

Abstract
CREBH is a liver-specific transcription factor that is localized in the endoplasmic reticulum (ER) membrane. Our previous work demonstrated that CREBH is activated by ER stress or inflammatory stimuli to induce an acute-phase hepatic inflammation. Here we demonstrate that CREBH is a key metabolic regulator of hepatic lipogenesis, fatty acid (FA) oxidation, and lipolysis under metabolic stress. Saturated FA, insulin signals, or an atherogenic high-fat diet can induce CREBH activation in the liver. Under the normal chow diet, CrebH knockout mice display a modest decrease in hepatic lipid contents but an increase in plasma triglycerides (TG). After feeding an atherogenic high-fat diet, massive accumulation of hepatic lipid metabolites and significant increase in plasma TG levels were observed in the CrebH knockout mice. Along with the hypertriglyceridemia phenotype, the CrebH null mice displayed significantly reduced body weight gain, diminished abdominal fat, and increased non-alcoholic steatohepatitis (NASH) activities under the atherogenic high-fat diet. Gene expression analysis and chromatin-immunoprecipitation (ChIP) assay indicated that CREBH is required to activate expression of the genes encoding functions involved in de novo lipogenesis, TG and cholesterol biosynthesis, FA elongation and oxidation, lipolysis, and lipid transport. Supporting the role of CREBH in lipogenesis and lipolysis, forced expression of an activated form of CREBH protein in the liver significantly increases accumulation of hepatic lipids but reduces plasma TG levels in mice. All together our study shows that CREBH plays a key role in maintaining lipid homeostasis by regulating expression of the genes involved in hepatic lipogenesis, FA oxidation, and lipolysis under metabolic stress. The identification of CREBH as a stress-inducible metabolic regulator has important implications in the understanding and treatment of metabolic disease.
Introduction
The liver plays a key role in controlling lipid homeostasis through regulation of lipid biosynthesis, oxidation, and transport. In response to different metabolic conditions, hepatic lipid metabolism is tightly and coordinately regulated by nuclear receptors, transcription factors, and cellular enzymes (1). Metabolic signals, such as fatty acids (FA), glucose, insulin, or inflammatory cytokines, can regulate the activity and/or abundance of key transcription factors to modulate hepatic lipid metabolism (2, 3). Many hepatic transcription factors, such as sterol regulatory element binding protein-1c (SREBP-1c), liver X receptor (LXRα), peroxisome proliferator-activated receptors (PPARs), and carbohydrate-responsive element-binding protein (ChREBP), have been identified as important regulators of gene expression involved in gluconeogenesis, lipogenesis, FA oxidation, and lipid transport.
The endoplasmic reticulum (ER) is an organelle that plays important roles in stress response and cell metabolism. ER-transmembrane signaling molecules regulate lipid metabolism and/or glucose biosynthesis in response to a variety of stress stimuli (4-7). CREBH is an ER-transmembrane transcription factor of the CREB/ATF6 family that is expressed in gastrointestinal tract tissues including the liver, pyloric stomach, duodenum, and ileum (8-10). Previously we demonstrated that CREBH is activated through regulated-intramembrane proteolysis (RIP) in response to ER stress, pro-inflammatory cytokines, or bacterial endotoxin lipopolysaccharide (LPS) (11). Pro-inflammatory cytokines, such as TNFα, IL6, and IL1β, can induce both expression and activation of CREBH in liver hepatocytes. Interestingly, CREBH activates expression of genes encoding major liver acute-phase response proteins or phosphoenolpyruvate carboxykinase 1 (PCK1), but not those involved in the classical ER stress response (9, 11). More recent studies have shown that CREBH is inducible by free FA or fasting and regulated by peroxisome proliferator-activated receptor α (PPARα) (7, 12, 13). Gain- and loss- of function studies suggested that CREBH regulates hepatic gluconeogenesis by activating expression of the Pck1 and glucose-6-phosphatase (G6Pase) genes in the liver (7).
In this study, we show that CREBH is a key metabolic regulator of lipid homeostasis. Under conditions of metabolic stress, CREBH is activated to function as a transcription factor to activate the genetic programs required for lipogenesis, FA metabolism, and lipolysis. CREBH activity is required to maintain lipid equilibrium in the liver, plasma, and abdominal fat tissue. The identification of CREBH as a key hepatic lipid regulator has important implications in understanding and treatment of metabolic diseases.
Materials and Methods
Mouse experiments
The CrebH knockout mice with exons 4-7 of the CrebH gene deleted were previously described (10). CrebH null and wild-type control mice on a C57Bl/6J background at 3-months of age were used for the metabolic diet study. The AHF diet, also called Paigen diet (16% fat, 41% carbohydrate, 1.25% cholesterol, and 0.5% sodium cholate by weight), was purchased from Harlan Laboratories, Inc (TD.88051). All the animal experiments were approved by the Wayne State University IACUC committee and carried out under the institutional guidelines for ethical animal use.
Measurement of mouse lipid contents
Liver tissue and blood plasma samples were prepared from the mice under normal chow, after fasting, or after the AHF diet. To determine hepatic TG levels, liver tissue was homogenized in PBS followed by centrifugation. The supernatant was mixed with 10% Triton-100 in PBS for TG measurement using a commercial kit (BioAssay Systems, Hayward, CA). Mouse blood plasma samples were subjected to quantitative analyses of plasma TG, total cholesterol, HDL, and LDL as previously described (6). Levels of the ketone body metabolite 3-hydroxybutyric acid in the plasma were determined using commercial kits (BioAssay Systems).
Isolation of primary hepatocytes from the CrebH null and wild-type mice
To isolate murine primary hepatocytes, livers from both CrebH null and wild-type mice were first perfused by HBSS supplemented with 8 mM HEPES (pH 7.35) and 1mM sodium pyruvate followed by collagenase digestion. Hepatocytes were pelleted by centrifugation at 50g for 2 min, washed three times with DMEM media, and then seeded in the collagen-coated plates with Williams E media.
For a full description of materials and methods used in this work, see Supplemental Information.
Results
Metabolic stress signals induce CREBH cleavage
Previously we demonstrated that activation of CREBH, indicated by its cleavage, can be induced by ER stress or inflammatory stimuli (11). To further explore pathophysiological challenges that trigger CREBH cleavage, we cultured a human hepatoma cell line Huh-7 expressing full-length human CREBH protein fused with N-terminal three tandem copies of FLAG epitope protein. The design of three copies of FLAG tag has been used to study stimuli-regulated mechanisms for cleavage of CREBH and another ER stress sensor ATF6 by immunoblotting (11, 14). We exposed Huh-7 cells expressing flag-tagged full-length CREBH protein to a variety of biochemical or pathophysiological stimuli including ER stress-eliciting reagent tunicamycin (TM), saturated fatty acid palmitate (PA), and insulin. In the absence of challenges, hepatoma cells expressing full-length CREBH displayed basal levels of CREBH protein cleavage (Figure 1A). This was likely due to ER stress caused by over-expression of exogenous full-length CREBH. Consistent with our previous study, TM treatment increased CREBH cleavage (Figure 1A). Among the pathophysiological stimuli tested, we found that insulin and PA can induce CREBH cleavage. To confirm this finding, we isolated murine primary hepatocytes and stimulated them with insulin and PA. Both insulin and PA can induce CREBH cleavage in a time-dependent manner (Figure 1B-C). Additionally, we examined CREBH activation in the liver of wild-type mice fed an atherogenic high-fat (AHF) diet, which is known to induce atherosclerosis and/or non-alcoholic fatty liver disease (NAFLD) in murine models (15, 16). Higher levels of cleaved CREBH protein were detected in the livers of the mice under the AHF diet, compared to that under the normal chow diet (Figure 1D).
Figure 1. Metabolic stress signals can activate CREBH.

(A) HuH-7 cells were transfected with plasmid DNA vector expressing full-length human CREBH protein fused with N-terminal 3 copies of Flag tag. At 48 hours after the transfection, the cells were treated with PBS (control), tunicamycin (TM, 5μg/ml), insulin (100nM), or PA (10μM) for 6 hours. The non-transfected cells were included as negative controls. Western blot analysis was performed to detect CREBH cleavage by using an anti-flag antibody. CREBH (f), full-length CREBH; CREBH (a), activated/cleaved CREBH. Levels of GAPDH were determined for protein loading controls. (B-C) Primary hepatocytes isolated from wild-type mice were treated with insulin (100nM) for 10, 30, 40, 60, and 360 min or PA (10μM) for 0.5, 1, 2, 4 and 8 hours. Western blot analysis was performed to detect endogenous CREBH cleavage using a polyclonal anti-CREBH antibody. (D) Western blot analysis of CREBH cleavage in the liver tissue samples from wild-type mice under the normal chow (NC) or AHF diet for 6 months. For A-D, The values below the gels represent the activated form of CREBH protein signal intensities after normalization to full-length CREBH signal intensities. (E) Quantitative real-time RT-PCR analysis of expression of the CrebH mRNA in mouse primary hepatocytes treated with TM (5μg/ml), PA (10μM), or insulin (100nM) for 6 hours. Expression values were normalized to β-actin mRNA levels. Fold changes of mRNA are shown by comparing to that of non-treated control hepatocytes. Each bar denotes the mean ± SD (n=3 samples per treatment). (F) Quantitative real-time RT-PCR analysis of expression of the CrebH mRNA in the liver of age-matched male mice under normal chow diet, after 16 hours of fasting, or on the AHF diet for 6 months. Expression values were normalized to β-actin mRNA levels. Fold changes of mRNA levels are shown by comparing to one of the mice under normal chow diet. Each bar denotes the mean ± SEM (n=6 mice per group).
In addition to CREBH cleavage, we examined expression of the murine CrebH mRNA in response to ER stress or metabolic signals. Although TM, insulin, and PA can induce CREBH protein cleavage, they only marginally induced expression of the CrebH mRNA (Figure 1E). Furthermore, we examined expression of the CrebH gene in the liver of mice under different metabolic conditions. Expression of the CrebH mRNA was significantly increased in response to 16-hours fasting, a condition that stimulates lipolysis, FA oxidation, and gluconeogenesis (17) (Figure 1F). Together, these data suggest that activation of CREBH is inducible by metabolic conditions, such as increased saturated FA, insulin signals, or the AHF diet.
Deletion of CrebH decreases hepatic lipid contents but increases plasma TG in mice fed normal chow
To explore pathophysiologic roles of CREBH, we bred CrebH knockout mice on C57Bl/6J strain background and studied them under different metabolic conditions. Although the CrebH null mice did not display any visible morphological or development defect (10, 11), they did exhibit a slight reduction in body weight, compared to the wild-type control mice (Figure 2A). Further analyses revealed that body fat mass in the CREBH null mice was reduced, compared to that in the control mice, under the normal chow diet (Figure 2B). To explore the cause of the body weight and fat mass reductions, we first measured food consumption of the CrebH null mice when fed the normal chow diet. The CrebH null and wild-type control mice consumed similar amounts of food (Figure 2C), thus excluding the possibility that food intake is a contributing factor to the body weight or fat mass reduction in the CrebH null mice. Based on these initial analyses, we suspected that the reduced fat mass and body weight reflected an imbalance in lipogenesis, FA oxidation, lipolysis, and/or lipid transport in the CrebH null mice (1, 3). Indeed, the CrebH null mice displayed reduced levels of hepatic TG, plasma cholesterol, high-density lipoproteins (HDL), and low-density lipoproteins (LDL), compared to the wild-type mice (Figure 2D-E). Surprisingly, the levels of plasma TG in the CrebH null mice were higher than that in the control mice (Figure 2D). To further evaluate the involvement of CREBH in maintaining lipid homeostasis, we fasted the CrebH null and wild-type control mice for 16 hours. Supporting the roles of CREBH in lipolysis, the CrebH null mice maintained much higher levels of plasma TG than the control mice under nutrient starvation, a condition that stimulates lipolysis (Figure 2F). To determine whether the impaired lipid profile in the CrebH null mice was associated with FA oxidation, we measured levels of ketone body 3-hydroxybutyric acid (BOH), a product of FA oxidation in the plasma. After the fasting, levels of BOH were slightly reduced in the CrebH null mice, compared to that of the control mice (Figure 2F).
Figure 2. The CrebH null mice show a reduction in whole body fat mass, hepatic TG, and plasma cholesterol but an increase in plasma TG.

(A) Body weights of the CrebH null and wild-type control mice of 5-months old on normal chow diet. Each bar denotes the mean ± SEM (n=6 mice per group). (B) Whole body fat mass of CrebH null and wild-type control mice of 5-months old on normal chow diet. Each bar denotes the mean ± SEM (n=6 mice per group). * P<0.05. (C) Daily food intakes of the CrebH null and wild-type mice of 5-months old under the normal chow diet. The daily food intake was determined based on the measurement of food consumption of the CrebH null and control mice in one week. Each bar denotes the mean ± SEM (n=8 wild-type mice or 7 knockout mice). (D) Levels of hepatic and plasma TG in the CrebH null and wild-type mice of 5-months old under the normal chow diet. Each bar denotes the mean ± SEM (n=6 mice per group). * P<0.05. (E) Levels of plasma total cholesterol, HDL, and LDL in the CrebH null and wild-type control mice of 5-months old under the normal chow diet. Each bar denotes the mean ± SEM (n=6 mice per group). * P<0.05; ** P<0.01. (F) Levels of plasma TG and 3-hydroxybutyric acid (BOH) in the CrebH null and wild-type mice after 16 hours of fasting. Each bar denotes the mean ± SEM (n=6 mice per group). ** P<0.01.
CrebH null mice develop profound hepatic steatosis and hypertriglyceridemia when fed the AHF diet
To further delineate the role of CREBH in hepatic lipid metabolism, we fed the CrebH null and wild-type control mice with the AHF diet. After 6 months on the AHF diet, the body weight gain in the CrebH null mice was much less than that in the control mice (Figure 3A). Strikingly, the CrebH null mice developed enlarged fatty livers with pale color under the AHF diet (Figure 3B). After the AHF diet-feeding, the liver mass of the CrebH null mice was approximately 2 times of that of the wild-type control mice. Correlating with reduced body weight gain and enlarged fatty livers, the sizes of abdominal fat tissues in the CrebH null mice were significantly smaller than those in the control mice fed the AHF diet (Figure 3B). Additionally, we observed oily blood plasma with a milky color in the AHF diet-fed CrebH null mice (Figure 3C).
Figure 3. CrebH null mice display reduced body weight gain, massive steatosis, diminished abdominal fat, and hypertriglyceridemia under the AHF diet.

(A) Photograph of representative CrebH null and wild-type control mice after the AHF diet for 6 months (left), and the body weights of the CrebH null and wild-type control mice under normal chow diet or after the AHF diet (right). Each bar denotes the mean ± SEM (n=6 mice per group). ** P<0.01. (B) Liver and abdominal fat tissue of the wild-type control and CrebH null mice were visualized in situ (left), and liver mass of the wild-type and CrebH null mice after 6 months on the AHF diet (right). Each bar denotes the mean ± SEM (n=6). ** P<0.01. (C) Photograph of blood plasma samples from representative CrebH null and wild-type mice at 6 months after the AHF diet. (D) Oil-red O staining of cytosolic lipids in the liver tissue sections of representative CrebH null and wild-type control mice after the AHF diet for 6 months (magnification: 600x). (E-F) Levels of plasma TG, total cholesterol, HDL, and LDL, and levels of hepatic TG in the CrebH null and wild-type control mice after the AHF diet for 6 months. Each bar denotes the mean ± SEM (n=6). ** P<0.01.
To determine whether the phenotypes in the body weight, liver and fat mass, as well as oily plasma of the CrebH null mice are relevant to impaired lipid metabolism, we performed oil-red O lipid staining to visualize the morphology and distribution of hepatic lipids in the liver tissue of the CrebH null mice after the AHF diet. The livers from the wild-type mice displayed macrovesicular steatosis characterized by engorgement of the hepatocytes with lipid droplets (Figure 3D). In contrast, massive lipid contents, characterized by non-structural lipid smears, were detected in the liver tissues of the AHF diet-fed CrebH null mice, indicating impaired hepatic lipid metabolism in the absence of CREBH. Furthermore, levels of plasma TG in the CrebH null mice were significantly higher than those in the control mice after the AHF diet (Figure 3E), consistent with the oily plasma phenotype. However, levels of plasma lipoproteins, including total plasma cholesterol, HDL, and LDL, were slightly decreased in the CrebH null mice after the AHF diet (Figure 3E). The plasma lipid profiles of the AHF diet-fed CrebH null mice were consistent with those observed with the CrebH null mice fed normal chow or after fasting (Figure 2D-F), implicating a prominent hypertriglyceridemia phenotype caused by CrebH deletion. Additionally, levels of hepatic TG in the CrebH null mice were much higher than those in the control mice after the AHF diet (Figure 3F).
Deletion of CrebH leads to impaired fatty acid metabolism and severe steatohepatitis in mice after the AHF diet
To evaluate the abnormal lipid mass in the liver of the AHF-fed CrebH null mice, we performed lipidomic analysis of eicosanoids and docosanoids in the liver tissues of CrebH null and wild-type control mice fed the AHF diet. Eicosanoids and docosanoids are highly bioactive metabolites synthesized through oxidation of 20- and 22-carbon polyunsaturated FA (PUFA), respectively (18). Supporting a role of CREBH in FA oxidation, levels of PUFA metabolites, including hydroxyeicosatetraenoic acids (HETEs), lipoxins (LXs), prostaglandins (15-keto PGE1 and 15-keto PGE2), hydroxy docosahexaenoic acids (HDoHEs), and eicosatrienoic acids (DiHETEs), were significantly reduced in the liver tissues of the CrebH null mice fed the AHF diet, compared to those of wild-type mice (Figure 4A and Supplemental S1). In particular, the synthesis of epoxy FA catalyzed by cytochrome P450 (Cyp) enzymes, including HETEs, EpETrEs, and DiHETEs, appeared to be defective in the CrebH null mice. The downstream metabolism of eicosanoids by the prostaglandin dehydrogenases inactivates the inflammatory lipid metabolites (18). Deficiency in this inactivation pathway is indicated by the significantly diminished levels of 15-keto PGE2, 15-keto PGE1, and 5-OxoETE in the CrebH null liver. Interestingly, metabolism of PUFA to the primary bioactive lipids, such as PGE2 and TXA2 (measured as TXB2), is not significantly altered, suggesting a potential pro-inflammatory effect of CrebH deletion via a decrease in the inactivating metabolism rather than increasing the inflammatory mediators (19). Together, these results implicate that CrebH deletion impairs FA metabolism in the liver, which may partially contribute to abnormal accumulation of hepatic lipid contents and liver inflammation in the AHF diet-fed CrebH null mice.
Figure 4. Deletion of CrebH leads to impaired FA metabolism and profound NASH in the mice under the AHF diet.

(A) Lipidomic analysis of eicosanoids and docosanoids in the liver tissues of the CrebH null and wild-type control mice after the AHF diet for 6 months. Levels of lipid metabolites that were decreased or increased in the CrebH null liver, compared to that in the wild-type control liver (P-value cutoff was <0.05), were shown. Each bar denotes the mean ± SEM (n=4 mice per group). (B) Histological examination (hematoxylin eosion staining) of liver tissue sections of the CrebH null and wild-type mice after the AHF diet for 6 months (magnification: 200x). (C) Sirius staining of collagen deposition in the liver tissue of the wild-type and CrebH null mice after the AHF diet (magnification: 200x). (D) Histological scoring for NASH activities in the livers of the CrebH null and wild-type mice after the AHF diet for 6 months. The Grade scores were calculated based on the scores of steatosis, hepatocyte ballooning, lobular and portal inflammation, and Mallory bodies. The Stage scores were based on the liver fibrosis. Mean ± SEM (n=4) values are shown. P-values were calculated by Mann-Whitney U-test.
Next, we evaluated NAFLD-associated activities in the CrebH null mice. Significant liver inflammation and fibrosis in the AHF diet-fed CrebH null animals were evidenced by increased hepatocyte ballooning, lobular and portal inflammation, Mallory bodies, as well as collagen deposition (Figure 4B-C). Using a histological scoring system for NAFLD (20, 21), we revealed that the CrebH null mice, not the wild-type mice, developed profound NASH after the AHF diet (Figure 4D). Supporting the histological analysis, levels of alanine aminotranferease (ALT) and aspartate aminotransferase (AST), the key indicators of hepatotoxicity, were increased in the CrebH null mice after the AHF diet (Supplemental S2A-B). Moreover, expression of two NASH-associated pro-inflammatory cytokines (22), TNFα and IL6, was also significantly increased in the liver of the AHF diet-fed CrebH null mice (Supplemental S2C-D). Additionally, we assessed the effects of CrebH deletion on glucose/insulin homeostasis. CrebH deletion led to decreased blood glucose levels in the mice after overnight fasting, consistent with a previous study (Supplemental S3A)(7). We confirmed that expression of the gene encoding the gluconeogenesis regulator PCK1 was defective in the CrebH null mice upon overnight fasting (Supplemental S3B) (7, 9). Glucose tolerance test (GTT) showed that the CrebH null mice displayed similar patterns in decreasing exogenously-injected glucose, compare to the control mice (Supplemental S3C-D). However, insulin tolerance test (ITT) revealed that the AHF diet-fed CrebH null mice were not responsive to insulin in decreasing blood glucose (Supplemental S3F), consistent with the increased NASH activities in the CrebH null mice after the AHF diet (Figure 4D).
CREBH is required for transcriptional activation of genes involved in lipogenesis, FA and cholesterol metabolism, and lipolysis
To understand the molecular basis underlying the lipid phenotypes observed with the CrebH null mice, we performed gene microarray analysis using total liver mRNA from the CrebH null and wild-type control mice fed either normal chow or the AHF diet. Through this analysis, we identified a subset of lipid metabolism-associated genes whose expression was down-regulated in the CrebH null mice fed normal chow or the AHF diet (Supplemental S4 and S5). To verify the microarray analysis results, we performed quantitative real-time RT-PCR analysis and confirmed that deletion of CrebH in the liver decreased expression of 5 groups of genes encoding functions critical for lipid metabolism (Figure 5): (1) lipogenic regulators, including ChREBP, LXRα, PPARγ-coactivator (PGC)-1α, PGC-1β, and fat-specific protein 27 (FSP27) (Figure 5A); (2) TG synthesis enzymes, including fatty acid synthase (FASN), acetyl CoA carboxylase 1 (ACC1), ACC2, CoA desaturase 1 (SCD1), and diacylglycerol acetyltransferase 2 (DGAT2) (Figure 5B); (3) enzymes or regulators in lipolysis and lipid transport, including apolipoprotein (Apo) C2, Apo4, ApoA5, Apof, lipolysis-stimulated lipoprotein receptor (LSR), lecithin-cholesterol acyltransferase (LCAT), sterol carrier protein 2 (SCP2), and acyl-CoA thioesterase 4 (Acot4) (Figure 5C); (4) FA elongation enzymes, including elongation of very long chain fatty acids protein (Elovl) 2, Elovl5, Elovl6, and peroxisomal trans-2-enoyl-CoA reductase (Pecr) (Figure 5D); (5) FA oxidation or cholesterol biosynthesis enzymes, including carnitine palmitoyltransferase 1A (CPT1α), CYP4a10, CYP4a14, CYP2b9, CYP2b13, fatty acid desaturase 1 (FADS1), FADS2, palmitoyl-CoA oxidase1 (ACOX1), PPARα, 24-dehydrocholesterol reductase (Dhcr24), and long-chain-fatty-acid-CoA ligase 1 (Acsl1) (Figure 5E). The identified gene expression profile is consistent with the phenotypes observed with the CrebH null mice. First, decreased expression of the lipogenic regulators and TG synthesis enzymes is consistent with reduced de novo lipogenesis observed in the CrebH null mice fed normal chow (Figure 2). Second, decreased expression of enzymes or regulators in FA elongation and oxidation or cholesterol biosynthesis may be responsible, at least partially, for abnormal accumulation of hepatic lipid metabolites in the AHF-fed CrebH null mice (Figures 3D and 4A). Finally, defective expression of enzymes required for lipolysis and lipid transport may account for hypertriglyceridemia, reduced fat mass and body weight gain, and massive steatosis in the CrebH null mice fed the AHF diet or normal chow (Figures 2 and 3).
Figure 5. Deletion of CrebH leads to defective expression of genes involved in lipogenesis, FA and cholesterol metabolism, and lipolysis.

Total RNAs were isolated from liver tissues of the CrebH null and wild-type control mice under the normal chow diet or after the AHF diet for 6 months, and subjected to quantitative real-time RT-PCR analysis of expression of the genes involved in hepatic lipid metabolism. Expression values were normalized to β-actin mRNA levels. Fold changes of mRNA levels are shown by comparing to one of the control mouse under the normal chow diet or the AHF diet. Each bar denotes the mean ± SEM (n=4). * P<0.05; ** P<0.01.
To determine whether some or all the lipid-associated genes that were down-regulated in the CrebH null mice are direct CREBH targets, we performed chromatin-immunoprecipitation (ChIP) experiments to test the potential of CREBH in binding to the promoter regions of the lipid-associated genes regulated by CREBH (Figure 5). We expressed an activated form of CREBH with flag tag or a GFP control protein in primary hepatocytes isolated from CrebH null mice using an adenoviral-based expression system. The ChIP analysis indicated that CREBH can bind to the promoter regions of the genes encoding: TG synthesis enzymes including ACC1, ACC2, and FASN; lipolysis co-activators including ApoC2 and ApoA4; and enzymes involved in FA elongation including Elovl2, Elov5, and Elovl6 (Figure 6A-B). To assure CREBH-mediated transcriptional regulation of these genes, we performed gene expression analysis with the animals over-expressing the activated CREBH in the liver. Quantitative real-time RT-PCR analysis indicated that the activated CREBH can dramatically up-regulate expression of its target genes confirmed by ChIP analysis as well as other genes involved in lipogenesis, lipolysis, and FA metabolism (Figure 6C-E). Additionally, we examined expression of the proteins encoded by the CREBH-targeted genes in the animals over-expressing the activated CREBH or GFP. Western blot analysis confirmed that levels of SCD1, PGC1, and ApoA4 proteins in the liver tissue or blood serum of the mice expressing the activated CREBH were significantly increased, compared to the mice expressing GFP (Figure 6F). Together these results suggested that CREBH can directly bind to and activate expression of some of the key genes encoding functions involved in lipogenesis, FA metabolism, and lipolysis.
Figure 6. CREBH binds to and activate expression of the genes involved in hepatic lipid metabolism.

(A-B) ChIP analysis of CREBH-binding activity to the promoter regions of the genes involved in hepatic lipid metabolism. Primary hepatocytes isolated from the CrebH null liver were infected with adenovirus expressing GFP or an activated CREBH protein with N-terminal fused flag tag. PCR was performed to identify potential CREBH-binding regions in the genes whose expression was defective in the CrebH null liver. Mock ChIP was included as a negative control (N-ctl). The PCR reactions with the genomic DNA isolated from sonicated cell lysates were included as positive controls (P-ctl). (C-E) Gene expression analysis in the liver of the mice over-expressing the activated CREBH. Total RNAs from the mice infected with adenovirus expressing GFP or the activated CREBH for 72 hours were subjected to quantitative real-time RT-PCR analysis of gene expression. Expression values were normalized to β-actin mRNA levels. Fold changes of mRNA levels are shown by comparing to one of the mice over-expressing GFP. Each bar denotes the mean ± SEM (n=3). ** P<0.01. (F) Western blot analysis of protein levels for CREBH in the nuclear extract (NE), SCD1 and PGC1 in the liver tissue lysates, and ApoA4 in the blood plasma of mice over-expressing GFP or the activated CREBH. Levels of GAPDH and Albumin were included as loading controls for liver tissue lysate and blood plasma samples, respectively.
CREBH activity facilitates de novo lipogenesis and lipolysis
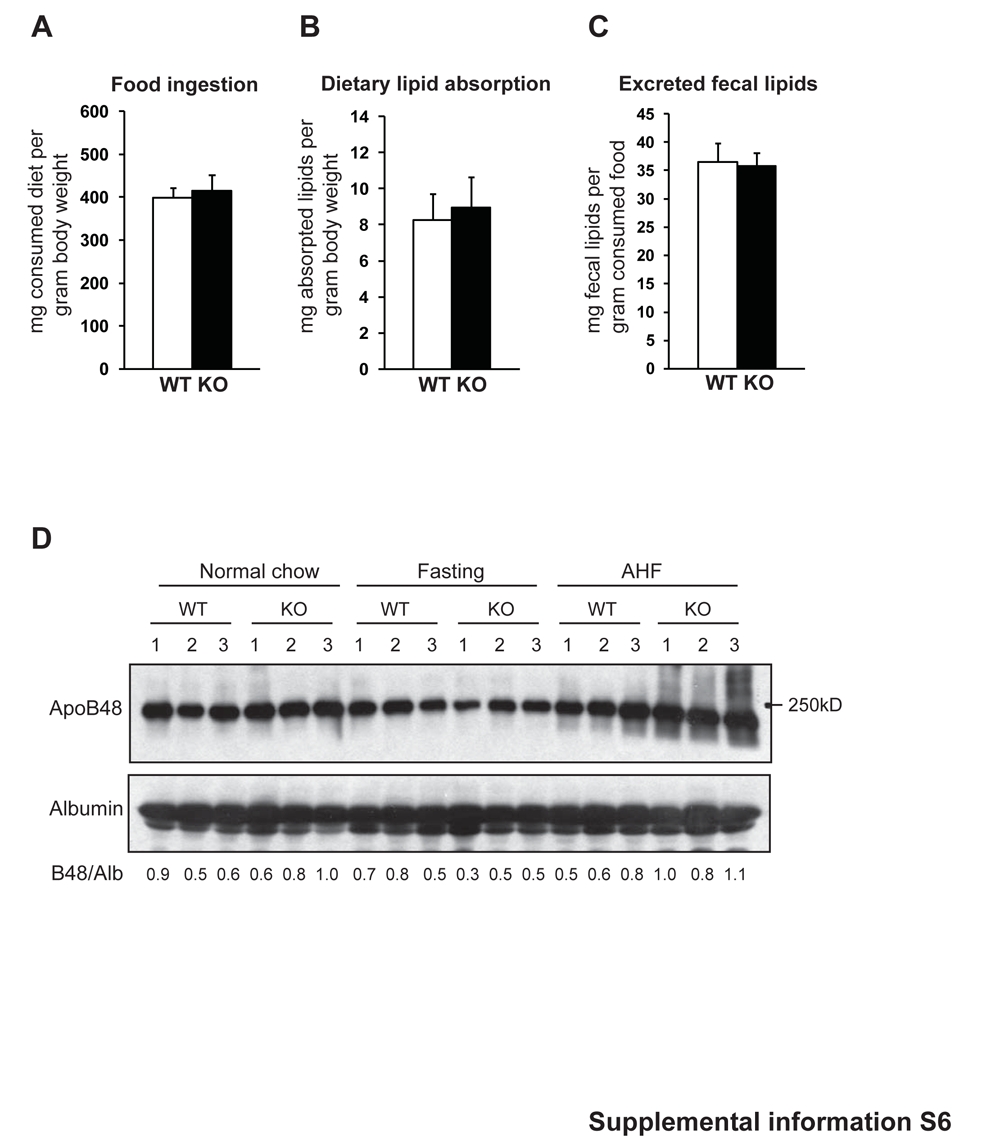
To verify the roles of CREBH in lipid metabolism, we performed functional analyses for CREBH using both in vitro and in vivo systems. First, we challenged primary hepatocytes from CrebH null and wild-type mice with ER stress-inducing reagent TM or the saturated fatty acid PA. As a response to acute liver injury induced by TM or feeding with PA, lipogenesis and lipid deposition are increased in hepatocytes (6, 23). Without stress challenges, a modest reduction in cytosolic lipid contents, as characterized by oil-red O staining, was detected in the CrebH null primary hepatocytes, compared to the wild-type hepatocytes (Figure 7A). After 12 hours of incubation with TM or PA, wild-type hepatocytes developed severe steatosis. In contrast, only small amounts of cytosolic lipids were accumulated in the CrebH null hepatocytes after the incubation with either TM or PA (Figure 7A), thus confirming the involvement of CREBH in hepatic lipogenesis. Second, to verify the role of CREBH in lipolysis, we performed fat tolerance test to track the ability of the CrebH null mice in clearance of exogenously-infused TG. The CrebH null and control animals were fasted for 16 hours followed by oral gavage of pure olive oil that is composed mainly of TG. After dietary TG absorption, the CrebH null mice failed to decrease plasma TG levels (Figure 7B). At the 8 hours after olive oil infusion, the wild-type mice decreased the plasma TG levels to the basal levels. In contrast, the plasma TG levels in the knockout mice were kept as high as that after olive oil absorption, suggesting the defect of plasma TG lipolysis in the absence of CREBH. Because expression of ApoC2, a component of mature chylomicron, is decreased in the CrebH null mice, we wondered whether CrebH deletion could affect dietary lipid absorption in mice. Analyses of food uptake and dietary fat absorption suggested no significant differences in food ingestion, dietary lipid absorption, and excreted fecal lipids between the CrebH null and wild-type mice (Supplemental S6A-C). This was further evidenced by the comparable levels of plasma ApoB48, the key indicator of chylomicron, detected in the CrebH null and wild-type mice (Supplemental S6D).
Figure 7. CREBH is required for lipogenesis and lipolysis.

(A) Oil-red O staining of cytosolic lipids in the CrebH null and wild-type control primary hepatocytes after the treatment of vehicle, TM (5μg/ml), or PA (100μM) for 12 hours (magnification: 400x). (B) Fat tolerance test for the CrebH null and wild-type control mice. Mice were fasted for 16 hours, followed by oral gavage of pure olive oil at the dose of 12 μl/g body weight. Plasma TG levels were determined immediately before and at 2, 4, 6, and 8 hours after olive oil loading. The mean ± SEM (n=5) values were shown.
Having established essential roles for CREBH in de novo lipogenesis and lipolysis, we next tested whether CREBH can regulate these lipid-associated processes by expressing an activated form of human CREBH in the mouse liver (11). The activated CREBH protein was over-expressed in mouse liver tissue through tail vein infusion of adenovirus expressing the activated CREBH into wild-type mice. Oil-red O staining of liver tissue sections revealed a dramatic accumulation of lipid droplets in the liver of the mice over-expressing the activated CREBH protein (Figure 8A). Consistently, levels of hepatic TG were significantly increased in the mice over-expressing the activated CREBH (Figure 8B), supporting the role of CREBH in promoting de novo lipogenesis. In contrast, levels of plasma TG were significantly decreased in the mice expressing the activated form of CREBH, compared to that of the control mice expressing GFP (Figure 8C), confirming the role of CREBH in facilitating lipolysis.
Figure 8. Forced expression of the activated CREBH in the liver increases lipogenesis and lipolysis.

(A) Oil-red O staining of cytosolic lipids in the liver tissue sections of the mice over-expressing GFP or the activated form of CREBH (magnification: 400 ×). Frozen liver tissue sections were prepared from the mice at 72 hours after the infection with adenovirus expressing GFP or the activated CREBH through tail vein injection. (B-C) Levels of hepatic TG and plasma TG in the liver of the mice over-expressing GFP or the activated CREBH. Each bar denotes the mean ± SEM (n=3). ** P<0.01. (D) A working model for the role of CREBH in regulating lipid homeostasis under metabolic stress
Discussion
In this study, we demonstrated that the ER-resident transcription factor CREBH regulates multiple lipid-associated pathways, including lipogenesis, FA and cholesterol metabolism, and lipolysis, to maintain lipid homeostasis under metabolic stress conditions (Figure 8D). With the CrebH knockout mice, we identified complex lipid phenotypes caused by CrebH deletion. Under the normal chow, the CrebH null mice displayed reduced lipogenesis, as evidenced by lower levels of hepatic TG and plasma cholesterols, reduced whole body fat mass, and decreased expression of regulators and enzymes required for TG and cholesterol biosynthesis (Figures 2 and 5). Simultaneously, CrebH deletion caused a defect in TG lipolysis, as the CrebH null mice displayed higher levels of plasma TG and reduced expression of key activators of lipolysis, compared to the wild-type control mice (Figures 2D and 5C). The involvement of CREBH in maintaining lipid homeostasis was further evidenced by the phenotypes and gene expression profile in the CrebH null mice fed the AHF diet. The AHF-fed CrebH null mice displayed hypertriglyceridemia, severe NASH, reduced body weight gain, diminished abdominal fat, as well as insulin resistance (Figures 3 and 4 and Supplemental S3). Lipid staining and lipidomic analysis revealed that the enlarged livers of the CrebH null mice accumulated non-structural lipid mass and significantly altered profile of lipid metabolites (Figures 3D and 4A). This phenotype is likely caused by the defects in FA and cholesterol metabolism in the absence of CREBH. In support of the role of CREBH in promoting lipogenesis and lipolysis, the CrebH null primary hepatocytes showed the defect in accumulating cytosolic lipid droplets in response to stress/injuries (Figure 7A), and the CrebH null mice failed to decrease exogenously-infused TG in the blood stream (Figure 7B). Indeed, during the review process of this manuscript, a report was published that confirmed the requirement of CREBH for lipolysis (24).
Among the list of the CREBH target genes, ApoC2 is a key activator of lipolysis. In response to nutrient overload, ApoC2 is secreted into plasma where it activates lipoprotein lipase to hydrolyze TG, thus providing a source of free FA for cells. Mutations in this gene cause hyperlipoproteinemia, characterized by hypertriglyceridemia and xanthomas (25). Defective expression of ApoC2, and two other lipolysis co-activators LCAT and ApoA4, likely accounts for hypertriglyceridemia and diminished peripheral fat mass observed in the CrebH null mice, especially after the AHF diet. Because ApoC2 is a component of mature chylomicron (26), the vehicle of dietary lipids in the blood stream, we suspected the ApoC2 defect may affect dietary lipid absorption in the CrebH null mice. However, our analyses suggested no significant differences in food consumption and dietary lipid absorption between the CrebH null and wild-type mice (Figures 2C and Supplemental S6A-C). Apparently, decreased expression of ApoC2 does not affect nascent chylomicron to deliver dietary TG from the small intestine in the CrebH null mice. Instead, the ApoC2 defect leads to a failure in triggering the activity of lipoprotein lipase to break down dietary TG in the blood stream. As a consequence, chylomicron remnant in which the dietary TG store is not completely distributed will retain in the liver. This may partially contribute to hepatic TG accumulation in the liver of the AHF diet-fed CrebH null animals. It should be noted that the CrebH null mice exhibited outwardly opposite phenotypes in hepatic lipid accumulation upon normal chow or AHF diet (Figures 2 and 3). Under normal chow diet, both lipogenesis and lipolysis are crucial in determining hepatic and plasma TG levels, and therefore, the roles of CREBH in both lipogenesis and lipolysis are visible, as the CrebH null mice displayed lower hepatic TG levels but higher plasma TG levels, compared to the control mice (Figure 2). However, under the AHF diet, FA elongation and oxidation, lipolysis, and lipid transport, but not de novo lipogenesis, are prevalent programs that determine lipid levels in the liver and other periphery organs (2, 3). The defects of FA oxidation, cholesterol metabolism, and lipolysis in the CrebH null mice likely overrode the defect of de novo lipogenesis, leading to profound hepatic steatosis and hypertriglyceridemia in the CrebH null mice after the AHF diet (Figure 3).
In summary, our work implicates that CREBH plays key roles in maintaining lipid homeostasis by regulating multiple key metabolic pathways in the liver (Figure 8D). Disruption of CREBH activity leads to complex lipid-associated phenotypes, including hepatic steatosis, hypertriglyceridemia, and reduction in fat mass and body weight. In contrast, over-expression of the activated CREBH in the liver can significantly increase de novo hepatic lipogenesis but reduce plasma TG levels simultaneously. These findings may have important implication in pharmaceutical intervention towards controlling lipid homeostasis by modulating CREBH activity. Our work has also raised many immediate and important questions. For example, what is the precise mechanism by which CREBH controls the synthesis of lipogenic enzymes or regulators? Does CREBH act alone or in partnership with other known transcription factors such as SREBP, ChREBP, and LXRα? Additionally, other ER stress-induced transcription factors, such as ATF6 and X-box binding protein 1 (XBP1), also regulate hepatic lipid homeostasis (5, 27). It is interesting to elucidate whether CREBH can function with ATF6 and/or XBP1 in regulating lipid homeostasis. All these questions deserve future research effort on CREBH.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgement
Portions of this work were supported by American Heart Association Grants 0635423Z and 09GRNT2280479 (KZ), National Institutes of Health (NIH) grants DK090313 and ES017829 (KZ), National Center for Research Resources (NCRR) grant S10RR027926 (KRM), and NIH grants HL057346, DK042394 and HL052173 (RJK). We thank Drs James Granneman, Todd Leff, and Emilio Mottillo for their critical comments and technical help.
Abbreviations
- ER
endoplasmic reticulum
- CREBH
cAMP responsive element binding protein, hepatocytes-specific
- NAFLD
non-alcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- TG
triglycerides
- FA
fatty acids
- AHF
atherogenic high-fat
- PA
palmitate
- TM
tunicamycin
- ChIP
chromatin immunoprecipitation
- CYP
cytochrome P450 enzymes
- Apo
apolipoprotein
References
- 1.Musso G, Gambino R, Cassader M. Recent insights into hepatic lipid metabolism in non-alcoholic fatty liver disease (NAFLD). Prog Lipid Res. 2009;48:1–26. doi: 10.1016/j.plipres.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 2.Jump DB, Botolin D, Wang Y, Xu J, Christian B, Demeure O. Fatty acid regulation of hepatic gene transcription. J Nutr. 2005;135:2503–2506. doi: 10.1093/jn/135.11.2503. [DOI] [PubMed] [Google Scholar]
- 3.Postic C, Girard J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered mice. J Clin Invest. 2008;118:829–838. doi: 10.1172/JCI34275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89:331–340. doi: 10.1016/s0092-8674(00)80213-5. [DOI] [PubMed] [Google Scholar]
- 5.Rutkowski DT, Wu J, Back SH, Callaghan MU, Ferris SP, Iqbal J, Clark R, et al. UPR pathways combine to prevent hepatic steatosis caused by ER stress-mediated suppression of transcriptional master regulators. Dev Cell. 2008;15:829–840. doi: 10.1016/j.devcel.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang K, Wang S, Malhotra J, Hassler JR, Back SH, Wang G, Chang L, et al. The unfolded protein response transducer IRE1alpha prevents ER stress-induced hepatic steatosis. EMBO J. 2011;30:1357–1375. doi: 10.1038/emboj.2011.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee MW, Chanda D, Yang J, Oh H, Kim SS, Yoon YS, Hong S, et al. Regulation of hepatic gluconeogenesis by an ER-bound transcription factor, CREBH. Cell Metab. 2010;11:331–339. doi: 10.1016/j.cmet.2010.02.016. [DOI] [PubMed] [Google Scholar]
- 8.Omori Y, Imai J, Watanabe M, Komatsu T, Suzuki Y, Kataoka K, Watanabe S, et al. CREB-H: a novel mammalian transcription factor belonging to the CREB/ATF family and functioning via the box-B element with a liver-specific expression. Nucleic Acids Res. 2001;29:2154–2162. doi: 10.1093/nar/29.10.2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chin KT, Zhou HJ, Wong CM, Lee JM, Chan CP, Qiang BQ, Yuan JG, et al. The liver-enriched transcription factor CREB-H is a growth suppressor protein underexpressed in hepatocellular carcinoma. Nucleic Acids Res. 2005;33:1859–1873. doi: 10.1093/nar/gki332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luebke-Wheeler J, Zhang K, Battle M, Si-Tayeb K, Garrison W, Chhinder S, Li J, et al. Hepatocyte nuclear factor 4alpha is implicated in endoplasmic reticulum stress-induced acute phase response by regulating expression of cyclic adenosine monophosphate responsive element binding protein H. Hepatology. 2008;48:1242–1250. doi: 10.1002/hep.22439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang K, Shen X, Wu J, Sakaki K, Saunders T, Rutkowski DT, Back SH, et al. Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell. 2006;124:587–599. doi: 10.1016/j.cell.2005.11.040. [DOI] [PubMed] [Google Scholar]
- 12.Danno H, Ishii KA, Nakagawa Y, Mikami M, Yamamoto T, Yabe S, Furusawa M, et al. The liver-enriched transcription factor CREBH is nutritionally regulated and activated by fatty acids and PPARalpha. Biochem Biophys Res Commun. 2010;391:1222–1227. doi: 10.1016/j.bbrc.2009.12.046. [DOI] [PubMed] [Google Scholar]
- 13.Gentile CL, Wang D, Pfaffenbach KT, Cox R, Wei Y, Pagliassotti MJ. Fatty acids regulate CREBh via transcriptional mechanisms that are dependent on proteasome activity and insulin. Mol Cell Biochem. 2010;344:99–107. doi: 10.1007/s11010-010-0533-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shen J, Prywes R. ER stress signaling by regulated proteolysis of ATF6. Methods. 2005;35:382–389. doi: 10.1016/j.ymeth.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 15.Paigen B, Morrow A, Holmes PA, Mitchell D, Williams RA. Quantitative assessment of atherosclerotic lesions in mice. Atherosclerosis. 1987;68:231–240. doi: 10.1016/0021-9150(87)90202-4. [DOI] [PubMed] [Google Scholar]
- 16.Matsuzawa N, Takamura T, Kurita S, Misu H, Ota T, Ando H, Yokoyama M, et al. Lipid-induced oxidative stress causes steatohepatitis in mice fed an atherogenic diet. Hepatology. 2007;46:1392–1403. doi: 10.1002/hep.21874. [DOI] [PubMed] [Google Scholar]
- 17.Finn PF, Dice JF. Proteolytic and lipolytic responses to starvation. Nutrition. 2006;22:830–844. doi: 10.1016/j.nut.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 18.Serhan CN. Novel eicosanoid and docosanoid mediators: resolvins, docosatrienes, and neuroprotectins. Curr Opin Clin Nutr Metab Care. 2005;8:115–121. doi: 10.1097/00075197-200503000-00003. [DOI] [PubMed] [Google Scholar]
- 19.Tai HH, Ensor CM, Tong M, Zhou H, Yan F. Prostaglandin catabolizing enzymes. Prostaglandins Other Lipid Mediat. 2002;68-69:483–493. doi: 10.1016/s0090-6980(02)00050-3. [DOI] [PubMed] [Google Scholar]
- 20.Brunt EM, Janney CG, Di Bisceglie AM, Neuschwander-Tetri BA, Bacon BR. Nonalcoholic steatohepatitis: a proposal for grading and staging the histological lesions. Am J Gastroenterol. 1999;94:2467–2474. doi: 10.1111/j.1572-0241.1999.01377.x. [DOI] [PubMed] [Google Scholar]
- 21.Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 22.Sakaguchi S, Takahashi S, Sasaki T, Kumagai T, Nagata K. Progression of alcoholic and non-alcoholic steatohepatitis: common metabolic aspects of innate immune system and oxidative stress. Drug Metab Pharmacokinet. 2011;26:30–46. doi: 10.2133/dmpk.dmpk-10-rv-087. [DOI] [PubMed] [Google Scholar]
- 23.Wobser H, Dorn C, Weiss TS, Amann T, Bollheimer C, Buttner R, Scholmerich J, et al. Lipid accumulation in hepatocytes induces fibrogenic activation of hepatic stellate cells. Cell Res. 2009;19:996–1005. doi: 10.1038/cr.2009.73. [DOI] [PubMed] [Google Scholar]
- 24.Lee JH, Giannikopoulos P, Duncan SA, Wang J, Johansen CT, Brown JD, Plutzky J, et al. The transcription factor cyclic AMP-responsive element-binding protein H regulates triglyceride metabolism. Nat Med. 2011;17:812–815. doi: 10.1038/nm.2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Breckenridge WC, Little JA, Steiner G, Chow A, Poapst M. Hypertriglyceridemia associated with deficiency of apolipoprotein C-II. N Engl J Med. 1978;298:1265–1273. doi: 10.1056/NEJM197806082982301. [DOI] [PubMed] [Google Scholar]
- 26.Hussain MM. A proposed model for the assembly of chylomicrons. Atherosclerosis. 2000;148:1–15. doi: 10.1016/s0021-9150(99)00397-4. [DOI] [PubMed] [Google Scholar]
- 27.Lee AH, Scapa EF, Cohen DE, Glimcher LH. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science. 2008;320:1492–1496. doi: 10.1126/science.1158042. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.