

Abstract
Evidence suggests that the role played by the adipocyte-derived hormone leptin in female reproductive physiology is mediated in part by neurons located within the ventral premammillary nucleus (PMV). Leptin activates PMV neurons; however, the intracellular signaling pathway and channel(s) involved remain undefined. Notably, leptin's excitatory and inhibitory effects within hypothalamic and brainstem nuclei share the intracellular signaling cascade phosphoinositide 3 kinase (PI3K). Therefore, we assessed whether PI3K signaling is required for the acute effect of leptin to alter cellular activity of PMV neurons that express leptin receptors (LepR PMV neurons). Leptin caused a rapid depolarization in the majority of LepR PMV neurons in patch-clamp recordings of hypothalamic slices, while a subset of LepR PMV neurons were hyperpolarized in response to leptin. Data were obtained from both male and female mice and results demonstrate that the acute effect of leptin on LepR PMV neurons was identical for both sexes. Pharmacological inhibition of PI3K prevented the acute leptin-induced change in neuronal activity of LepR PMV neurons, indicating a PI3K-dependent mechanism of leptin action. Similarly, mice with genetically disrupted PI3K signaling in LepR PMV neurons failed to alter cellular activity in response to leptin. Moreover, the leptin-induced depolarization was dependent on a putative TRPC channel. In contrast, the leptin-induced-hyperpolarization required the activation of a putative Katp channel. Collectively, these results suggest that PI3K signaling in LepR PMV neurons is essential for leptin-induced alteration in cellular activity, and these data may suggest a cellular correlate in which leptin contributes to the initiation of reproductive development.
Introduction
The adipocyte-derived hormone leptin has a profound influence on energy and glucose homeostasis. Leptin has also been implicated in pubertal development and fertility (Clayton et al., 1997; Mantzoros et al., 1997; Quinton et al., 1999). Leptin's role in sexual development is highlighted by the fact that leptin deficiency or lack of the leptin receptor (LepR) results in a failure of sexual maturity (Coleman, 1978; Zhang et al., 1994; Tartaglia et al., 1995). Moreover, leptin is required for pubertal development in normal female mice and rescues the infertility of ob/ob mice (Ahima et al., 1996, 1997), probably signaling directly in the brain at what time the body is ready for sexual maturation (de Luca et al., 2005). Leptin also increases luteinizing hormone (LH) secretion during negative energy balance in many species, including humans (Ahima et al., 1996; Nagatani et al., 1998; Watanobe et al., 1999; Chan et al., 2003; Welt et al., 2004). Together these data suggest that leptin may be acting directly in the brain to initiate reproductive development. Importantly, recent evidence suggests that leptin may act directly within the ventral premammillary nucleus (PMV) to control many parameters of the reproductive physiology (Clayton et al., 1997; Mantzoros et al., 1997; Quinton et al., 1999; Donato et al., 2009, 2011). Interestingly, leptin has recently been shown to activate ∼75% of the neurons that express leptin receptors within the PMV (Leshan et al., 2009). Leshan et al. (2009) further demonstrated that PMV neurons that express leptin receptor directly innervate gonadotropin-releasing hormone (GnRH) neurons, highlighting a potential role of PMV neurons in stimulating LH secretion from the pituitary gland. Together these data demonstrate that although great strides have been made in understanding the effects of leptin on PMV neurons as they relate to mammalian fertility and pubertal development, the intracellular signaling pathway and channel(s) underlying leptins acute effects within the PMV remain undefined.
The arcuate nucleus has received significant attention with respect to the acute effects of leptin on cellular activity and may serve as a model system for the possible leptin-induced effects on cellular activity in other CNS nuclei (e.g., PMV). Importantly, leptin's excitatory and inhibitory effects within the hypothalamic and brainstem nuclei share the intracellular signaling cascade phosphoinositide 3 kinase (PI3K) (Spanswick et al., 1997; Cowley et al., 2001; van den Top et al., 2004; Williams and Smith, 2006; Williams et al., 2007; Hill et al., 2008b). In the arcuate nucleus, leptin depolarizes arcuate POMC neurons via a PI3K-dependent activation of a putative TRPC channel (Hill et al., 2008b; Qiu et al., 2010), while at the same time leptin hyperpolarizes arcuate NPY/AgRP neurons via a PI3K-dependent activation of a Katp channel (Spanswick et al., 1997; van den Top et al., 2004). We therefore tested the hypothesis that PI3K signaling in PMV neurons that express the leptin receptor, LepR PMV neurons, is necessary for normal leptin responsiveness in the PMV.
Materials and Methods
Subjects.
The mice in this study were housed in the University of Texas Southwestern Medical Center Animal Resource Center, in a light- (12 h on/12 h off) and temperature- (21–23°C) controlled environment. They were fed standard chow diet (Harlan Teklad Global Diet) and had ad libitum access to water. All experiments were performed in accordance with the guidelines established by the National Institute of Health Guide for the Care and Use of Laboratory Animals, as well as with those established by the University of Texas Institutional Animal Care and Use Committee.
Leptin administration.
Male (8–10 weeks) pathogen-free C57BL/6 mice were housed in a light-dark (12 h on/off; lights on at 7:00 A.M.) and temperature-controlled environment with food and water available ad libitum. C57BL6 mice were injected with recombinant murine leptin intraperitoneal (n = 3, 5.0 mg/kg; provided by A. F. Parlow, Harbor-UCLA Medical Center, Torrance, California; through the National Hormone and Peptide Program) or pyrogen-free saline (n = 3, Sigma). All injections were given between 11:00 A.M. and 12:00 P.M. After 45 min, they were perfused and brains were dissected and processed as follows.
Histology.
To assess whether the PI3K catalytic subunits p110α and p110β are expressed in neurons that express LepR, we crossed the LepR-IRES-Cre mice with the reporter mice that express βGal [B6.129S4-Gt(ROSA)26-Sortm1Sor/J; The Jackson Laboratory] in a Cre-dependent manner (Srinivas et al., 2001) to obtain the LepR-reporter mice. In this study, we used adult (90–100 d old) male mice homozygous for Cre-recombinase. The LepR-Cre reporter mice and those treated with leptin or saline were perfused with 10% buffered formalin, and brains were dissected, cryoprotected overnight, and cut in the frontal plane into 25 μm sections on a freezing microtome. Five series were collected into antifreeze solution prepared with diethyl pyrocarbonate (DEPC)-treated water and were stored at −20°C.
In situ hybridization.
The generation and validation of Pik3ca (p110α) riboprobe have been described in previous study ((Hill et al., 2009). To produce the Pik3cb (p110β) riboprobe, PCR fragments were amplified with ExTaq DNA polymerase (Takara) from cDNA generated with the SuperScript II Reverse Transcriptase (Invitrogen) for RT-PCR, using total mouse hypothalamic RNA as template. The Pik3cb probe is equivalent to positions 2625 to 3102 of GenBank accession number NM_029094.3 (primer forward: 5′-cgtcatcctctcagagctct-3′; primer reverse: 5′-gttgtcactgtgcctgtcacca-3′).
Antisense and sense 35S- or 33P-labeled probes were generated with MAXIscript In Vitro Transcription Kits (Ambion) with 35S-UTP or 33P-UTP. The nucleotide mixture was then digested with DNase, and the labeled probes were purified and collected by using resin spin columns (GE Healthcare). The labeled probes were diluted (106 dpm/ml) in a hybridization solution containing 50% formamide, 10 mm Tris-HCl, pH 7.5 (Invitrogen), 1% sheared salmon sperm DNA (Sigma-Aldrich), 5 mg of tRNA (Invitrogen), 2.5% total yeast RNA (Sigma), 100 mm dithiothreitol, 10% dextran sulfate, 0.6 m NaCl, 0.5 mm EDTA, pH 8.0, 0.1% SDS, 0.1% sodium thiosulfate, and 1× Denhardt's solution (Sigma). Hybridization solution and a coverslip were applied to each slide, and sections were incubated overnight, at 57°C. On the following day, sections were incubated in 0.02% RNase A (Roche Applied Bioscience) solution and submitted to stringency washes. Sections were then dehydrated and enclosed in x-ray film cassettes with Kodak BioMax MR-2 film (Carestream Molecular Imaging) for 3 d. Subsequently, the slides were dipped in Kodak NTB autoradiographic emulsion (Carestream Molecular Imaging), dried, and stored at 4°C for 15 d. Slides were developed with Dektol developer and Fixer (Kodak/Carestream Molecular Imaging), counterstained with thionin, dehydrated, cleared in xylenes, and coverslipped with Permaslip (Newcomer Supply). Control procedures included hybridization with sense probes.
Dual-label immunohistochemistry/in situ hybridization.
The procedure was a modification of that described previously (Liu et al., 2003). Series of brain sections containing the hypothalamus of LepR-IRES-Cre/LacZ mice or of mice treated with leptin or saline were rinsed with DEPC-treated PBS, pH 7.0, for 1 h and in 0.1% sodium borohydride (Sigma) in DEPC-PBS for 15 min. Sections were then incubated for 10 min in 0.25% acetic anhydride in 0.1 m TEA. The Fos and p110α 35S-labeled riboprobes or the p110β33P-labeled riboprobe were diluted to 106 cpm/ml in hybridization buffer as described and applied to series of hypothalamic sections. Sections were hybridized overnight at 50°C. The following day, tissue was rinsed four times in 4× sodium chloride/sodium citrate (SSC) and was incubated in 0.02% RNase A (Roche Applied Bioscience) diluted in 0.5 m NaCl, 10 mm Tris-HCl, pH 8.0, and 1 mm EDTA for 30 min at 37°C. Sections were submitted to stringency wash in 0.1× SSC for 60 min at 55°C. Series of sections of mice treated with leptin or saline were incubated in anti-pSTAT3 antisera raised in rabbit (1:2000, Cell Signaling Technology) for 48 h and processed for standard immunoperoxidase reaction using ABC kit (Vector). Series of sections from LepR-IRES-Cre/LacZ mice were incubated in anti-βGalactosidase (1:10,000, Abcam, cat# ab9361) antisera and processed for standard immunoperoxidase reaction using ABC kit (Vector). Tissue was mounted onto SuperFrost Plus slides (Fisher Scientific) and dehydrated in increasing concentrations of ethanol. After air drying, slides were placed in x-ray film cassettes with BMR-2 film (Kodak) for 1–3 d. Slides were then dipped in NTB2 photographic emulsion (Kodak), dried overnight, and stored in desiccant-containing foil-wrapped slide boxes at 4°C for 1–3 weeks. Slides were developed with Dektel developer (Kodak), dehydrated in increasing concentration of ethanol, cleared in xylenes, and coverslipped with Permaslip. Sections were analyzed with a Zeiss Axioplan microscope.
Estimates of cell counts.
The estimates of cell counts were performed as previously described (Elias et al., 1999). Briefly, the relative number of dual-labeled neurons was counted in one side and one rostrocaudal level of specific nuclei. We quantified the colocalization of pSTAT3-ir and Fos mRNA in the PMV and the colocalization of βGal-ir and p110α or p110β mRNA in the arcuate nucleus, the dorsomedial nucleus of the hypothalamus, and the PMV using a 10× and 20× objectives. Only cells containing silver grains 3× above background levels overlaying a brown (DAB stained) nucleus (pSTAT3-ir) or cytoplasm (βGal-ir) were considered positive. The data were not corrected for double counting because the objects we were counting did not change in size, and section thickness did not vary between groups. Any systematic error should be identical for all groups. Hence, as all double-label studies are inherently qualitative, our results are meant to provide relative data, but are not meant to be accurate estimates of absolute cell counts. Data are presented as mean ± SEM.
Production of photomicrographs.
Photomicrographs were produced by capturing images with a digital camera (Axiocam, Zeiss) mounted directly on the microscope (Zeiss Axioplan). Image-editing software (Adobe Photoshop CS3) was used to combine photomicrographs into plates. Only the sharpness, contrast, and brightness were adjusted.
Electrophysiology.
Whole-cell patch-clamp recordings were performed on 113 LepR PMV neurons from 65 mice (36 male and 29 female) maintained in hypothalamic slice preparations, and data analysis was performed as previously described (Hill et al., 2008b; Williams et al., 2010). An additional 12 LepR PMV neurons were recorded from 6 mice (2 female and 4 male) deficient for both p110α and p110β in LepR neurons with concomitant EYFP or tdtomato reporter expression. Briefly, 4- to 16-week-old male or female mice were deeply anesthetized with intraperitoneal injection of 7% chloral hydrate and transcardially perfused with a modified ice-cold artificial CSF (ACSF) (described below), in which an equiosmolar amount of sucrose was substituted for NaCl. The mice were then decapitated, and the entire brain was removed, and immediately submerged in ice-cold, carbogen-saturated (95% O2 and 5% CO2) ACSF (126 mm NaCl, 2.8 mm KCl, 1.2 mm MgCl2, 2.5 mm CaCl2, 1.25 mm NaH2PO4, 26 mm NaHCO3, and 5 mm glucose). A brain block containing the hypothalamus was made. Coronal sections (250 μm) were cut with a Leica VT1000S Vibratome and then incubated in oxygenated ACSF at room temperature for at least 1 h before recording. Slices were transferred to the recording chamber and allowed to equilibrate for 10–20 min before recording. The slices were bathed in oxygenated ACSF (32°C–34°C) at a flow rate of ∼2 ml/min. The pipette solution for whole-cell recording was modified to include an intracellular dye (Alexa Fluor 594, Alexa Fluor 488, or Alexa Fluor 350) for whole-cell recording: 120 mm K-gluconate, 10 mm KCl, 10 mm HEPES, 5 mm EGTA, 1 mm CaCl2, 1 mm MgCl2, and 2 mm MgATP, 0.03 mm Alexa Fluor 594, Alexa Fluor 488, or Alexa Fluor 350 hydrazide dye, pH 7.3. Epifluorescence was briefly used to target fluorescent cells, at which time the light source was switched to infrared differential interference contrast imaging to obtain the whole-cell recording (Zeiss Axioskop FS2 Plus equipped with a fixed stage and a QuantEM:512SC electron-multiplying charge-coupled device camera). Electrophysiological signals were recorded using an Axopatch 700B amplifier (Molecular Devices), low-pass filtered at 2–5 kHz, and analyzed offline on a PC with pCLAMP programs (Molecular Devices). Recording electrodes had resistances of 2.5–5 MΩ when filled with the K-gluconate internal solution. Input resistance was assessed by measuring voltage deflection at the end of the response to a hyperpolarizing rectangular current pulse steps (500 ms of −10 to −50 pA). For some experiments measuring ramp current–voltage (I–V) relationships, the K-gluconate internal solution was replaced with Cs-gluconate, and the extracellular solution contained sodium, potassium, Ih (HCN), and calcium channel blockers (126 mm NaCl, 5 mm 4-aminopyridine, 2.8 mm KCl, 1.2 mm MgCl2, 100 μm CdCl2, 2.5 mm CaCl2, 1 mm CsCl2, 1.25 mm NaH2PO4, 26 mm NaHCO3, 1 μm TTX, and 5 mm glucose). Membrane potential values were compensated to account for junction potential (−8 mV).
Leptin (100 nm; provided by A. F. Parlow, through the National Hormone and Peptide Program) was added to the ACSF for specific experiments. Solutions containing leptin were typically perfused for 2–4 min. A drug effect was required to be associated temporally with peptide application, and the response had to be stable within a few minutes. A neuron was considered depolarized or hyperpolarized if a change in membrane potential was at least 2 mV in amplitude. After recording, slices were fixed with 4% formalin in PBS at 4°C overnight. After washing in PBS, slices were mounted onto slides, covered in Vectashield (Vector Laboratories), and coverslipped to reduce photo-oxidation during visualization with fluorescent light. Cells were then visualized with ApoTome imaging system (Imager Z1; Zeiss) to identify post hoc the anatomical location of the recorded neuron.
Drugs.
TTX, SKF96365, and 2-APB were obtained from Tocris Bioscience. All solutions were made according to manufacturer's specifications. Stock solutions of SKF96365 and 2-APB were made by dissolution in DMSO (Sigma). The concentration of DMSO in the external solution was <0.1%. Stock solutions of leptin were made by dissolution in d-PBS (Invitrogen). Stock solutions of TTX were made by dissolution in deionized water.
Data analysis.
Statistical data are expressed as mean ± SEM, where n represents the number of cells/animals studied. The significance of differences between groups was evaluated using unpaired two-tailed Student's t test with a confidence level of p < 0.05 (*). Degrees of freedom (DF) for t statistics are marked as t(DF).
Results
PMV neurons in rats and mice express LepRs and are directly engaged by circulating leptin as peripheral leptin administration induces the expression of SOCS-3 mRNA, pSTAT3 and Fos protein in PMV neurons (Elmquist et al., 1997, 1998; Elias et al., 2000, 2001; Leshan et al., 2009). To identify LepR-expressing neurons in the PMV, mice expressing Cre under the control of the endogenous Lepr promoter (LepRb-IRES-Cre mice) (DeFalco et al., 2001) were crossed to various reporter mice that carry a Cre-inducible R26R-EYFP (The Jackson Laboratory #006148; Fig. 1A), R26R-LacZ (The Jackson Laboratory, #003309), or R26R-tdtomato (The Jackson Laboratory, #007908) allele. In our reporter mice (LepR-LacZ), we previously demonstrated that 94 ± 3% of all LacZ-immunoreactive neurons in the PMV expressed pSTAT3 following leptin administration; thus, LepR-LacZ expression identifies neurons that are a potential direct target of leptin (Scott et al., 2009). Additionally, using dual-label immunohistochemistry/in situ hybridization, we found that a high proportion of pSTAT3-immunoreactive neurons within the PMV also express Fos mRNA (76.8 ± 11.9% neurons; t(2) = 11.19; n = 3; Fig. 1B). Fos expression has been widely accepted as an indirect marker for neuronal activation; thus, we hypothesized that leptin directly stimulates a high percentage of PMV neurons expressing LepR.
Figure 1.
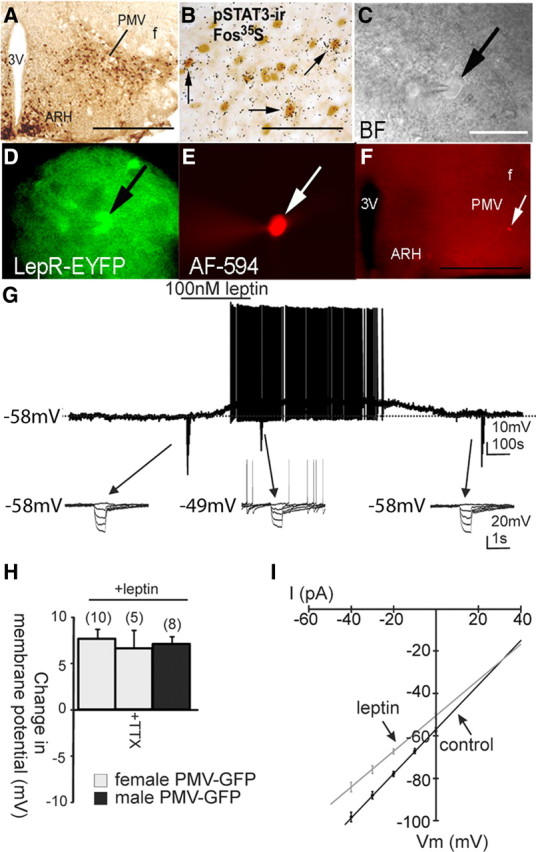
Leptin causes depolarization of PMV neurons that express leptin receptors. A–E, Identification of PMV-LepR-EYFP cells for whole-cell patch-clamp recordings. A, Distribution of GFP immunoreactivity (GFP-ir) in the PMV of LepR-EYFP mice. B, Immunohistochemistry coupled with in situ hybridization histochemistry (ISHH) demonstrates that some LepR-PMV neurons express Fos-mRNA following intraperitoneal leptin. The neurons containing clusters of silver grains were hybridized with 35S-labeled Fos riboprobe. The pSTAT3-immunoreactive neurons contain brown nuclear reaction product. Single arrows indicate cells positive for both Fos-ISHH and pSTAT3-IR. C, Bright-field illumination of a PMV-LepR-EYFP neuron during acquisition of a whole-cell recording (arrow indicates the targeted cell). D, The same neuron under fluorescent (FITC) illumination. E, Image showing the complete dialysis of Alexa Fluor-594 (AF-594) from the intracellular pipette at the end of the recording. F, Post hoc identification of recorded neuron within the PMV and in relation to anatomical landmarks (ARH, arcuate nucleus; 3V, third ventricle; f, fornix). G, Current-clamp recording of cell depicted in previous panels (C–F) demonstrating that leptin (100 nm) application results in a depolarization of most PMV LepR-EYFP neurons. The depolarization reversed 20 min following washout of leptin. Downward deflections represent voltage deflections to hyperpolarizing current injections. H, Histogram showing the depolarization of PMV LepR-EYFP neurons in response to leptin. I, I–V plot from a group (n = 14) of PMV-LepR-EYFP neurons illustrates a characteristic reduction in input resistance during the leptin-induced depolarization. Arrows indicate responses before and during leptin application (reversal = −24 mV). Scale bars: A, F, 400 μm; B, 80 μm; (in C) C–E, 50 μm.
Effects of leptin on LepR PMV membrane potential
To better assess the acute effects of leptin on PMV neurons, whole-cell patch-clamp recordings were performed on LepR-EYFP neurons anatomically confined to the PMV (Figs. 1A,C–F, 2A,B). LepR PMV neurons were recorded at rest in current-clamp mode. Alternatively, neurons recorded in voltage-clamp mode were transiently monitored for changes in resting membrane potential by periodically removing voltage clamp (i.e., switch to I = 0) to monitor resting membrane potential. Neurons were dialyzed with Alexa Fluor 594 or Alexa Fluor 350 hydrazide dye throughout the recording. Recorded neurons were identified as EYFP-labeled PMV neurons via real-time visualization under fluorescence microscopy and via post hoc identification of Alexa Fluor dye (Fig. 1C–F). In current-clamp mode, leptin (100 nm) caused a depolarization from rest in 10 of 13 PMV-EYFP neurons in female mice (77%, +7.7 ± 0.9 mV; resting membrane potential, −57.5 ± 1.6 mV; n = 10; Fig. 1G) and 8 of 11 PMV-EYFP neurons in slices from male mice (75%, +7.1 ± 0.8 mV; resting membrane potential, −56.5 ± 1.2 mV; n = 8). The remaining three neurons in each group were hyperpolarized from rest (−7.0 ± 2.4 mV; in females, n = 3; and −6.7 ± 1.2 mV; in males, n = 3; Fig. 2). Similar to the effects of leptin observed in hypothalamic (Spanswick et al., 1997, 2000; Hill et al., 2008b) and brainstem nuclei (Williams and Smith, 2006; Williams et al., 2007), subsequent application of tolbutamide (200 μm) completely reversed the leptin-induced hyperpolarization in LepR PMV neurons (n = 3), suggesting that leptin hyperpolarizes a subset of LepR PMV neurons via activation of a Katp channel. The differential responses suggests that at least two populations of leptin-responsive PMV neurons exist, one that is directly activated and one that is directly inhibited by leptin. These data demonstrate that the effect of leptin on the acute cellular activity of LepR PMV neurons was identical for both male and female mice. Thus, unless otherwise indicated, the data from both sexes will be reported together for the remainder of the manuscript.
Figure 2.
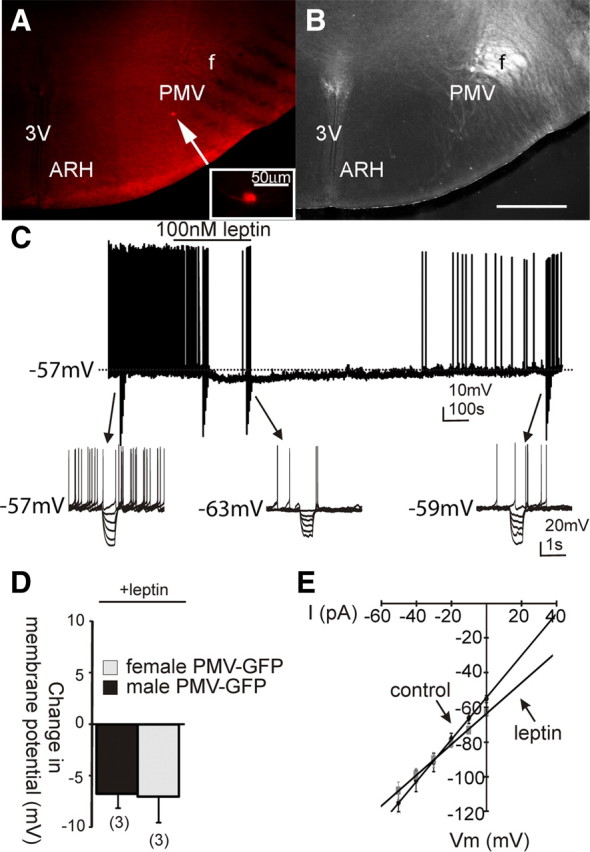
Leptin causes a hyperpolarization in a subset of PMV LepR-EYFP neurons. A, Image illustrating post hoc identification of a recorded neuron that hyperpolarized in response to leptin. Recorded PMV LepR-EYFP neuron was dialyzed with Alexa Fluor-594 throughout recording. The arrow indicates the targeted cell. B, Same slice under dark-field optics, for reference. C, Current-clamp recording of the same cell as in A demonstrating a leptin-induced (100 nm) hyperpolarization observed in a subpopulation of all PMV LepR-EYFP neurons. The hyperpolarization reversed 15 min after washout of leptin. D, Histogram depicting the hyperpolarization observed in male and female PMV LepR-EYFP neurons after leptin application. E, I–V plot from a group (n = 6) of PMV LepR-EYFP neurons illustrates a reduction in input resistance observed during the leptin-induced hyperpolarization. Arrows indicate responses before and during leptin application (reversal = −88 mV). Scale bars: A, B, 200 μm.
Similar to reports describing acute leptin activation in other regions of the hypothalamus (Cowley et al., 2001; Dhillon et al., 2006; Hill et al., 2008b), the depolarization was long lasting in most of the cells such that the depolarization did not reverse during the recording (n = 14, from male and female mice). However, four cells completely recovered membrane potential within 20–30 min after washout (Fig. 1G). In the presence of 2 μm TTX, leptin depolarized all PMV-EYFP cells tested (6.6 ± 1.9 mV; n = 5; Fig. 1H), indicative of a direct membrane depolarization independent of action-potential-mediated synaptic transmission. Conversely, all non-EYFP neurons tested were unaffected by leptin (1.2 ± 0.4 mV, n = 5), suggesting that the effects of leptin are specific to neurons that express leptin receptors.
A subset (n = 22) of LepR PMV neurons were recorded in voltage clamp under normal recording conditions at a membrane potential of −50 mV, and changes in whole-cell current after leptin (100 nm) application were observed. Application of leptin resulted in an inward current in 15 of 22 neurons (−11.5 ± 1.2 pA, n = 15; Fig. 3). Of the remaining neurons, leptin application resulted in an outward current in six neurons (+9.9 ± 2.9, n = 6), while one cell was unresponsive to superfusion of leptin. Together these data demonstrate that leptin predominately activates an inward current contributing to the excitation of LepR PMV neurons. However, a subset of LepR PMV neurons are inhibited by a leptin-activated outward conductance, putative Katp channel, suggesting that the effects of leptin in the PMV mirror those observed in the arcuate POMC and NPY/AgRP neuronal populations, respectively.
Figure 3.

Leptin excites LepR-PMV neurons via the activation of TRPC channels. A, Leptin application produced an inward current in the majority of LepR-PMV neurons. Holding potential was −50 mV. B, Current–voltage relationships were examined in the same cell from A by applying voltage ramps (−130 to 10 mV in 1.4 s, 100 mV/s). Subtraction of the control (black) response from that in the presence of leptin (gray) reveals the I–V curve (slope conductance). Extrapolation of the linear portion of the slope conductance demonstrates the I–V relation of the leptin-induced current, which reversed (Erev) at −20 mV. C, I–V responses of a separate cell were obtained with Cesium in the intracellular pipette and in the presence of 4-AP (5 mm), cesium chloride (1 mm), cadmium chloride (100 μm), and TTX (1 μm). D, The I–V relationship obtained from subtracting the control from the leptin-induced current in C. E, F, Absence of a leptin-induced change in membrane potential in LepR-PMV neurons that are pretreated with either 2APB (100 μm) or SKF96365 (100 μm), respectively. G, Histogram illustrating the leptin-induced responses of identified LepR-PMV neurons recorded in the presence of 2APB and SKF96365.
Leptin effects on the neuronal excitability of LepR PMV neurons
LepR PMV neurons that were depolarized in response to leptin were subjected to a rectangular current step protocol (400 ms; ±10–50 pA) to obtain an I–V plot. In male and female mice, the depolarization was accompanied by a 20% decrease in whole-cell input resistance, such that the input resistance was reduced from 1059 ± 281 MΩ in control ACSF to 852 ± 211 MΩ in leptin (n = 14; t(13) = 5.71; p = 0.000137, paired two-tailed t test; Fig. 1I). Extrapolation of the slope conductance in control and leptin-containing ACSF revealed a reversal potential of −24.5 ± 3.5 mV (n = 14; Fig. 1I), which suggests a putative mixed-cation channel involved in the leptin-induced depolarization of PMV cells. Six neurons that were hyperpolarized in response to leptin (100 nm) were subjected to the same rectangular current step protocol to obtain an I–V plot. All six neurons exhibited a decrease in the whole-cell input resistance, suggestive of an increased conductance (1205.2 ± 77.9 MΩ in control ACSF; 1034.3 ± 61.2 MΩ in leptin; n = 6; t(5) = 5.54; p < 0.05, paired t test; Fig. 2E). The I–V plot revealed a reversal potential of the leptin-induced hyperpolarization that was near the equilibrium potential for K+ (−88.0 ± 5.6 mV; n = 6; Fig. 2E), supporting a leptin-induced activation of a potassium conductance, putative Katp channel, in this subset of PMV neurons.
The leptin-induced depolarization of LepR PMV neurons is concomitant with an activated conductance with a reversal potential indicative of a putative mixed-/nonselective-cation channel. To better assess changes in membrane conductance, a subset of LepR PMV neurons were transiently recorded in voltage clamp. Current–voltage relationships were examined by applying voltage ramps (−130 mV to 10 mV in 1.4 s, 100 mV/s) from a holding potential of −50 mV in five neurons that were also depolarized in response to leptin. Extrapolation of the linear portion of the net current revealed a reversal potential (−25.5 ± 3.9 mV, n = 5; Fig. 3B). To better isolate the leptin-activated conductance, changes in membrane conductance were further examined in a subset of LepR PMV neurons in which cesium was used as the primary cation in the recording pipette and the potassium channel antagonist 4-AP (5 mm), the calcium channel blocker cadmium chloride (100 μm), the Ih blocker cesium chloride (1 mm), and the voltage-gated sodium channel antagonist TTX (1 μm) were added to the extracellular medium. Application of leptin resulted in an inward current at −50 mV of −11.7 ± 1.7 pA in six of nine neurons tested (n = 6; Fig. 3C,D). The subtracted current revealed a net current with a reversal potential of −28.4 ± 3.7 mV (n = 6, Fig. 3D). The remaining three neurons were unaffected by leptin in this recording condition. Collectively, these data suggest that leptin activates a mixed-/nonselective-cation whole-cell conductance independent of afferent inputs in LepR PMV neurons.
Leptin excites LepR PMV neurons via activation of a TRPC channel
Leptin-induced inward currents in POMC neurons have recently been attributed to the activation of TRPC channels (Qiu et al., 2010). Given the electrophysiological properties of the leptin-activated current observed in the present study (Fig. 3C,D), we hypothesized that TRPC channels may also mediate the acute excitatory effects of leptin on LepR PMV neurons. To directly assess the role of TRPC channels in the leptin-dependent depolarization of LepR PMV neurons, we used the TRPC channel antagonists, SKF96365 (100 μm) and 2-APB (100 μm) (Qiu et al., 2010). Preapplication of SKF96365 completely prevented the depolarization of LepR PMV neurons by leptin in 11 of 12 neurons examined (0.1 ± 0.4 mV, n = 11; Fig. 3F). Interestingly, the one remaining neuron was hyperpolarized in the presence of SKF96365, suggesting that the leptin-induced hyperpolarization occurs independent of a TRPC channel. Moreover, as observed in control conditions, tolbutamide (200 μm) completely reversed the hyperpolarization, further supporting the involvement of a Katp channel in the leptin-induced hyperpolarization of LepR PMV neurons. Similarly, all neurons were unresponsive to leptin when pretreated with 2-APB (−0.5 ± 0.4 mV; n = 11; Fig. 3E). These data support the involvement of TRPC channels in the leptin-induced neuronal activation of LepR PMV neurons while a putative Katp channel is responsible for the leptin-induced inhibition of LepR PMV neurons.
PMV neurons that putatively express LepR coexpress PI3K catalytic subunits
Leptin activates an ATP-sensitive K+ conductance via a PI3K-mediated mechanism in hypothalamic cell lines and hypothalamic brain slices (Spanswick et al., 1997, 2000; Mirshamsi et al., 2004), including NPY neurons of the arcuate nucleus (van den Top et al., 2004) and neurons in the dorsal vagal complex (Williams and Smith, 2006; Williams et al., 2007). Similarly, a PI3K-mediated mechanism has been implicated in the leptin-induced depolarization of arcuate POMC neurons(Hill et al., 2008b). In numerous cell types, PI3K acts in a heterodimeric form consisting of a single 85 kDa regulatory (p85α or p85β) and a single 110 kDa catalytic (p110α, p110β, or p110δ) subunit. A recent report suggests that the acute effects of leptin and insulin are dependent on either p110α and/or p110β in arcuate POMC and NPY/AgRP neurons (Al-Qassab et al., 2009). To assess whether LepR PMV neurons coexpress PI3K catalytic subunits Pik3ca (p110α) and Pik3cb (p110β), we performed dual-label in situ hybridization immunohistochemistry in hypothalamic sections of LepR-LacZ mice. We found that both subunits display similar expression patterns throughout the hypothalamus (Table 1). Both genes were particularly abundant in nuclei related to energy homeostasis, including the ventromedial, the dorsomedial, and the arcuate nuclei (Hill et al., 2009), and also in the PMV. We also noticed that Pik3ca displays higher expression levels compared to Pik3cb throughout the hypothalamus (data not shown). Because Pik3cb showed lower expression levels, we used a radioisotope with higher activity (33P-UTP versus 35S-UTP used for Pik3ca) to determine the degree of colocalization with the LepR reporter gene (Fig. 4A–F). In general, we found a higher percentage of colocalization between the LepR reporter gene and Pik3ca compared to Pik3cb (Table 1). In the PMV, of the total neurons that putatively express LepR, 90% coexpress Pik3ca while 65% coexpress Pik3cb.
Table 1.
Percentage of colocalization between βGalactosidase-immunoreactive (βGal-ir) neurons and p110α or p110β mRNA
| Hypothalamic nuclei | Atlas levels | Total βGal-ir |
% doubles/total βGal-ir |
||
|---|---|---|---|---|---|
| P110α | P110β | P110α | P110β | ||
| Arc | 48 | 80.3 ± 11.1 | 87.0 ± 5.3 | 87 ± 3.0 | 64 ± 8.6 |
| DMHv | 48 | 77.5 ± 23.5 | 71.3 ± 8.1 | 92 ± 1.9 | 58 ± 11 |
| PMV | 50 | 77.0 ± 1.0 | 129.8 ± 6.7 | 88 ± 2.0 | 66 ± 12 |
Values represent mean number of cells ± SEM (n = 3–4). Arc, Arcuate nucleus; DMHv, dorsomedial nucleus of the hypothalamus, ventral part. The atlas level designation corresponds to those described by Paxinos and Franklin (2001).
Figure 4.
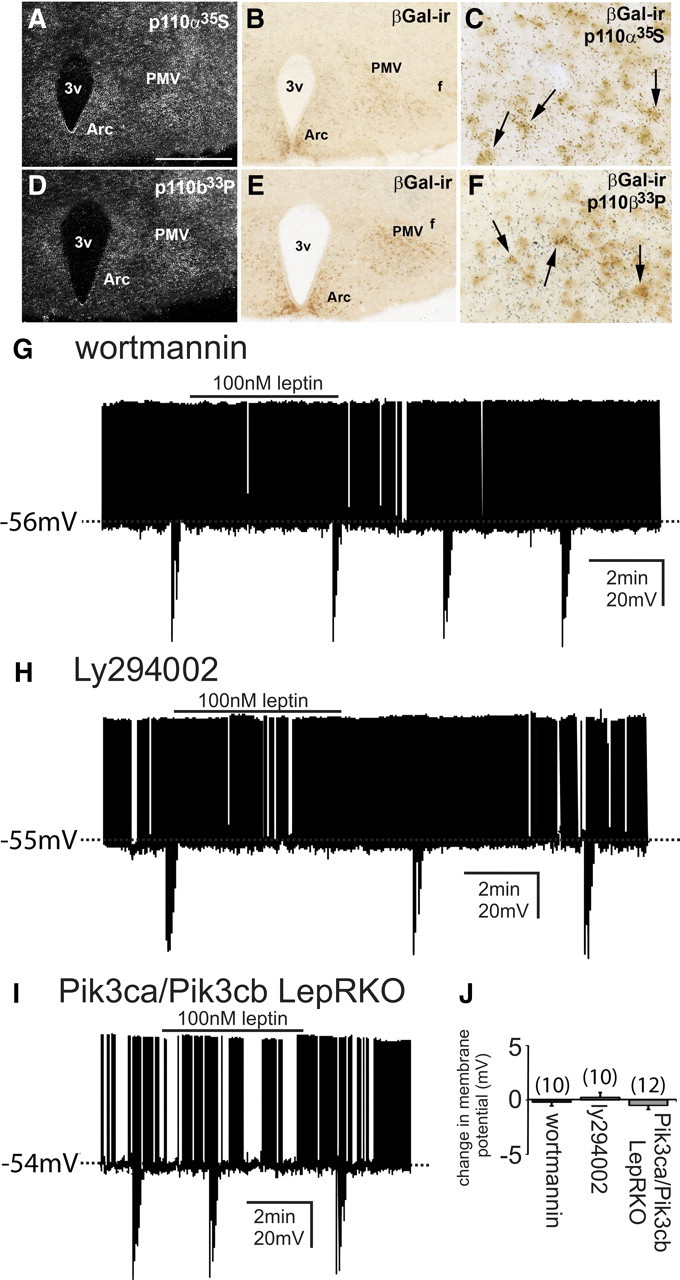
Leptin alters the neuronal activity of LepR-PMV neurons via a PI3K-dependent mechanism. A–F, Colocalization of p110α-mRNA and p110β-mRNA with PMV-LepR-LacZ immunoreactivity. A, D, Clusters of silver grains demonstrate the distribution of 35S-labeled p110α-mRNA and 33P-labeled p110β-mRNA, respectively. B, E, Same sections from A and D illustrating the distribution of βGal immunoreactivity (βGal-ir) in the PMV of LepR-LacZ mice. C, F, High-powered image depicting the colocalization of 35S-labeled p110α-mRNA and 33P-labeled p110β-mRNA with βGal-ir in the PMV of LepR-LacZ mice. Single arrows indicate cells positive for both p110α-ISHH and βGal-ir (C) and p110β-ISHH and βGal-ir (F). G–I, Current-clamp recordings demonstrated that disruption of PI3K signaling occluded the leptin-induced depolarization and hyperpolarization of LepR-PMV neurons from rest. G, H, Absence of a leptin-induced change in membrane potential in LepR-PMV neurons that are pretreated with either wortmannin (100 nm) or LY294002 (10 μm), respectively. I, Leptin failed to alter the membrane potential of LepR-PMV neurons from Pik3ca/Pik3cb LepRKO mice. J, Histogram illustrating the leptin-induced responses of identified LepR-PMV neurons recorded in the presence of either wortmannin or LY294002 or from Pik3ca/Pik3cb LepRKO mice. Scale bar: A, D, 400 μm; B, E, C, F, 80 μm.
Mechanisms of the leptin-induced change in neuronal activity of LepR PMV neurons
To determine whether PI3K is responsible for the leptin-induced changes in neuronal excitability of LepR PMV neurons, slices were pretreated with the selective PI3K inhibitors, wortmannin and LY294002. When pretreated with wortmannin (100 nm), leptin failed to either depolarize or hyperpolarize all PMV neurons targeted (−0.2 ± 0.4 mV; n = 10; Fig. 4G). Similarly, the PI3K inhibitor LY294002 (10 μm) blocked the leptin-induced alteration in the membrane potential in all LepR PMV neurons examined (0.3 ± 0.4 mV; n = 10; Fig. 4H), suggesting the involvement of PI3K in both the leptin-induced depolarization and hyperpolarization of LepR PMV neurons.
Targeted deletion of PI3K catalytic subunits in LepR PMV neurons disrupts the acute leptin effects on neuronal activity
Although both wortmannin and LY294002 are selective inhibitors of PI3K signaling at the concentrations used in the current study, both inhibitors have also been shown to inhibit multiple protein kinases (Cross et al., 1995; Nakanishi et al., 1995; Davies et al., 2000). Therefore, to further delineate the involvement of PI3K in the acute effect of leptin to alter the cellular activity of PMV neurons, we generated mice with impaired PI3K signaling in LepR PMV neurons. To generate mice with impaired PI3K signaling in LepR-PMV neurons, we obtained mice carrying a conditional mutation in the PI3K catalytic subunits Pik3ca (p110α) and Pik3cb (p110β) (Zhao et al., 2006; Jia et al., 2008). Transgenic mice expressing Cre driven by Lepr regulatory elements (DeFalco et al., 2001) were crossed to mice in which loxP sites flank exon 1 of the p110α gene pik3ca (Zhao et al., 2006). The resulting (Pik3ca LepRKO) mice were then crossed with mice in which loxP sites flank exon 2 of the p110β gene pik3cb (Jia et al., 2008). By interbreeding the resulting mice (Pik3ca/Pik3cb LepRKO) with EYFP or tdtomato mice, we were able to produce mice deficient for both p110α and p110β in LepR neurons with concomitant EYFP or tdtomato reporter expression. Leptin application failed to influence the membrane potential in all LepR PMV neurons examined from Pik3ca/Pik3cb LepRKO mice (−0.5 ± 0.4 mV; n = 12; Fig. 4I). Similarly, leptin also failed to influence any change in holding current in LepR PMV neurons from Pik3ca/Pik3cb LepRKO mice (−1.0 ± 0.8 pA; n = 10). These data suggest that PI3K signaling is required for the leptin-induced changes in neuronal excitability of LepR-expressing PMV neurons.
Discussion
Our present results demonstrated that leptin acutely activates the majority of LepR PMV neurons while a subset of LepR PMV neurons are inhibited by leptin. Similar to the effects of leptin observed in hypothalamic and brainstem nuclei (Spanswick et al., 1997; Williams and Smith, 2006; Williams et al., 2007; Hill et al., 2008b; Al-Qassab et al., 2009), the acute actions of leptin within the PMV require PI3K signaling. In addition, we demonstrated that a putative TRPC channel underlies the stimulatory activity of leptin within the PMV, while a leptin-induced activation of a putative Katp channel inhibits LepR PMV neurons. These results further define the acute activity of leptin within the PMV, and provide a potential cellular mechanism for a leptin-induced regulation of reproductive function.
Divergent signaling cascades activated by leptin: a role for PI3K in the PMV
Similar to a recent report, we found that ∼75% of all LepR PMV neurons are depolarized in response to leptin (Leshan et al., 2009). Leshan et al. (2009) had also suggested that the remaining neurons were unresponsive to leptin; however, we found the majority of the remaining neurons were hyperpolarized in response to leptin. Perhaps the hyperpolarizing effect of leptin is less surprising given that we targeted PMV neurons that specifically express the leptin receptor. As a result, one would expect leptin to activate multiple signaling cascades specifically in these neurons, ultimately leading to a change in cellular activity. Moreover the acute effects of leptin in LepR PMV neurons mirrors previously observed effects of leptin within arcuate POMC and NPY/AgRP neurons. The leptin receptor, a member of the interleukin-6 receptor family of type I cytokine receptors, is predominately noted for activating the JAK-STAT signaling pathway in mediating effects on energy and glucose homeostasis (Williams et al., 2009, 2011). However, recent studies have demonstrated that leptin activates multiple signaling cascades with each arm contributing to different facets of energy and glucose homeostasis and fertility and pubertal development (Hill et al., 2008a; Williams et al., 2009, 2011). For instance, Bates et al. (2003) demonstrated that disruption of leptin-activated STAT3 by a targeted mutation of the leptin receptor causes dramatic hyperphagia and decreased energy expenditure, resulting in profound obesity similar to db/db mice. However, whereas db/db mice are infertile, the s/s mutant mice are fertile, suggesting that another signaling cascade activated by leptin is responsible for the effects of leptin on fertility. Similarly, a neuronal (CaMKIIα-Cre)-specific deletion of Shp2, a Src homology 2-containing tyrosine phosphatase linked to the enhancement of the Erk pathway, resulted in early-onset obesity and several diabesity metabolic profile characteristics similar to that observed in the db/db and s/s mice (Zhang et al., 2004). Analogous to the s/s mice, the neuronal Shp2−/− remained fertile, ultimately supporting a role of Shp2 (Erk pathway) in regulating energy and glucose homeostasis, while Shp2 is not required for the regulation of fertility or pubertal development in neurons. Although complete loss of either catalytic or regulatory subunits of PI3K is lethal, recent studies have begun to investigate the role of PI3K in the regulation of the reproductive function via similar targeted strategies as described above (Fruman et al., 2000; Bi et al., 2002; Acosta-Martinez et al., 2009). Future studies will have to further delineate the physiological effects of leptin action within the PMV; however, the current study supports a role for an acute leptin-induced regulation of the PMV via a PI3K-dependent mechanism.
PI3K is required for the acute effects of leptin within the PMV
A recent study described that acute activation of arcuate POMC neurons results in a reduction in food intake (Aponte et al., 2011). Interestingly, the acute ability of leptin to reduce food intake has been directly linked to the PI3K-induced activation of a mixed cation, putative TRPC, channel in arcuate POMC neurons (Hill et al., 2008b; Qiu et al., 2010). In the current study, we demonstrated that the leptin-induced depolarization of LepR PMV neurons was associated with an inward current that had a calculated reversal potential supporting the activation of a nonselective (mixed) cation conductance. The leptin-induced depolarization/inward current was present when cesium was used in the intracellular pipette and when slices were pretreated with potassium, voltage-gated sodium, and calcium channel blockers. However the leptin-induced depolarization/inward current was absent in the presence of the PI3K antagonists wortmannin and LY294002 and when Pik3ca and Pik3cb were selectively ablated in LepR PMV neurons (Pik3ca/Pik3cb LepRKO). Importantly, the TRPC channel antagonists SKF96365 and 2APB also prevented the leptin-induced depolarization/inward current in LepR PMV neurons. Together, we have now linked a leptin-induced PI3K-activated putative TRPC conductance in the PMV. Similarly, a subset of LepR PMV neurons was hyperpolarized in response to leptin. The hyperpolarization was concomitant with an increased conductance that had a calculated reversal potential close to EK, and the leptin-induced hyperpolarization/outward current was occluded in the presence of tolbutamide or with the use of cesium in the intracellular pipette. LepR PMV neurons were still hyperpolarized when pretreated with TRPC antagonists SKF96365 and 2APB. However, the hyperpolarization was absent in the presence of the PI3K antagonists wortmannin and LY294002, and selective deletion of the catalytic subunits of PI3K in LepR PMV neurons prevented the leptin-induced hyperpolarization. Together these data suggest that the leptin-induced hyperpolarization in LepR PMV neurons requires PI3K and ultimately activates a Katp channel, which mirrors the activity observed in arcuate NPY neurons. Although it is unlikely that the activation of a leptin-induced TRPC or Katp conductance in the PMV results in a change of feeding behavior, there is growing evidence that the acute activity of leptin within the PMV may have profound effects in the coordinated control of the reproductive function (Donato et al., 2009, 2011; Leshan et al., 2009). However, it is difficult to understand the effects on physiology without first delineating which neural circuits and to what extent these circuits are affected by acute leptin signaling.
Modeling a potential role for leptin and the PMV in reproductive development
The PMV projects to many areas implicated in reproductive control (i.e., AVPV, MPO, and directly to GnRH neurons) (Canteras et al., 1992; Elias et al., 2000; Rondini et al., 2004; Boehm et al., 2005; Leshan et al., 2009). Leptin administration to leptin-deficient subjects or ob/ob mice induces gonadotropin release and restores fertility (Barash et al., 1996; Chehab et al., 1996; Farooqi et al., 1999, 2002). Importantly, our laboratory recently demonstrated that the PMV is a key site linking changing levels of leptin with gonadotropin release and the coordinated control of reproduction (Donato et al., 2009). Thus, the current study may reveal a cellular correlate in which leptin, acting as a permissive factor, directly regulates the reproductive function via the activation of a PI3K-dependent change in PMV cellular activity resulting in the release of gonadotropins.
In conclusion, our results suggest that the PI3K pathway, through its control of PMV neuronal activity, plays a role in the detection of short-term alterations in leptin levels as occurs during the postnatal “leptin surge” (Mantzoros et al., 1997; Ahima et al., 1998) or during food deprivation (Ahima et al., 1996). It remains to be seen whether these data support a role for the leptin-PI3K pathway within the PMV to directly regulate different aspects of the reproductive physiology. Undoubtedly, cre-mediated deletion of PI3K subunits in targeted neuronal subtypes will be a powerful tool for formally examining these possibilities.
Footnotes
This work was supported by NIH Grant HD061539 and Regent's Scholar Research Award (University of Texas Southwestern Medical Center at Dallas) grants (to C.F.E.), American Diabetes Association Grant 7-07-MN-17 (to J.-W.S.), NIH Grant K01DK087780 (to K.W.W.), and NIH Grants R01DK53301, R01MH61583, R01DK088423, and RL1DK081185 (to J.K.E.). This work was also supported by NIH Grants PL1 DK081182 and UL1RR024923.
The authors declare no competing financial interests.
References
- Acosta-Martínez M, Luo J, Elias C, Wolfe A, Levine JE. Male-biased effects of gonadotropin-releasing hormone neuron-specific deletion of the phosphoinositide 3-kinase regulatory subunit p85alpha on the reproductive axis. Endocrinology. 2009;150:4203–4212. doi: 10.1210/en.2008-1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahima RS, Prabakaran D, Mantzoros C, Qu D, Lowell B, Maratos-Flier E, Flier JS. Role of leptin in the neuroendocrine response to fasting. Nature. 1996;382:250–252. doi: 10.1038/382250a0. [DOI] [PubMed] [Google Scholar]
- Ahima RS, Dushay J, Flier SN, Prabakaran D, Flier JS. Leptin accelerates the onset of puberty in normal female mice. J Clin Invest. 1997;99:391–395. doi: 10.1172/JCI119172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahima RS, Prabakaran D, Flier JS. Postnatal leptin surge and regulation of circadian rhythm of leptin by feeding. Implications for energy homeostasis and neuroendocrine function. J Clin Invest. 1998;101:1020–1027. doi: 10.1172/JCI1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Qassab H, Smith MA, Irvine EE, Guillermet-Guibert J, Claret M, Choudhury AI, Selman C, Piipari K, Clements M, Lingard S, Chandarana K, Bell JD, Barsh GS, Smith AJ, Batterham RL, Ashford ML, Vanhaesebroeck B, Withers DJ. Dominant role of the p110beta isoform of PI3K over p110alpha in energy homeostasis regulation by POMC and AgRP neurons. Cell Metab. 2009;10:343–354. doi: 10.1016/j.cmet.2009.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aponte Y, Atasoy D, Sternson SM. AGRP neurons are sufficient to orchestrate feeding behavior rapidly and without training. Nat Neurosci. 2011;14:351–355. doi: 10.1038/nn.2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barash IA, Cheung CC, Weigle DS, Ren H, Kabigting EB, Kuijper JL, Clifton DK, Steiner RA. Leptin is a metabolic signal to the reproductive system. Endocrinology. 1996;137:3144–3147. doi: 10.1210/endo.137.7.8770941. [DOI] [PubMed] [Google Scholar]
- Bates SH, Stearns WH, Dundon TA, Schubert M, Tso AW, Wang Y, Banks AS, Lavery HJ, Haq AK, Maratos-Flier E, Neel BG, Schwartz MW, Myers MG., Jr STAT3 signalling is required for leptin regulation of energy balance but not reproduction. Nature. 2003;421:856–859. doi: 10.1038/nature01388. [DOI] [PubMed] [Google Scholar]
- Bi L, Okabe I, Bernard DJ, Nussbaum RL. Early embryonic lethality in mice deficient in the p110beta catalytic subunit of PI 3-kinase. Mamm Genome. 2002;13:169–172. doi: 10.1007/BF02684023. [DOI] [PubMed] [Google Scholar]
- Boehm U, Zou Z, Buck LB. Feedback loops link odor and pheromone signaling with reproduction. Cell. 2005;123:683–695. doi: 10.1016/j.cell.2005.09.027. [DOI] [PubMed] [Google Scholar]
- Canteras NS, Simerly RB, Swanson LW. Projections of the ventral premammillary nucleus. J Comp Neurol. 1992;324:195–212. doi: 10.1002/cne.903240205. [DOI] [PubMed] [Google Scholar]
- Chan JL, Heist K, DePaoli AM, Veldhuis JD, Mantzoros CS. The role of falling leptin levels in the neuroendocrine and metabolic adaptation to short-term starvation in healthy men. J Clin Invest. 2003;111:1409–1421. doi: 10.1172/JCI17490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chehab FF, Lim ME, Lu R. Correction of the sterility defect in homozygous obese female mice by treatment with the human recombinant leptin. Nat Genet. 1996;12:318–320. doi: 10.1038/ng0396-318. [DOI] [PubMed] [Google Scholar]
- Clayton PE, Gill MS, Hall CM, Tillmann V, Whatmore AJ, Price DA. Serum leptin through childhood and adolescence. Clin Endocrinol (Oxf) 1997;46:727–733. doi: 10.1046/j.1365-2265.1997.2081026.x. [DOI] [PubMed] [Google Scholar]
- Coleman DL. Obese and diabetes: two mutant genes causing diabetes-obesity syndromes in mice. Diabetologia. 1978;14:141–148. doi: 10.1007/BF00429772. [DOI] [PubMed] [Google Scholar]
- Cowley MA, Smart JL, Rubinstein M, Cerdán MG, Diano S, Horvath TL, Cone RD, Low MJ. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001;411:480–484. doi: 10.1038/35078085. [DOI] [PubMed] [Google Scholar]
- Cross MJ, Stewart A, Hodgkin MN, Kerr DJ, Wakelam MJ. Wortmannin and its structural analogue demethoxyviridin inhibit stimulated phospholipase A2 activity in Swiss 3T3 cells. Wortmannin is not a specific inhibitor of phosphatidylinositol 3-kinase. J Biol Chem. 1995;270:25352–25355. doi: 10.1074/jbc.270.43.25352. [DOI] [PubMed] [Google Scholar]
- Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFalco J, Tomishima M, Liu H, Zhao C, Cai X, Marth JD, Enquist L, Friedman JM. Virus-assisted mapping of neural inputs to a feeding center in the hypothalamus. Science. 2001;291:2608–2613. doi: 10.1126/science.1056602. [DOI] [PubMed] [Google Scholar]
- de Luca C, Kowalski TJ, Zhang Y, Elmquist JK, Lee C, Kilimann MW, Ludwig T, Liu SM, Chua SC., Jr Complete rescue of obesity, diabetes, and infertility in db/db mice by neuron-specific LEPR-B transgenes. J Clin Invest. 2005;115:3484–3493. doi: 10.1172/JCI24059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhillon H, Zigman JM, Ye C, Lee CE, McGovern RA, Tang V, Kenny CD, Christiansen LM, White RD, Edelstein EA, Coppari R, Balthasar N, Cowley MA, Chua S, Jr, Elmquist JK, Lowell BB. Leptin directly activates SF1 neurons in the VMH, and this action by leptin is required for normal body-weight homeostasis. Neuron. 2006;49:191–203. doi: 10.1016/j.neuron.2005.12.021. [DOI] [PubMed] [Google Scholar]
- Donato J, Jr, Silva RJ, Sita LV, Lee S, Lee C, Lacchini S, Bittencourt JC, Franci CR, Canteras NS, Elias CF. The ventral premammillary nucleus links fasting-induced changes in leptin levels and coordinated luteinizing hormone secretion. J Neurosci. 2009;29:5240–5250. doi: 10.1523/JNEUROSCI.0405-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donato J, Jr, Cravo RM, Frazão R, Gautron L, Scott MM, Lachey J, Castro IA, Margatho LO, Lee S, Lee C, Richardson JA, Friedman J, Chua S, Jr, Coppari R, Zigman JM, Elmquist JK, Elias CF. Leptin's effect on puberty in mice is relayed by the ventral premammillary nucleus and does not require signaling in Kiss1 neurons. J Clin Invest. 2011;121:355–368. doi: 10.1172/JCI45106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias CF, Aschkenasi C, Lee C, Kelly J, Ahima RS, Bjorbaek C, Flier JS, Saper CB, Elmquist JK. Leptin differentially regulates NPY and POMC neurons projecting to the lateral hypothalamic area. Neuron. 1999;23:775–786. doi: 10.1016/s0896-6273(01)80035-0. [DOI] [PubMed] [Google Scholar]
- Elias CF, Kelly JF, Lee CE, Ahima RS, Drucker DJ, Saper CB, Elmquist JK. Chemical characterization of leptin-activated neurons in the rat brain. J Comp Neurol. 2000;423:261–281. [PubMed] [Google Scholar]
- Elias CF, Lee CE, Kelly JF, Ahima RS, Kuhar M, Saper CB, Elmquist JK. Characterization of CART neurons in the rat and human hypothalamus. J Comp Neurol. 2001;432:1–19. doi: 10.1002/cne.1085. [DOI] [PubMed] [Google Scholar]
- Elmquist JK, Ahima RS, Maratos-Flier E, Flier JS, Saper CB. Leptin activates neurons in ventrobasal hypothalamus and brainstem. Endocrinology. 1997;138:839–842. doi: 10.1210/endo.138.2.5033. [DOI] [PubMed] [Google Scholar]
- Elmquist JK, Bjørbaek C, Ahima RS, Flier JS, Saper CB. Distributions of leptin receptor mRNA isoforms in the rat brain. J Comp Neurol. 1998;395:535–547. [PubMed] [Google Scholar]
- Farooqi IS, Jebb SA, Langmack G, Lawrence E, Cheetham CH, Prentice AM, Hughes IA, McCamish MA, O'Rahilly S. Effects of recombinant leptin therapy in a child with congenital leptin deficiency. N Engl J Med. 1999;341:879–884. doi: 10.1056/NEJM199909163411204. [DOI] [PubMed] [Google Scholar]
- Farooqi IS, Matarese G, Lord GM, Keogh JM, Lawrence E, Agwu C, Sanna V, Jebb SA, Perna F, Fontana S, Lechler RI, DePaoli AM, O'Rahilly S. Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J Clin Invest. 2002;110:1093–1103. doi: 10.1172/JCI15693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruman DA, Mauvais-Jarvis F, Pollard DA, Yballe CM, Brazil D, Bronson RT, Kahn CR, Cantley LC. Hypoglycaemia, liver necrosis and perinatal death in mice lacking all isoforms of phosphoinositide 3-kinase p85 alpha. Nat Genet. 2000;26:379–382. doi: 10.1038/81715. [DOI] [PubMed] [Google Scholar]
- Hill JW, Elmquist JK, Elias CF. Hypothalamic pathways linking energy balance and reproduction. Am J Physiol Endocrinol Metab. 2008a;294:E827–832. doi: 10.1152/ajpendo.00670.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JW, Williams KW, Ye C, Luo J, Balthasar N, Coppari R, Cowley MA, Cantley LC, Lowell BB, Elmquist JK. Acute effects of leptin require PI3K signaling in hypothalamic proopiomelanocortin neurons in mice. J Clin Invest. 2008b;118:1796–1805. doi: 10.1172/JCI32964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JW, Xu Y, Preitner F, Fukuda M, Cho YR, Luo J, Balthasar N, Coppari R, Cantley LC, Kahn BB, Zhao JJ, Elmquist JK. Phosphatidyl inositol 3-kinase signaling in hypothalamic proopiomelanocortin neurons contributes to the regulation of glucose homeostasis. Endocrinology. 2009;150:4874–4882. doi: 10.1210/en.2009-0454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia S, Liu Z, Zhang S, Liu P, Zhang L, Lee SH, Zhang J, Signoretti S, Loda M, Roberts TM, Zhao JJ. Essential roles of PI(3)K-p110beta in cell growth, metabolism and tumorigenesis. Nature. 2008;454:776–779. doi: 10.1038/nature07091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leshan RL, Louis GW, Jo YH, Rhodes CJ, Münzberg H, Myers MG., Jr Direct innervation of GnRH neurons by metabolic- and sexual odorant-sensing leptin receptor neurons in the hypothalamic ventral premammillary nucleus. J Neurosci. 2009;29:3138–3147. doi: 10.1523/JNEUROSCI.0155-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Kishi T, Roseberry AG, Cai X, Lee CE, Montez JM, Friedman JM, Elmquist JK. Transgenic mice expressing green fluorescent protein under the control of the melanocortin-4 receptor promoter. J Neurosci. 2003;23:7143–7154. doi: 10.1523/JNEUROSCI.23-18-07143.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantzoros CS, Flier JS, Rogol AD. A longitudinal assessment of hormonal and physical alterations during normal puberty in boys. V. Rising leptin levels may signal the onset of puberty. J Clin Endocrinol Metab. 1997;82:1066–1070. doi: 10.1210/jcem.82.4.3878. [DOI] [PubMed] [Google Scholar]
- Mirshamsi S, Laidlaw HA, Ning K, Anderson E, Burgess LA, Gray A, Sutherland C, Ashford ML. Leptin and insulin stimulation of signalling pathways in arcuate nucleus neurones: PI3K dependent actin reorganization and KATP channel activation. BMC Neurosci. 2004;5:54. doi: 10.1186/1471-2202-5-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagatani S, Guthikonda P, Thompson RC, Tsukamura H, Maeda KI, Foster DL. Evidence for GnRH regulation by leptin: leptin administration prevents reduced pulsatile LH secretion during fasting. Neuroendocrinology. 1998;67:370–376. doi: 10.1159/000054335. [DOI] [PubMed] [Google Scholar]
- Nakanishi S, Catt KJ, Balla T. A wortmannin-sensitive phosphatidylinositol 4-kinase that regulates hormone-sensitive pools of inositolphospholipids. Proc Natl Acad Sci U S A. 1995;92:5317–5321. doi: 10.1073/pnas.92.12.5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin C. The mouse brain in stereotaxic coordinates. Ed 3. Sydney: Academic; 2001. [Google Scholar]
- Qiu J, Fang Y, Rønnekleiv OK, Kelly MJ. Leptin excites proopiomelanocortin neurons via activation of TRPC channels. J Neurosci. 2010;30:1560–1565. doi: 10.1523/JNEUROSCI.4816-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinton ND, Smith RF, Clayton PE, Gill MS, Shalet S, Justice SK, Simon SA, Walters S, Postel-Vinay MC, Blakemore AI, Ross RJ. Leptin binding activity changes with age: the link between leptin and puberty. J Clin Endocrinol Metab. 1999;84:2336–2341. doi: 10.1210/jcem.84.7.5834. [DOI] [PubMed] [Google Scholar]
- Rondini TA, Baddini SP, Sousa LF, Bittencourt JC, Elias CF. Hypothalamic cocaine- and amphetamine-regulated transcript neurons project to areas expressing gonadotropin releasing hormone immunoreactivity and to the anteroventral periventricular nucleus in male and female rats. Neuroscience. 2004;125:735–748. doi: 10.1016/j.neuroscience.2003.12.045. [DOI] [PubMed] [Google Scholar]
- Scott MM, Lachey JL, Sternson SM, Lee CE, Elias CF, Friedman JM, Elmquist JK. Leptin targets in the mouse brain. J Comp Neurol. 2009;514:518–532. doi: 10.1002/cne.22025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spanswick D, Smith MA, Groppi VE, Logan SD, Ashford ML. Leptin inhibits hypothalamic neurons by activation of ATP-sensitive potassium channels. Nature. 1997;390:521–525. doi: 10.1038/37379. [DOI] [PubMed] [Google Scholar]
- Spanswick D, Smith MA, Mirshamsi S, Routh VH, Ashford ML. Insulin activates ATP-sensitive K+ channels in hypothalamic neurons of lean, but not obese rats. Nat Neurosci. 2000;3:757–758. doi: 10.1038/77660. [DOI] [PubMed] [Google Scholar]
- Srinivas S, Watanabe T, Lin C-S, William CM, Tanabe Y, Jessell TM, Costantini F. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol. 2001;1:4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartaglia LA, Dembski M, Weng X, Deng N, Culpepper J, Devos R, Richards GJ, Campfield LA, Clark FT, Deeds J, Muir C, Sanker S, Moriarty A, Moore KJ, Smutko JS, Mays GG, Wool EA, Monroe CA, Tepper RI. Identification and expression cloning of a leptin receptor, OB-R. Cell. 1995;83:1263–1271. doi: 10.1016/0092-8674(95)90151-5. [DOI] [PubMed] [Google Scholar]
- van den Top M, Lee K, Whyment AD, Blanks AM, Spanswick D. Orexigen-sensitive NPY/AgRP pacemaker neurons in the hypothalamic arcuate nucleus. Nat Neurosci. 2004;7:493–494. doi: 10.1038/nn1226. [DOI] [PubMed] [Google Scholar]
- Watanobe H, Suda T, Wikberg JE, Schiöth HB. Evidence that physiological levels of circulating leptin exert a stimulatory effect on luteinizing hormone and prolactin surges in rats. Biochem Biophys Res Commun. 1999;263:162–165. doi: 10.1006/bbrc.1999.1331. [DOI] [PubMed] [Google Scholar]
- Welt CK, Chan JL, Bullen J, Murphy R, Smith P, DePaoli AM, Karalis A, Mantzoros CS. Recombinant human leptin in women with hypothalamic amenorrhea. N Engl J Med. 2004;351:987–997. doi: 10.1056/NEJMoa040388. [DOI] [PubMed] [Google Scholar]
- Williams KW, Smith BN. Rapid inhibition of neural excitability in the nucleus tractus solitarii by leptin: implications for ingestive behaviour. J Physiol. 2006;573:395–412. doi: 10.1113/jphysiol.2006.106336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams KW, Zsombok A, Smith BN. Rapid inhibition of neurons in the dorsal motor nucleus of the vagus by leptin. Endocrinology. 2007;148:1868–1881. doi: 10.1210/en.2006-1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams KW, Scott MM, Elmquist JK. From observation to experimentation: leptin action in the mediobasal hypothalamus. Am J Clin Nutr. 2009;89:985S–990S. doi: 10.3945/ajcn.2008.26788D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams KW, Margatho LO, Lee CE, Choi M, Lee S, Scott MM, Elias CF, Elmquist JK. Segregation of acute leptin and insulin effects in distinct populations of arcuate proopiomelanocortin neurons. J Neurosci. 2010;30:2472–2479. doi: 10.1523/JNEUROSCI.3118-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams KW, Scott MM, Elmquist JK. Modulation of the central melanocortin system by leptin, insulin, and serotonin: co-ordinated actions in a dispersed neuronal network. Eur J Pharmacol. 2011;660:2–12. doi: 10.1016/j.ejphar.2010.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang EE, Chapeau E, Hagihara K, Feng GS. Neuronal Shp2 tyrosine phosphatase controls energy balance and metabolism. Proc Natl Acad Sci U S A. 2004;101:16064–16069. doi: 10.1073/pnas.0405041101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- Zhao JJ, Cheng H, Jia S, Wang L, Gjoerup OV, Mikami A, Roberts TM. The p110alpha isoform of PI3K is essential for proper growth factor signaling and oncogenic transformation. Proc Natl Acad Sci U S A. 2006;103:16296–16300. doi: 10.1073/pnas.0607899103. [DOI] [PMC free article] [PubMed] [Google Scholar]