in similar cellular compartments. As reported previously, LRRK2 is enriched in the LP1 and LP2 fraction (Biskup et al., 2006; Shin et al., 2008), which also contains high levels of ArfGAP1. The integrity of our subcellular fractionation was verified by immunoblotting with PGK1, Histone, and VAMP2, which segregate into appropriate subcellular fractions (Fig. 1F). To determine the domain of ArfGAP1 that interacts with LRRK2, coimmunoprecipitation of LRRK2–Myc was performed with V5–ArfGAP1 and compared with ArfGAP1 lacking the ArfGAP domain (Δ64–ArfGAP1) and the ArfGAP (1–64 aa) domain alone. LRRK2–Myc fails to interact with Δ64–ArfGAP1 (Fig. 2A). To ascertain the domain of LRRK2 that interacts with ArfGAP1, different domains of the LRRK2 spanning the entire LRRK2 protein were evaluated for their ability to interact with V5–ArfGAP1. The WD40 of LRRK2 predominantly interacts with ArfGAP1, and the kinase domain of LRRK2 also interacts with ArfGAP1 to a small extent (Fig. 2B). The ArfGAP (1–64 aa) domain alone is sufficient for binding to LRRK2 and binds to the same LRRK2 fragments as full-length ArfGAP1 (Fig. 2A,C).
Figure 1.

ArfGAP1 interacts with LRRK2. A, ArfGAP1 interacts with LRRK2 in vitro. Cell lysates from HEK 293T cells cotransfected with LRRK2–Myc and V5–ArfGAP1 were subjected to coimmunoprecipitation with anti-Myc, followed by anti-V5 immunoblotting or by anti-Myc to show an equivalent amount of input. B, ArfGAP1 interacts with LRRK2 in vivo. Whole-brain lysates prepared from WT and LRRK2 KO mice were subjected to coimmunoprecipitation with anti-ArfGAP1 and anti-IgG antibodies, followed by anti-LRRK2 and anti-ArfGAP1 immunoblotting. C, Whole-brain lysates prepared from WT and LRRK2 KO mice were subjected to coimmunoprecipitation with anti-LRRK2 (Neuromab), followed by anti-LRRK2 and anti-ArfGAP1 immunoblotting. D, ArfGAP1 interacts with LRRK2 familial mutants. Cell lysates from HEK 293 cells cotransfected with V5–ArfGAP1 and WT or familial mutants LRRK2–Myc (R1441C/G, G2019S, and I2020T) were subjected to coimmunoprecipitation with anti-Myc, followed by anti-V5 immunoblotting or anti-Myc to show an equivalent amount of input. E, Quantification of LRRK2 binding to ArfGAP1 relative to WT LRRK2. Data are representative of three independent experiments. Bars represent the mean ± SEM. Data were analyzed for statistical significance by two-tailed unpaired Student's t test relative to WT LRRK2. No significant difference was found between WT and mutant LRRK2 in terms of binding to ArfGAP1. F, Subcellular distribution of LRRK2 and ArfGAP1 of mouse whole-brain lysates. P1, Nuclei and large debris; S1, supernatant of the homogenate at low-speed centrifugation; P2, crude synaptosomes and membranous organelles such as Golgi, endoplasmic reticulum, and plasma membrane; S2, supernatant of S1 subjected to medium-speed centrifugation; P3, light membranes/microsome-enriched part; S3, cytosol, which corresponds to the supernatant of S2 subjected to high-speed centrifugation; LP1, synaptosomal membranes; LP2, synaptic vesicle-enriched fraction; LS2, soluble synaptosomal proteins; WB, Western blot.
Figure 2.

LRRK2 interacts with the ArfGAP domain of ArfGAP1, and ArfGAP1 interacts with the WD40 and kinase domain of LRRK2. A, LRRK2 interacts with the ArfGAP domain of ArfGAP1. Cell lysates from HEK 293T cells cotransfected with LRRK2–Myc and V5–ArfGAP1, V5–Δ64–ArfGAP1, or V5–ArfGAP (1–64 aa) were subjected to coimmunoprecipitation with anti-Myc, followed by anti-V5 immunoblotting or by anti-Myc to show an equivalent amount of input. A schematic representation of ArfGAP1, Δ64–ArfGAP1, and ArfGAP (1–64 aa) is shown. B, ArfGAP1 interacts with the WD40 and kinase domain of LRRK2. Cell lysates from HEK 293T cells cotransfected with V5–ArfGAP1 and flag-tagged full-length (FL) LRRK2 and LRRK2 fragments (F1–F8) were subjected to coimmunoprecipitation with anti-V5, followed by anti-flag immunoblotting or by anti-V5 to show an equivalent amount of input. A schematic representation of the different LRRK2 fragments used is shown. C, ArfGAP (1–64 aa) domain interacts with the WD40 and kinase domain of LRRK2. Cell lysates from HEK 293T cells cotransfected with V5–ArfGAP (1–64 aa) and flag-tagged LRRK2 fragments were subjected to coimmunoprecipitation with anti-V5, followed by anti-flag immunoblotting or by anti-V5 to show an equivalent amount of input. WB, Western blot.
ArfGAP1 is a GAP for LRRK2 that regulates LRRK2 toxicity
To ascertain whether ArfGAP1 acts as a GAP for LRRK2, the effect of ArfGAP1 on LRRK2 GTP hydrolysis was measured by a colorimetric assay for free Pi release after GTP hydrolysis (Xiong et al., 2010). For these studies, we used recombinant LRRK2 to avoid the potential copurification of LRRK2-associated GAPs and GEFs that may be present after IP of LRRK2 (Xiong et al., 2010). Under these conditions, recombinant ArfGAP1 enhances the GTP hydrolysis of recombinant WT LRRK2 by ∼2.5-fold. As reported previously, GTP hydrolysis activity of recombinant R1441C LRRK2 is reduced compared with recombinant WT LRRK2 (Lewis et al., 2007; Li et al., 2007). Surprisingly, ArfGAP1 enhances the GTP hydrolysis of R1441C LRRK2 by almost twofold. Unexpectedly, recombinant G2019S LRRK2 also has reduced GTP hydrolysis activity that is also enhanced nearly twofold by ArfGAP1. As expected, ArfGAP1 enhances the GTP hydrolysis activity of ARF1 (Fig. 3A). Δ64–ArfGAP1, which fails to bind LRRK2, has no effect on WT, R1441C, or G2019S LRRK2 or ARF1 GTP hydrolysis (Fig. 3A). The effect of ArfGAP1 on LRRK2 toxicity was monitored by transient transfection of ArfGAP1 and LRRK2 into primary murine cortical cultures. Toxicity was assessed by measuring neuronal morphology and TUNEL labeling (Fig. 3B–E). Toxicity of WT, R1441C, and G2019S LRRK2 are rescued by coexpression of ArfGAP1, whereas Δ64–ArfGAP1 is mildly protective. Unexpectedly, ArfGAP1 itself has intrinsic toxicity to cortical neurons. Δ64–ArfGAP1 blocks ArfGAP1 toxicity (Fig. 3B–E). Coexpression of LRRK2 and ArfGAP1 did not have significant effects on exogenous LRRK2 expression levels, and the coexpression did not lead to any substantial cellular redistribution (Fig. 3F,G). Thus, LRRK2 and ArfGAP1 expressed individually are toxic to cortical neurons, but, when coexpressed, LRRK2 and ArfGAP1 protect against each other's neurotoxicity. The partial rescue of LRRK2 toxicity by Δ64–ArfGAP1 suggests that LRRK2 toxicity may involve ArfGAP1. To test this hypothesis, ArfGAP1 was knocked down by lenti-shRNA to ArfGAP1 (LV.shRNA–ArfGAP1). The levels of ArfGAP1 are reduced by ∼90% by LV.shRNA–ArfGAP1 (Fig. 4A,B). WT LRRK2 and G2019S LRRK2 toxicity are significantly reduced after knockdown of ArfGAP1 expression (Fig. 4C,D). Exogenous LRRK2 expression levels and the cellular localization are not significantly changed with downregulation of ArfGAP1 (Fig. 4E,F). To assess the effect of LRRK2 deficiency on ArfGAP1 toxicity, ArfGAP1 was overexpressed in WT and LRRK2 KO neurons. No significant difference was observed on ArfGAP1 toxicity in WT and LRRK2 KO neurons (Fig. 4G,H). Thus, ArfGAP1 toxicity is unlikely to directly involve LRRK2.
Figure 3.

ArfGAP1 is a GAP for LRRK2 that regulates LRRK2 toxicity. A, ArfGAP1 increases GTP hydrolysis of WT and mutant LRRK2. GTP hydrolysis was determined by measuring the concentration of free Pi release from GTP for recombinant WT, G2019S (GS), R1441C (RC) LRRK2, and Arf1 incubated with recombinant ArfGAP1 or Δ64–ArfGAP1. Arf1 is a positive control. GTP hydrolysis activity is expressed as Pi release as a percentage of WT LRRK2 activity, with each bar representing the mean ± SEM from three independent experiments (*p < 0.05, **p < 0.01). B, ArfGAP1 and LRRK2 protect against each other's neuronal toxicity. Representative high-power fluorescent images (GFP) showing mouse primary cortical neurons cotransfected with LRRK2 and ArfGAP1–GFP or Δ64–ArfGAP1–GFP at a 10:1 molar ratio. Neuronal viability was analyzed at 48 h after transfection (DIV 12) with nonviable neurons exhibiting no obvious neurite process and positive TUNEL staining (arrow). C–E, Quantification of neuronal viability. Bars indicate the viability of GFP-positive neurons (n = 200) for each transfection condition expressed as a percentage of control neurons cotransfected with GFP and pcDNA3.1. Data represent the mean ± SEM from three independent experiments. Data were analyzed for statistical significance by two-way ANOVA (*p < 0.05, **p < 0.01). F, Exogenous LRRK2 expression levels were not significantly changed with overexpression of ArfGAP1. Cell lysates from primary cortical neurons cotransfected with LRRK2–Myc and ArfGAP1–GFP or Δ64–ArfGAP1–GFP were subjected to immunoblotting with anti-Myc or with anti-actin to show an equivalent amount of input. Data represent the mean ± SEM from three independent experiments. ns, Nonsignificant. G, Exogenous LRRK2 cellular localization was not significantly changed with overexpression of ArfGAP1. Primary cortical neurons cotransfected with LRRK2–Myc and ArfGAP1–GFP or Δ64–ArfGAP1–GFP were stained with anti-Myc and anti-GFP antibodies, followed by detection of secondary antibodies conjugated to Alexa Fluor-488 (GFP, green) and Alexa Fluor-594 (LRRK2, red). Enlarged images (highlighted by the white dashed box in the left LRRK2 panels) for exogenous LRRK2 staining are shown on the right panel for LRRK2. Scale bars: 20 μm; high-power images, 5 μm. WB, Western blot.
Figure 4.

Knockdown of ArfGAP1 expression reduces LRRK2 toxicity. A, Total cell lysates from NIH 3T3 cells transduced by GFP-tagged LV.shRNA GAPDH, LV.shRNA ArfGAP1-1, LV.shRNA ArfGAP1-2, and LV.shRNA ArfGAP1-3 for 5 d were subjected to immunoblotting with anti-ArfGAP1 and anti-tubulin antibodies. B, Quantification of ArfGAP1 levels normalized (nanograms) to the percentage of cells treated with LV.shRNA GAPDH. Data are representative of three independent experiments. Bars represent the mean ± SEM. Data were analyzed for statistical significance by Student's t test (*p < 0.05). LV.shRNA–ArfGAP1-2 was chosen for future studies. C, Representative fluorescent images (red) showing mouse primary cortical neurons transduced with GFP-tagged LV.shRNA GAPDH or LV.shRNA ArfGAP1 for 3 d (>95% neurons are GFP positive) before cotransfection with LRRK2 and mCherry at a 10:1 molar ratio. Neuronal viability was analyzed 48 h after transfection (DIV 12) with nonviable neurons exhibiting no obvious neurite process (arrow) (GS, G2019S; DA, D1994A). D, Quantification of neuronal viability. Bars indicate the viability of mCherry-positive neurons (n = 200) for each transfection condition expressed as a percentage of control neurons (mCherry with pcDNA3.1 empty vector). Data represent the mean ± SEM from three independent experiments. Data were analyzed for statistical significance by the Student's t test (*p < 0.05). E, Exogenous LRRK2 expression levels were not significantly changed with downregulation of ArfGAP1. Cell lysates were collected from primary cortical neurons transduced with LV.shRNA GAPDH or LV.shRNA ArfGAP1 for 3 d before cotransfection with LRRK2 and mCherry at a 10:1 molar ratio for 48 h. Cell lysates were subjected to immunoblotting with anti-Myc or with anti-actin to show an equivalent amount of input. Data represent the mean ± SEM from three independent experiments. ns, Nonsignificant. F, Exogenous LRRK2 cellular localization was not significantly changed with downregulation of ArfGAP1. Primary cortical neurons transduced with LV.shRNA GAPDH or LV.shRNA ArfGAP1 for 3 d before cotransfection with LRRK2 and mCherry at a 10:1 molar ratio for 48 h were stained with anti-Myc and anti-GFP antibodies, followed by detection of secondary antibodies conjugated to Alexa Fluor-488 (GFP, green) and Alexa Fluor-405 (LRRK2, blue). Enlarged images (highlighted by the white dashed box in the left LRRK2 panels) for exogenous LRRK2 staining are shown on the right panel for LRRK2. Scale bars: 20 μm; high-power images, 5 μm. G, No significant difference was observed on ArfGAP1 toxicity in WT and LRRK2 KO neurons. Representative high-power fluorescent images (GFP) showing WT and LRRK2 KO mouse primary cortical neurons transfected with ArfGAP1–GFP. Neuronal viability was analyzed at 48 h after transfection (DIV 12) with nonviable neurons exhibiting no obvious neurite process and positive TUNEL staining (arrow). H, Quantification of neuronal viability. Bars indicate the viability of GFP-positive neurons (n = 200) for each transfection condition expressed as a percentage of control neurons cotransfected with GFP alone. Data represent the mean ± SEM from three independent experiments. WB, Western blot.
LRRK2 phosphorylates ArfGAP1, resulting in inhibition of the GAP activity of ArfGAP1
The ability of LRRK2 and ArfGAP1 to cancel each other's toxicity suggested that LRRK2 could regulate the activity of ArfGAP1. Accordingly, we determined whether LRRK2 could phosphorylate and regulate the activity of ArfGAP1. WT LRRK2 phosphorylates ArfGAP1. This phosphorylation is twofold greater with G2019S LRRK2 (Fig. 5A,B). Accompanying the phosphorylation of ArfGAP1 by WT and G2019S LRRK2 is a reduction in the autophosphorylation of WT and G2019S LRRK2 (Fig. 5A,C). We added MBP, a generic kinase substrate to assess LRRK2 autophosphorylation and kinase activity compared with LRRK2 alone and LRRK2 with ArfGAP1. We find that ArfGAP1 does not nonspecifically reduce LRRK2 kinase activity because MBP does not affect LRRK2 autophosphorylation and kinase activity (Fig. 5A,C). ArfGAP1 also reduces LRRK2 phosphorylation of MBP (Fig. 5A,D). The phosphorylation of ArfGAP1 by LRRK2 was subjected to mass spectrometry to determine the sites of phosphorylation. There was 95.3% coverage, and serines (S155, S246, and S284) and threonine (T189, T216, and T292) are phosphorylated by LRRK2 (Fig. 5E). Mutational analysis of these serine and threonine amino acids to alanine reveals that no single amino acid is the predominant phospho-amino acid. Mutation of all six phosphorylated amino acids is required to substantially reduce LRRK2-mediated phosphorylation of ArfGAP1 (Fig. 5F,G). The functional consequences of LRRK2 phosphorylation of ArfGAP1 was monitored by assessing the effects of ArfGAP1 on ARF1 GTP hydrolysis. WT and G2019S LRRK2 significantly reduce the ability of ArfGAP1 to enhance ARF1 GTP hydrolysis activity, whereas a kinase-dead LRRK2 has no effect. An ArfGAP1 mutant with all six potential LRRK2 phosphorylated amino acids mutated to alanine (ArfGAP1-6A) enhances ARF1 GTP hydrolysis activity, which is not altered by WT or G2019S LRRK2 (Fig. 5H). Δ64–ArfGAP1 reduces ArfGAP1 enhancement of ARF1 GTP hydrolysis activity, suggesting that Δ64–ArfGAP1 may function as a dominant negative. To assess the phosphorylation state of ArfGAP1 by LRRK2 in vivo, the phosphorylation levels of ArfGAP1 were detected in WT and LRRK2 KO mouse brain lysates. ArfGAP1 has significantly lower phosphorylation levels in LRRK2 KO mouse brain (29.64 ± 13.34) versus WT mouse brain (100 ± 10.54) (Fig. 5I).
Figure 5.
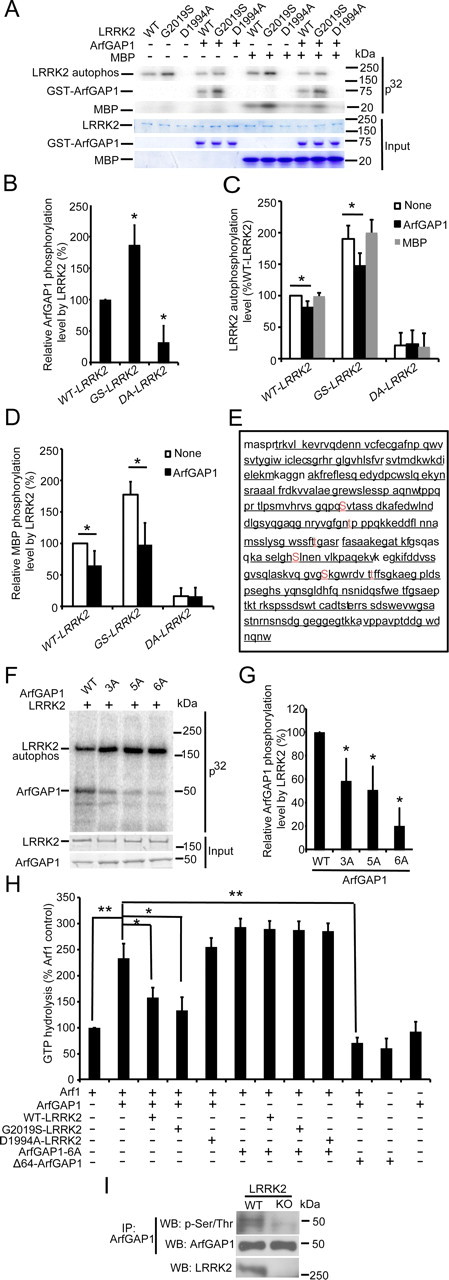
LRRK2 phosphorylates ArfGAP1, resulting in inhibition of the GAP activity of ArfGAP1. A, LRRK2 phosphorylates ArfGAP1. A total of 40 ng of WT, G2019S (GS), and D1994A (DA) LRRK2 recombinant proteins were incubated with or without recombinant ArfGAP1 or MBP protein and subjected to an in vitro kinase assay. Equivalent input is shown by Coomassie blue staining for LRRK2, ArfGAP1, and MBP. B, Quantification of relative ArfGAP1 phosphorylation level by LRRK2 via densitometric analysis. Bars indicate ArfGAP1 phosphorylation level as a percent of ArfGAP1 phosphorylation level by WT LRRK2, with each bar representing the mean ± SEM from three independent experiments (*p < 0.01). C, Quantification of LRRK2 autophosphorylation level via densitometric analysis. Bars indicate LRRK2 autophosphorylation level incubated with or without ArfGAP1 or MBP as a percentage of WT LRRK2 alone, with each bar representing the mean ± SEM from three independent experiments (*p ≤ 0.05). D, Quantification of relative MBP phosphorylation level by LRRK2 with or without ArfGAP1 via densitometric analysis. Bars indicate MBP phosphorylation level as a percentage of MBP phosphorylation level by WT LRRK2, with each bar representing the mean ± SEM from three independent experiments (*p < 0.05). E, ArfGAP1 is phosphorylated by LRRK2 at serines (S155, S246, and S284) and threonine (T189, T216, and T292). Mass spectrometric analysis reveals 95.3% sequence coverage of ArfGAP1. Phosphorylated sites are indicated in red. F, Mutations of three, five, or all six phospho-amino acids to alanine (ArfGAP1-3A, ArfGAP1-5A, and ArfGAP1-6A) substantially reduce LRRK2-mediated phosphorylation of ArfGAP1. A total of 40 ng of recombinant G2019S LRRK2 protein was incubated with recombinant WT or mutant ArfGAP1 proteins and subjected to an in vitro kinase assay. Equivalent input is shown by Coomassie blue staining. G, Quantification of relative ArfGAP1 phosphorylation level by LRRK2 via densitometric analysis. Bars indicate ArfGAP1 phosphorylation level as a percentage of WT ArfGAP1, with each bar representing the mean ± SEM from three independent experiments (*p < 0.05). H, LRRK2 phosphorylation of ArfGAP1 inhibits the ability of ArfGAP1 to enhance ARF1 GTP hydrolysis. Glutathione beads bound with WT or six-point mutation ArfGAP1 (ArfGAP1-6A) recombinant proteins were incubated with WT, G2019S, and D1994A LRRK2 recombinant proteins and subjected to an in vitro kinase assay without [γ-32P]ATP, followed by stringent washes to remove LRRK2. The beads were subsequently incubated with Arf1 recombinant protein and subjected to an in vitro GTPase assay by measuring the concentration of free Pi release from GTP for Arf1. Δ64–ArfGAP1 was incubated with ArfGAP1 and Arf1 and subjected to an in vitro GTPase assay as well. GTP hydrolysis activity is expressed as Pi release as a percentage of Arf1 alone, with each bar representing the mean ± SEM from three independent experiments (*p < 0.05, **p < 0.01). I, ArfGAP1 has significantly lower phosphorylation levels in LRRK2 KO mouse brain (29.64 ± 13.34) versus WT mouse brain (100 ± 10.54). Whole-brain lysates prepared from WT and LRRK2 KO mice were subjected to IP with anti-ArfGAP1, followed by anti-phosphoserine/phosphothreonine and anti-ArfGAP1 immunoblotting.
ArfGAP1 and LRRK2 genetically interact and regulate each other's toxicity in vivo
Using WT and G2019S LRRK2 Drosophila that exhibit a parkinsonism-like phenotype (Liu et al., 2008), the effects of ArfGAP1 on LRRK2 were explored. ArfGAP1 and LRRK2 were expressed in dopaminergic neurons in which the UAS–WT–LRRK2 or UAS–G2019S–LRRK2 transgene and UAS–ArfGAP1 transgene along with UAS–GFP were driven by TH–GAL4 driver (Friggi-Grelin et al., 2003). Dopaminergic neuron number as revealed by GFP fluorescence was monitored in the six dopaminergic neuronal clusters that are normally present in Drosophila adult brain hemisphere (PAL, PPM1/2, PPM3, PPL1, and PPL2) (Budnik and White, 1988; Friggi-Grelin et al., 2003) (Fig. 6A). As reported previously (Liu et al., 2008; Ng et al., 2009), overexpression of WT and G2019S LRRK2 leads to a substantial reduction of dopamine neurons in PAL, PPM1/2, PPM3, and PPL1 compared with the GFP control. Coexpression of ArfGAP1 significantly prevents the loss of dopamine neurons in PAL and PPM1/2 but not PPM3 or PPL1. The expression of Δ64–ArfGAP1 also rescues WT or G2019S LRRK2 dopaminergic neuronal loss in the PPM1/2 but does not significantly rescue in the other dopaminergic clusters (Fig. 6B,C; Tables 1, 2). Similar to the results in cortical cultures (Fig. 3B–E), ArfGAP1 itself causes dopaminergic neuronal loss (Fig. 6B,C). A time course analysis of loss of the dopaminergic neuronal cluster PPM1/2 was conducted from 1 to 7 weeks of age, revealing a progressive loss of dopaminergic neurons as reported previously (Fig. 6D) (Liu et al., 2008). ArfGAP1 rescues the loss of dopaminergic neurons induced by WT or G2091S LRRK2 beginning at 5 weeks of age. The expression of Δ64–ArfGAP1 also rescues WT or G2019S LRRK2 dopaminergic neuronal loss in the PPM1/2 cluster. ArfGAP1 itself causes dopaminergic neuronal loss, which begins at 5 weeks of age (Fig. 6D). These data indicate that ArfGAP1 is not causing loss of dopamine neurons during development.
Figure 6.
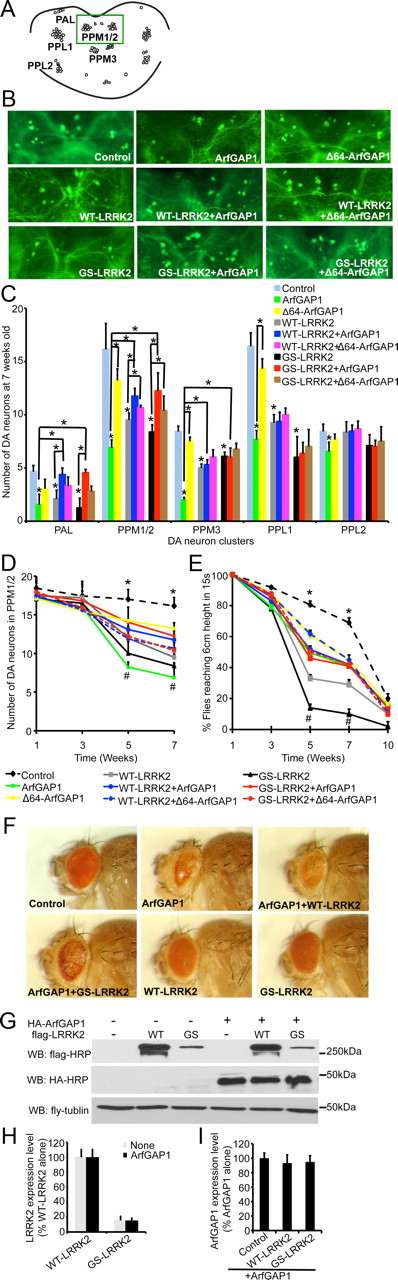
ArfGAP1 and LRRK2 genetically interact and regulate each other's toxicity in vivo. A, Diagram of dopaminergic neuronal clusters in the posterior areas of the adult fly brain. Green box shows the area selected for the images shown in B. B, Representative fluorescent images (GFP) of dopamine neurons in PPM1/2 clusters from 7-week-old flies of the indicated genotypes (green dots are neuron cell bodies). UAS–WT–LRRK2 or UAS–G2019S (GS) LRRK2, UAS–ArfGAP1 or UAS–Δ64–ArfGAP1 along with UAS–GFP were expressed in dopamine neurons by the TH–GAL4 driver. TH–GAL4>UAS–GFP was used as the control. C, Average numbers of dopamine neurons per DA cluster in 7-week- old flies of the indicated genotypes. Data represent the mean ± SEM from three independent experiments. Data were analyzed for statistical significance by two-way ANOVA (*p ≤ 0.05). D, Average numbers of dopamine neurons in PPM1/2 cluster of the flies of the indicated genotypes at different ages. Data represent the mean ± SEM from three independent experiments. Data were analyzed for statistical significance by two-way ANOVA (*p ≤ 0.05 statistically significant differences between the control and ArfGAP1, WT–LRRK2, or GS–LRRK2; #p ≤ 0.05 statistically significant differences between GS–LRRK2 and ArfGAP1+GS–LRRK2, GS–LRRK2 and Δ64–ArfGAP1+GS–LRRK2, ArfGAP1 and ArfGAP1+GS–LRRK2, WT–LRRK2 and ArfGAP1+WT–LRRK2, WT–LRRK2 and Δ64–ArfGAP1+WT–LRRK2, ArfGAP1 and ArfGAP1+WT–LRRK2, and ArfGAP1 and Δ64–ArfGAP1). E, Six groups of 20 flies from each genotype were subjected to the climbing assay every 2 weeks from 1 to 10 weeks. Data represent the mean ± SEM from three independent experiments. Data were analyzed for statistical significance by two-way ANOVA (*p < 0.05 statistically significant differences between the control and all other lines as indicated; #p < 0.05 statistically significant differences between GS–LRRK2 and ArfGAP1+GS–LRRK2, GS–LRRK2 and Δ64–ArfGAP1+GS–LRRK2, ArfGAP1 and ArfGAP1+GS–LRRK2, WT–LRRK2 and ArfGAP1+WT–LRRK2, WT–LRRK2 and Δ64–ArfGAP1+WT–LRRK2, ArfGAP1 and ArfGAP1+WT–LRRK2, ArfGAP1 and Δ64–ArfGAP1). F, Eye morphology of 1-week-old flies of the indicated genotypes. UAS–WT–LRRK2 or UAS–GS–LRRK2, UAS–ArfGAP1 were expressed in fly eyes by GMR–GAL4 driver. GMR–GAL4 is uses as the control. G, Coexpression of LRRK2 and ArfGAP1 in fly does not have significant effects on LRRK2 or ArfGAP1 expression levels. Lysates prepared form whole heads of 1-week-old flies of the indicated genotypes were subjected to immunoblotting with anti-flag–HRP, anti-HA–HRP, and anti-fly tubulin. H, Quantification of exogenous LRRK2 expression levels in fly heads. Data represent the mean ± SEM from three independent experiments. I, Quantification of exogenous ArfGAP1 expression levels in fly heads. Data represent the mean ± SEM from three independent experiments. WB, Western blot.
Table 1.
Average number of dopamine neurons per DA cluster in 7-week-old flies of the indicated genotypes
| Control | ArfGAP1 | Δ64–ArfGAP1 | WT–LRRK2 | WT–LRRK2 + ArfGAP1 | WT–LRRK2 + Δ64–ArfGAP1 | GS–LRRK2 | GS–LRRK2 + ArfGAP1 | GS–LRRK2 + Δ64–ArfGAP1 | |
|---|---|---|---|---|---|---|---|---|---|
| PAL | 4.7 ± 0.79 | 1.6 ± 1.48 | 3.0 ± 1.40 | 2.1 ± 0.81 | 4.4 ± 0.63 | 3.3 ± 0.82 | 1.3 ± 0.91 | 4.6 ± 0.32 | 2.8 ± 0.5 |
| PPM1/2 | 16.1 ± 2.4 | 6.9 ± 0.75 | 14.6 ± 2.09 | 9.4 ± 0.45 | 11.8 ± 0.73 | 10.7 ± 0.16 | 8.4 ± 0.69 | 12.3 ± 1.68 | 10.4 ± 1.36 |
| PPM3 | 8.4 ± 0.51 | 2 ± 0.24 | 7.5 ± 0.39 | 5 ± 0.39 | 5.3 ± 0.47 | 6.0 ± 0.68 | 6.1 ± 0.40 | 6.0 ± 0.87 | 6.8 ± 0.57 |
| PPL1 | 16.4 ± 2.62 | 7.7 ± 1.85 | 14.8 ± 2.41 | 9.3 ± 1.76 | 9.1 ± 1.87 | 10.0 ± 1.63 | 6.0 ± 2.94 | 6.4 ± 2.04 | 7.0 ± 2.60 |
| PPL2 | 8.4 ± 0.69 | 6.6 ± 0.85 | 7.7 ± 0.51 | 8.3 ± 0.99 | 8.1 ± 0.57 | 8.7 ± 0.73 | 7.1 ± 1.04 | 7.0 ± 0.60 | 7.5 ± 1.37 |
Data are representative of eight independent experiments.
Table 2.
p values of the data in Table 1 analyzed for statistical significance by two-way ANOVA
| Control versus ArfGAP1 | Control versus Δ64-ArfGAP1 | Control versus WT-LRRK2 | Control versus GS-LRRK2 | ArfGAP1 versus Δ64-ArfGAP1 | ArfGAP1 versus WT-LRRK2 + ArfGAP1 | ArfGAP1 versus GS-LRRK2 + ArfGAP1 | ArfGAP1 versus GS-LRRK2 + ArfGAP1 | WT-LRRK2 versus WT-LRRK2 + ArfGAP1 | WT-LRRK2 versus WT-LRRK2+ Δ64-ArfGAP1 | GS-LRRK2 versus GS-LRRK2 + ArfGAP1 | GS-LRRK2 versus GS-LRRK2+ Δ64-ArfGAP1 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PAL | p < 0.001 | p > 0.05 | p < 0.001 | p < 0.001 | p > 0.05 | p < 0.001 | p < 0.001 | p < 0.001 | p < 0.001 | p > 0.05 | p < 0.001 | p > 0.05 |
| PPM1/2 | p < 0.001 | p > 0.05 | p < 0.001 | p < 0.001 | p < 0.001 | p < 0.001 | p < 0.001 | p < 0.001 | p < 0.001 | p = 0.05 | p < 0.001 | p < 0.05 |
| PPM3 | p < 0.001 | p > 0.05 | p < 0.001 | p < 0.01 | p < 0.001 | p < 0.001 | p < 0.001 | p < 0.001 | p > 0.05 | p > 0.05 | p > 0.05 | p > 0.05 |
| PPL1 | p < 0.001 | p > 0.05 | p < 0.001 | p < 0.001 | p < 0.001 | p > 0.05 | p > 0.05 | p > 0.05 | p > 0.05 | p > 0.05 | p > 0.05 | p > 0.05 |
| PPL2 | p < 0.01 | p > 0.05 | p > 0.05 | p > 0.05 | p > 0.05 | p > 0.05 | p > 0.05 | p > 0.05 | p > 0.05 | p > 0.05 | p > 0.05 | p > 0.05 |
Locomotor activity was monitored by a climbing assay (negative geotaxis test). Nearly all control flies (TH–GAL4>UAS–GFP) at 5 and 7 weeks of age rapidly climb to the 6 cm mark, whereas WT LRRK2 and G2019S LRRK2 flies have significantly reduced climbing ability. ArfGAP1 and Δ64–ArfGAP1 rescues the climbing defect in the WT and G2019S LRRK2 flies. Expression of either ArfGAP1 or Δ64–ArfGAP1 mildly reduces the climbing ability of the flies (Fig. 6E).
To confirm that LRRK2 reciprocally regulates ArfGAP1 in vivo, retinal degeneration was monitored by light microscopy to reveal eye morphologies in flies expressing the GMR–GAL4 driver. Expression of ArfGAP1 leads to a profound eye phenotype that is rescued by coexpression of WT or G2019S LRRK2 (Fig. 6F). Coexpression of LRRK2 and ArfGAP1 in fly does not have significant effects on LRRK2 or ArfGAP1 expression levels (Fig. 6G–I).
Discussion
LRRK2 toxicity is generally thought to be driven by its kinase activity (West et al., 2005; Greggio et al., 2006; Smith et al., 2006; Guo et al., 2007). Using truncated fragments of LRRK2 containing the GTPase domain, we showed previously that enhancing LRRK2 GTP hydrolysis was protective against LRRK2 toxicity, whereas reducing LRRK2 GTP hydrolysis enhanced LRRK2 toxicity (Xiong et al., 2010). Although GTP binding appears to play a role in full-length LRRK2 toxicity, the role of GTP hydrolysis in the context of full-length LRRK2 is not known (Smith et al., 2006; Xiong et al., 2010). We confirm and extend these findings and show that the GTPase activity of full-length LRRK2 also plays an important role in LRRK2 toxicity both in vitro and in vivo. The familial mutations R1441C and R1441G, which are toxic, have decreased GTP hydrolysis, consistent with the idea that the GTPase activity of LRRK2 may play a role in LRRK2 toxicity (Lewis et al., 2007; Li et al., 2007). Unexpectedly, we show here that G2019S LRRK2 also has decreased GTP hydrolysis. Moreover, we identify ArfGAP1 as the GAP for LRRK2. Consistent with the notion that the GTPase activity of LRRK2 plays a role in LRRK2 toxicity is our observation that ArfGAP1 as a GAP for LRRK2 enhances both WT and mutant LRRK2 GTP hydrolysis and protects against LRRK2 toxicity in vitro and in vivo.
Unexpectedly, LRRK2 phosphorylates ArfGAP1 and inhibits its GAP activity, providing a reciprocal and novel mode of regulation between LRRK2 and ArfGAP1 (Fig. 7). The phosphorylation of ArfGAP1 by LRRK2 inhibits its GAP activity because the enhancement of GTP hydrolysis of ARF1 by ArfGAP1 is reduced in the presence of WT or G2019S LRRK2. Moreover, WT and G2019S autophosphorylation and kinase activity are significantly reduced in the presence of ArfGAP1. This reciprocal regulation leads to complex effects on cellular toxicity. WT and mutant LRRK2 toxicity is reduced by ArfGAP1. In addition, Δ64–ArfGAP1, which has no effect on LRRK2 GTP hydrolysis but inhibits ARF1 GTP hydrolysis, partially protects against LRRK2 toxicity, suggesting a possible direct role of ArfGAP1 in LRRK2 toxicity. Consistent with this idea is the observation that knockdown of ArfGAP1 partially protects against LRRK2 toxicity. In addition, the yeast homolog of ArfGAP1, GCS1, was identified in a suppressor screen in which the absence of GCS1 protects against LRRK2 toxicity in yeast (Xiong et al., 2010). Surprisingly, overexpression of ArfGAP1 alone was found to be toxic both in vitro and in vivo. The mechanism of ArfGAP1 toxicity requires additional study, but coexpression of LRRK2 inhibits ArfGAP1 toxicity in a manner similar to its inhibition of ArfGAP1 activity. Our findings in Drosophila confirm the reciprocal regulation of ArfGAP1 and LRRK2 activity.
Figure 7.

Model of reciprocal regulation between ArfGAP1 and LRRK2. Individually, ArfGAP1 or LRRK2 induces cell death. When expressed together, ArfGAP1 binds to LRRK2, promoting hydrolysis of GTP to GDP and decreased autophosphorylation and kinase activity of LRRK2. LRRK2 phosphorylates ArfGAP1, inhibiting its GAP activity. This reciprocal regulation leads to complex effects on cellular viability. Together, the actions of ArfGAP1 and LRRK2 promote cell survival.
In summary, we report the identification of ArfGAP1 as the first GAP for LRRK2 that regulates LRRK2 GTP hydrolysis and toxicity. In addition, ArfGAP1 is a substrate for LRRK2 whose activity is unexpectedly regulated by LRRK2. Together, our results indicate that the GTPase domain of LRRK2 contributes to LRRK2 toxicity. In theory, agents that act to increase LRRK2 GTP hydrolysis could be alternative therapeutic agents to kinase inhibitors for the treatment of Parkinson's disease. However, because of the complex regulation and the potential role of ArfGAP1 in LRRK2 toxicity revealed in this study, care would need to taken in the development of such agents.
Footnotes
This work was supported by National Institutes of Health NS38377 and the Michael J. Fox Foundation. Y.X. was an American Parkinson's Disease Association Postdoctoral Fellow. T.M.D. is the Leonard and Madlyn Abramson Professor in Neurodegenerative Diseases. We thank Dr. Wanli Smith (University of Maryland, Baltimore, MD) for the generous gift of the LRRK2 flies. We are grateful to Dr. Duojia Pan and Dr. Jianzhong Yu (Johns Hopkins University, Baltimore, MD) for fly technical advice. We acknowledge the joint participation by the Adrienne Helis Malvin Medical Research Foundation through its direct engagement in the continuous active conduct of medical research in conjunction with Johns Hopkins Hospital and Johns Hopkins University School of Medicine and Foundation's Parkinson's Disease Program M-1. We thank San-Xia Wang and Dr. Joo-ho Shin for assistance.
The authors declare no competing financial interests.
References
- Barr and Lambright, 2010.Barr F, Lambright DG. Rab GEFs and GAPs. Curr Opin Cell Biol. 2010;22:461–470. doi: 10.1016/j.ceb.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biskup et al., 2006.Biskup S, Moore DJ, Celsi F, Higashi S, West AB, Andrabi SA, Kurkinen K, Yu SW, Savitt JM, Waldvogel HJ, Faull RL, Emson PC, Torp R, Ottersen OP, Dawson TM, Dawson VL. Localization of LRRK2 to membranous and vesicular structures in mammalian brain. Ann Neurol. 2006;60:557–569. doi: 10.1002/ana.21019. [DOI] [PubMed] [Google Scholar]
- Budnik and White, 1988.Budnik V, White K. Catecholamine-containing neurons in Drosophila melanogaster: distribution and development. J Comp Neurol. 1988;268:400–413. doi: 10.1002/cne.902680309. [DOI] [PubMed] [Google Scholar]
- Cookson, 2010.Cookson MR. The role of leucine-rich repeat kinase 2 (LRRK2) in Parkinson's disease. Nat Rev Neurosci. 2010;11:791–797. doi: 10.1038/nrn2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng et al., 2011.Deng X, Dzamko N, Prescott A, Davies P, Liu Q, Yang Q, Lee JD, Patricelli MP, Nomanbhoy TK, Alessi DR, Gray NS. Characterization of a selective inhibitor of the Parkinson's disease kinase LRRK2. Nat Chem Biol. 2011;7:203–205. doi: 10.1038/nchembio.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- East and Kahn, 2011.East MP, Kahn RA. Models for the functions of Arf GAPs. Semin Cell Dev Biol. 2011;22:3–9. doi: 10.1016/j.semcdb.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friggi-Grelin et al., 2003.Friggi-Grelin F, Coulom H, Meller M, Gomez D, Hirsh J, Birman S. Targeted gene expression in Drosophila dopaminergic cells using regulatory sequences from tyrosine hydroxylase. J Neurobiol. 2003;54:618–627. doi: 10.1002/neu.10185. [DOI] [PubMed] [Google Scholar]
- Greggio et al., 2006.Greggio E, Jain S, Kingsbury A, Bandopadhyay R, Lewis P, Kaganovich A, van der Brug MP, Beilina A, Blackinton J, Thomas KJ, Ahmad R, Miller DW, Kesavapany S, Singleton A, Lees A, Harvey RJ, Harvey K, Cookson MR. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis. 2006;23:329–341. doi: 10.1016/j.nbd.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Guo et al., 2007.Guo L, Gandhi PN, Wang W, Petersen RB, Wilson-Delfosse AL, Chen SG. The Parkinson's disease-associated protein, leucine-rich repeat kinase 2 (LRRK2), is an authentic GTPase that stimulates kinase activity. Exp Cell Res. 2007;313:3658–3670. doi: 10.1016/j.yexcr.2007.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haebig et al., 2010.Haebig K, Gloeckner CJ, Miralles MG, Gillardon F, Schulte C, Riess O, Ueffing M, Biskup S, Bonin M. ARHGEF7 (Beta-PIX) acts as guanine nucleotide exchange factor for leucine-rich repeat kinase 2. PLoS One. 2010;5:e13762. doi: 10.1371/journal.pone.0013762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko et al., 2009.Ko HS, Bailey R, Smith WW, Liu Z, Shin JH, Lee YI, Zhang YJ, Jiang H, Ross CA, Moore DJ, Patterson C, Petrucelli L, Dawson TM, Dawson VL. CHIP regulates leucine-rich repeat kinase-2 ubiquitination, degradation, and toxicity. Proc Natl Acad Sci U S A. 2009;106:2897–2902. doi: 10.1073/pnas.0810123106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee et al., 2010.Lee BD, Shin JH, VanKampen J, Petrucelli L, West AB, Ko HS, Lee YI, Maguire-Zeiss KA, Bowers WJ, Federoff HJ, Dawson VL, Dawson TM. Inhibitors of leucine-rich repeat kinase-2 protect against models of Parkinson's disease. Nat Med. 2010;16:998–1000. doi: 10.1038/nm.2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis et al., 2007.Lewis PA, Greggio E, Beilina A, Jain S, Baker A, Cookson MR. The R1441C mutation of LRRK2 disrupts GTP hydrolysis. Biochem Biophys Res Commun. 2007;357:668–671. doi: 10.1016/j.bbrc.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li et al., 2007.Li X, Tan YC, Poulose S, Olanow CW, Huang XY, Yue Z. Leucine-rich repeat kinase 2 (LRRK2)/PARK8 possesses GTPase activity that is altered in familial Parkinson's disease R1441C/G mutants. J Neurochem. 2007;103:238–247. doi: 10.1111/j.1471-4159.2007.04743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu et al., 2008.Liu Z, Wang X, Yu Y, Li X, Wang T, Jiang H, Ren Q, Jiao Y, Sawa A, Moran T, Ross CA, Montell C, Smith WW. A Drosophila model for LRRK2-linked parkinsonism. Proc Natl Acad Sci U S A. 2008;105:2693–2698. doi: 10.1073/pnas.0708452105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu et al., 2011.Liu Z, Hamamichi S, Lee BD, Yang D, Ray A, Caldwell GA, Caldwell KA, Dawson TM, Smith WW, Dawson VL. Inhibitors of LRRK2 kinase attenuate neurodegeneration and Parkinson-like phenotypes in Caenorhabditis elegans and Drosophila Parkinson's disease models. Hum Mol Genet. 2011;20:3933–3942. doi: 10.1093/hmg/ddr312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mata et al., 2006.Mata IF, Wedemeyer WJ, Farrer MJ, Taylor JP, Gallo KA. LRRK2 in Parkinson's disease: protein domains and functional insights. Trends Neurosci. 2006;29:286–293. doi: 10.1016/j.tins.2006.03.006. [DOI] [PubMed] [Google Scholar]
- Ng et al., 2009.Ng CH, Mok SZ, Koh C, Ouyang X, Fivaz ML, Tan EK, Dawson VL, Dawson TM, Yu F, Lim KL. Parkin protects against LRRK2 G2019S mutant-induced dopaminergic neurodegeneration in Drosophila. J Neurosci. 2009;29:11257–11262. doi: 10.1523/JNEUROSCI.2375-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paisán-Ruíz et al., 2004.Paisán-Ruíz C, Jain S, Evans EW, Gilks WP, Simón J, van der Brug M, López de Munain A, Aparicio S, Gil AM, Khan N, Johnson J, Martinez JR, Nicholl D, Carrera IM, Pena AS, de Silva R, Lees A, Martí-Massó JF, Pérez-Tur J, Wood NW, Singleton AB. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron. 2004;44:595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- Sharp et al., 1995.Sharp AH, Loev SJ, Schilling G, Li SH, Li XJ, Bao J, Wagster MV, Kotzuk JA, Steiner JP, Lo A, Hedreen J, Sisodia S, Snyder SH, Dawson TM, Ryugo DK, Ross CA. Widespread expression of Huntington's disease gene (IT15) protein product. Neuron. 1995;14:1065–1074. doi: 10.1016/0896-6273(95)90345-3. [DOI] [PubMed] [Google Scholar]
- Shin et al., 2008.Shin N, Jeong H, Kwon J, Heo HY, Kwon JJ, Yun HJ, Kim CH, Han BS, Tong Y, Shen J, Hatano T, Hattori N, Kim KS, Chang S, Seol W. LRRK2 regulates synaptic vesicle endocytosis. Exp Cell Res. 2008;314:2055–2065. doi: 10.1016/j.yexcr.2008.02.015. [DOI] [PubMed] [Google Scholar]
- Smith et al., 2005.Smith WW, Pei Z, Jiang H, Moore DJ, Liang Y, West AB, Dawson VL, Dawson TM, Ross CA. Leucine-rich repeat kinase 2 (LRRK2) interacts with parkin, and mutant LRRK2 induces neuronal degeneration. Proc Natl Acad Sci U S A. 2005;102:18676–18681. doi: 10.1073/pnas.0508052102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith et al., 2006.Smith WW, Pei Z, Jiang H, Dawson VL, Dawson TM, Ross CA. Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat Neurosci. 2006;9:1231–1233. doi: 10.1038/nn1776. [DOI] [PubMed] [Google Scholar]
- Vigil et al., 2010.Vigil D, Cherfils J, Rossman KL, Der CJ. Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy? Nat Rev Cancer. 2010;10:842–857. doi: 10.1038/nrc2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West et al., 2005.West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL, Dawson TM. Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A. 2005;102:16842–16847. doi: 10.1073/pnas.0507360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West et al., 2007.West AB, Moore DJ, Choi C, Andrabi SA, Li X, Dikeman D, Biskup S, Zhang Z, Lim KL, Dawson VL, Dawson TM. Parkinson's disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum Mol Genet. 2007;16:223–232. doi: 10.1093/hmg/ddl471. [DOI] [PubMed] [Google Scholar]
- Xiong et al., 2010.Xiong Y, Coombes CE, Kilaru A, Li X, Gitler AD, Bowers WJ, Dawson VL, Dawson TM, Moore DJ. GTPase activity plays a key role in the pathobiology of LRRK2. PLoS Genet. 2010;6:e1000902. doi: 10.1371/journal.pgen.1000902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang et al., 2011.Zhang J, Wang Y, Chi Z, Keuss MJ, Pai YM, Kang HC, Shin JH, Bugayenko A, Wang H, Xiong Y, Pletnikov MV, Mattson MP, Dawson TM, Dawson VL. The AAA+ ATPase Thorase regulates AMPA receptor-dependent synaptic plasticity and behavior. Cell. 2011;145:284–299. doi: 10.1016/j.cell.2011.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimprich et al., 2004.Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, Stoessl AJ, Pfeiffer RF, Patenge N, Carbajal IC, Vieregge P, Asmus F, Müller-Myhsok B, Dickson DW, Meitinger T, Strom TM, Wszolek ZK, Gasser T. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]