

Abstract
With the continued failures of both early diagnosis and treatment options for pancreatic cancer, it is now time to comprehensively evaluate the role of the immune system on the development and progression of pancreatic cancer. It is important to develop strategies that harness the molecules and cells of the immune system to treat pancreatic cancer. This review will focus primarily on the role of immune cells in the development and progression of pancreatic ductal adenocarcinoma (PDAC). We will evaluate what is known about the interaction of immune cells with the tumor microenvironment and their role in tumor growth and metastasis. We will conclude with a brief discussion of therapy for pancreatic cancer and the potential role for immunotherapy. We hypothesize that the role of the immune system in tumor development and progression is tissue specific. Our hope is that better understanding of this process will lead to better treatments for this devastating disease.
Keywords: pancreatic cancer, immune response, immunotherapy, immune
Section I: Introduction
Pancreatic cancer has a fatality rate of 95%. Early detection is rare and the majority of patients present with locally advanced or metastatic disease with no real hope of an effective treatment. With the current treatment strategies, the median life expectancy is 6–10 and 3–6 months for patients presenting with locally advanced disease or metastatic disease, respectively.
For the purposes of this review we will be discussing the Immunology of PDAC, the major type of pancreatic cancer. We hypothesize that the role of the immune system in tumor development and progression is tissue specific. Our hope is that better understanding of the immunological aspects of PDAC will lead to better treatments for this devastating disease.
Basic Immunology Review
1. THE INNATE IMMUNE RESPONSE
Several cell types of the innate immune system can recognize “danger”, i.e., pathogens, tumors and damaged tissues. These include neutrophils, macrophages, dendritic cell (DCs), mast cells and natural killer (NK) cells. NK cells can also be involved in recognizing cells infected with intracellular pathogens that down-regulate major histocompatibility complex (MHC) antigens and express viral antigens or altered self-antigens. Activated cells of the innate immune system can release molecules such as cytokines [interferons (IFNs), interleukins (ILs), colony stimulating factors (CSF)] and chemokines (CC). These molecules can lead to cell migration, local and systemic inflammation, and ultimately alert the adaptive immune system.
2. THE ADAPTIVE IMMUNE RESPONSE
While the specificity of the innate immune response is limited to toll-like receptors (TLRs) and several other conserved molecules that NK cells recognize, the cells of the adaptive immune response have enormous diversity and can recognize tens of millions of antigenic determinants or epitopes. A DC that has taken up an antigen matures as it leaves the site of a wound or infection, and completes its journey to the regional lymph node. DCs can degrade large antigens and present peptides or lipids in human leukocyte antigen (HLA) molecules of either the Class I or Class II types or in CD1 molecules. In the first instance, CD8+ cytotoxic T cells (CTL or Tc) are activated and in the second, CD4+ T helper (Th) cells are activated. Mature Tc cells can kill infected cells directly. The activated Th cells can interact with naive B cells that also recognize the corresponding specific epitope(s) on the native molecule and provide co-stimulation for further differentiation. Both Tc and Th cells make cytokines that interact with B cells so that they eventually produce antibodies that are specific for epitopes on that antigen. Th cells are also responsible for controlling affinity maturation and isotype switching so that the antibodies produced are highly effective at eliminating a pathogen. This antibody can neutralize, opsonize and/or kill infected cells and pathogens.
3. TUMOR IMMUNOLOGY
Immune response against tumor cells can involve both innate and adaptive immune responses. An effective anti-tumor immune response involves recognition of tumor-associated antigens (TAAs) by the immune system and the generation of T or B cell responses that will kill the tumor cells but not damage life-sustaining normal tissue. Tc cells can kill the tumor cells directly. Paradoxically, both Tc and Th cells produce cytokines that can inhibit or enhance the growth of the tumor. They also help B cells differentiate into memory cells as well as plasma cells that make antibodies against the tumor. These antibodies can kill or opsonize the tumor cells, stimulate or inhibit their growth or actually block Tc cells from killing the tumor cells. As in other tumor models, the immune system in patients with PDAC appears to have several roles. One role is to prevent tumor development by recognizing and removing abnormal cells arising from normal pancreatic cells. In this situation the immune system exerts an “anti-tumor response”. But, the immune system can also provide a “pro-tumor response”, whereby components of the immune response can stimulate the growth of tumor cells directly or indirectly by dampening the anti-tumor immune response.
Section II: The Role of Immune System - “Anti versus Pro-Tumor Response”
PDAC is an exocrine tumor that develops from the epithelial cells that line pancreatic ducts. However, it is a complex environment composed of many cell types including immune cells, pancreatic stellate cells (fibroblasts), vascular endothelial cells, endocrine cells and nerve cells. These cells can interact with tumor cells to disrupt the normal tissue architecture to form the dense stroma and the dynamic environment found in PDAC. Although it is currently well accepted that the immune response is determined by an invading pathogen or “danger” signal, Matzinger et al. (2011) have recently provided a new perspective on how immune responses are determined. The authors suggest that the tissue rather than the pathogen itself determines the type of immune response. The idea that the tissues control the effector phase of the immune response has arisen from a better understanding of immunologic phenomena of immune-privilege sites, oral tolerance and oral vaccination1. This concept is slowly gaining support. The basic understanding of tissue-specific factors that control immune function may be critical in fully understanding the immune response not only for invading pathogens, but also for tumor development and progression. Exploration of this new hypothesis may shed light on the highly variable immune responses observed when tumors arise from different organs. However, unless we truly understand the epidemiology (i.e., genetic pre-disposition, chronic inflammation, viral infection) of a cancer we will never fully understand the role and complexity of the immune response to that tumor. Nevertheless we are making progress towards defining the role of the immune system in regulating the growth of malignant cells as recently reviewed by Schreiber et al. (2011)2.
Lymphocytes are considered the main effector cells for the anti-tumor immune response. The lymphocytic cell populations are predominantly found in the stroma surrounding the tumor mass with few or no lymphocytes in the actual tumor mass3,4. This surrounding stroma has a large population of CD4+ T lymphocytes and macrophages with a small population of B lymphocytes and plasma cells3. In patients with PDAC, no correlation was found between the numbers of tumor infiltrating lymphocytes (TILs) and the number of circulating lymphocytes. However, PDAC patients tend to have decreased numbers of circulating lymphocytes as compared to healthy individuals and individuals with chronic pancreatitis4.
The role of CD4+ T cells in PDAC immunity is poorly understood but depending upon the cytokine environment, CD4+ T cells can differentiate into Th1, Th2, Th17 or Treg cells. Th1 cells produce IL-2 and IFN-γ and induce B cells to make opsonizing antibodies. Th2 cells produce IL-4, IL-5 and IL-6 and induce B cells to make neutralizing antibodies. In cancer, as a general trend, the main immune response is mediated through Th2 cells. Currently, therapy for PDAC focuses on cellular immunity and “direct tumor cell killing” but humoral immunity could be just as important. Hence, understanding this complex balance between Th1 and Th2 responses in PDAC is crucial in developing better therapies for this disease. There are a few reports of the CD4+ T cell responses in PDAC. Tassi et al. (2008) compared CD4+ T cell responses in patients with PDAC to those of healthy donors and found that the former had impaired anti-carcinoembryonic antigen (CEA)-specific but not anti-viral specific CD4+ T cell immunity5. Interestingly, in healthy donors CEA -specific CD4+ T cell immunity was significantly higher and produced mainly granulocyte-macrophage CSF (GM-CSF) and IFN-γ, whereas CD4+ T cells from patients with PDAC produced IL-5. However, there was no difference in the anti-viral CD4+ T cell response between the two. This study suggests that in PDAC CD4+ T cell immunity is skewed towards a Th2 type immune response and that this is locally mediated at the tumor site5. On the other hand, some studies support a more systemic Th2-like cytokine expression profile following CD4+ T cell activation6. A possible explanation for this discrepancy may be best explained by the stage of disease of the patients in the studies. In the former study, the patients were at an earlier stage of disease either stage 1 or 2 but in the later study, the majority of patients where at later stages of disease either stage 3 or 4. We assume that in tumor progression the immune response is initially capable of eliminating tumor cells and is therefore most likely a Th1-skewed response, because this response is activated by intracellular “danger” signals (altered self-proteins produced by tumor cells) and leads to cell mediated immunity (IFN-γ and activation of macrophages) to eliminate tumor cells. However, it is also possible that an early Th2 response occurs, an adaptive immune response more effective at removing extracellular “danger signals” that lead to IL-4 production and neutralizing antibodies. Hence, the two responses could be competing with or potentially enhancing each other, ultimately leading to the development of Treg cells which dampen the response as a protective measure to prevent autoimmunity. Therefore, understanding the type and function of immune cells in PDAC as well as the time line of the immune response, will facilitate the development of immunotherapeutic strategies to use at different stages of disease. For example, if at early stage of disease in PDAC, both Th1 and Th2 responses are active there might be a more effective anti- tumor response. However, at later stages of disease if Th2 responses are more beneficial the best strategy might be to shift the balance towards a Th2 response. The role of the immune system in the development and progression of pancreatic cancer is a powerful and dynamic tool that we must understand and apply strategically to promote anti-tumor responses at specific stages of disease.
In contrast to the direct anti/pro-tumor activity of Th1 and Th2 CD4+ T cells, Th17 and Treg cells can regulate all T-cell responses. The differentiation of CD4+ T cells into either Th17 or Treg cells appears to involve a precarious balance between the transforming growth factor beta (TGF-β)- driven expression of forkhead box P3 (FoxP3) expression (which drives the development of Treg cells) and the production of TGF-β/IL-6 which favor the development of Th17 cells and inhibits the development of Treg cells. Th17 cells secrete IL17, a pro-inflammatory cytokine that mediates several effects on several different cell types7,8. In contrast, Treg cells inhibit the proliferation of T cells and dampen anti-tumor immunity.
Research on the Th17 CD4+ T cell lineage in PDAC is limited, but recent studies have shown that if the cytokine balance of the tumor environment is tipped in favor of the development of the Th17 cell lineage by inducing IL-6 or depleting the Treg cells an anti-tumor effect is achieved9,10. The role of Th17 cells in cancer is currently under investigation. There is no definitive answer as to whether Th17 cell enhance or inhibit tumor growth. However, it has been suggested that the role of Th17 cells may change depending on the cause, type, location and stage of the tumor11. If this were correct, it would further support the hypothesis that the role of the immune system in tumor development and progression is tissue specific and that an individual immune profile of each PDAC patient should guide therapy.
The role of Treg cells in PDAC is better understood. Both circulating Treg cellsand PDAC tissue-specific Treg cellsare significantly increased in patients with pancreatic cancer as compared to healthy controls. The presence of Treg cells in the tumor tissue correlates with the stage and progression of disease12,13. Treg cellsare known to induce tolerance against TAAs and suppress the anti-tumor activity of T cells14,15. In vivo studies suggest that a decrease or depletion of Treg cells in PDAC results in inhibited tumor growth and promotion of tumor-specific immune responses16,17 and that the increase in Treg cells in PDAC is dependent upon tumor derived TGF-β18,19.
PDAC has been characterized by the presence of few tumor-specific CD8+ T cells, B cells and tumor-reactive antibody producing plasma cells in the tumor mass. Antigens on PDAC cells can elicit either cellular or humoral immune responses, and in some instances, both. Tumor antigens that are recognized by the immune system and induce a response become tumor immunogens, and these are important for immune responses. However not all tumor antigens are immunogenic. A few of these PDAC cell antigens are being explored as targets in clinical trials. Immunogens in PDAC include α-enolase (ENOA)20, coactosin-like protein (CLP)21, mesothelin22, mucin 1 (MUC1)23–25, mutant kirsten rat sarcoma virus oncogene (KRAS)26–28, cadherin 3 (CDH3)/P-cadherin29, carcinoembryonic antigen (CEA)30, human epidermal growth factor receptor 2 (HER2/neu)31, prostate stem cell antigen (PSCA)32,33 and mutant p5334,35. The peripheral blood from PDAC patients contain a high frequency of functional tumor-reactive T cells that can ultimately lead to tumor antigen-specific T cell responses36. The bone marrow also contains tumor cell- reactive memory T cells36. Moreover, when evaluated together, the presence of both CD4+ and CD8+ T cells in malignant PDAC tissues correlates with a better prognosis than the presence of either alone37. Although this suggests that there is an anti-tumor response, unfortunately it is not enough. A possible explanation for the failure of the anti-tumor response may be provided by recent identification of the antibody independent functions of B cells. Thus, effector and regulatory B cells may regulate T cell immune responses by promoting the production of effector and memory CD4+ T cellsas well as the proliferation and survival of Treg cells38.
Natural killer (NK) cells are a subset of cytotoxic lymphocytes that only recently received attention for their role in tumor development. NK cells do not express unique antigen- specific receptors, but they play an important role in innate immunity and anti-tumor immunity39. They can induce target cell killing as a result of the complex integration of inhibitory and activating signals40. NK cells can produce IFN-α, tumor necrosis factor alpha (TNF-α), GM-CSF and IL-341. Initially in PDAC research, subsets of NK cells were not distinguished, but now they are divided into two phenotypically and functionally distinct types of cells. The majority expresses low densities of CD56 (CD56lo), secrete low levels of cytokines and exert potent effector cell cytotoxicity. In contrast, the minority group expresses high levels of CD56 (CD56hi) and IL-2 receptor alpha chains (CD25), secrete high levels of cytokines and are poorly cytotoxic40. NK cells in PDAC have been reported to mediate tumor cell lysis42 and high levels of NK cells lead to a better prognosis43. However, even in early stage of disease, NK cell activity is impaired and worsens with advancing disease44,45. Interestingly, CD56hi NK cells exhibited potent reactivity on several pancreatic cancer cell lines in addition to autologous tumor cells46 that were identified from a pancreatic cancer patient undergoing immunotherapy with ipilimumab (a therapeutic antibody against CTLA-4, a T cell co-inhibitory molecule)46. Although anti-CTLA-4 antibodies block the activity of CTLA-4 and sustained immune responses in T cells, this patient had an anti-tumor response with potent NK cell activity. This supports the possibility that the activation of NK cells as well as CD4/CD8+ T cells can lead to the killing of tumor cells. Several groups have evaluated the effect of modulating NK cell activity as well as CD4+ and CD8+ T cell activity by administering IL-2 to patients, to try and promote anti-tumor responses43,46–49.
Polymorphonuclear leukocytes or neutrophils are often a neglected cell type in the tumor microenvironment, but a better understanding of their impact in tumor development is beginning to emerge. Neutrophils are the most abundant type of leukocyte found in the blood and are not usually found in normal tissues. In response to the production of IL-8 and C5a during acute inflammation, neutrophils can produce reactive oxygen species (ROS), serine proteases, and metalloproteases to kill invading pathogens. The activity of neutrophils is thought to follow a linear progression and when recruited into the tumor microenvironment can induce both pro- and anti-tumor responses50. Although the active states of neutrophils are not clearly defined, it has been proposed that moderate neutrophil activity in the tumor microenvironment can promote tumor growth and invasion due to the production of ROS and proteases. In contrast, robust neutrophil activity can be toxic to tumor cells and promote an anti-tumor response50.
Only a few studies have evaluated the potential role of neutrophils in PDAC. In two separate studies, it was found that an elevated neutrophil/lymphocyte ratio is a predictor of decreased patient survival51,52. In in vitro studies, it was found that activated neutrophils promote the adhesion of PDAC cells to microvascular endothelium53 possibly promoting tumor migration and extravasation. Furthermore, in vivo studies have found that tumor-infiltrating neutrophils produce matrix metalloprotease type 9 (MMP-9) a potent vascular endothelial growth factor (VEGF)-independent angiogenic factor that mediates the initial angiogenic switch in PDAC54,55. Much more remains to be learned with regard to the role of neutrophils in PDAC. However, one might predict that by targeting tumor-associated neutrophils or their production of ROS and proteases, tumor invasion and growth might be inhibited.
Mast cells are typically studied in the context of type I hypersensitivity and autoimmunity. However, in a recent review by Khazaie et al. (2011) the role of mast cells as positive and negative regulators of the immune response in tumor development and progression is discussed56. Mast cells typically surround blood vessels and nerves and are activated by inflammation, cross-linking of IgE, or complement proteins. Following activation, mast cells can release several mediators including histamine, serine proteases, platelet activating factor and, importantly, VEGF57. In addition, mast cells can also produce cytokines typical of Th1 cells (GM-CSF, IFN-γ, and TNF-α) and Th2 cells (IL-4 and IL-13)56. Thus mast cells can play a pivotal role in both innate and adaptive immunity as well as modifying the tumor microenvironment by producing pro-inflammatory and angiogenic factors.
There are few studies involving the role of mast cells in PDAC. In one study by Esposito et al. (2004), mast cells were associated with lymph node metastasis as well as increased tumor microvessel density suggesting that their presence promotes an angiogenic phenotype58. However, this study did not find a correlation between mast cell number and patient survival58. In a subsequent study by Strouch et al. (2010) mast cell infiltration was significantly increased in pancreatic cancer as compared to normal controls and correlated with higher-grade tumors, as well as decreased recurrence-free and disease-specific survival59. In contrast to the previous study, Strouch et al. (2010) did not find a correlation between the number of mast cells and lymph node status. The discrepancies between these two studies may again be explained by the grade of tumor evaluated, or the heterogeneity between pancreatic tumors. Hence, in the former study, higher-grade metastatic tumors were studied, whereas in the later study all tumors were grade 3 or less. Strouch et al. (2010) also evaluated the in vitro mechanism by which mast cells can contribute to the poor prognosis in patients with PDAC. They found that in the absence of direct tumor cell contact, mast cells, mediated tumor cell migration, proliferation and invasion via MMPs59. These studies provide evidence that mast cells are emerging as promoters of angiogenesis and tumor progression in patients with PDAC.
Gabrilovich et al. (2009) have recently reviewed myeloid-derived suppressor cells (MDSCs) and their role as regulatory cells in the immune system60. MDSCs consist of myeloid progenitor cells and immature myeloid cells with immunosuppressive activity in cancer and other diseases60. In cancer, MDSCs are characterized by the expression of CD33 and the lack of expression of markers for mature myeloid or lymphoid cells61. Increased numbers of MDSCs have been associated with high levels of GM-CSF62 or VEGF63 in the circulation. However, these MDSCs do not differentiate in a normal way64. Once activated, MDSCs can serve as immunosuppressive cells by up-regulating arginase 1, nitric oxide synthase and increasing nitric oxide production from M-MDSCs and ROS production by G-MDSCs60,65–67. MDSCs can also inhibit the function of T cells in several ways that are not yet entirely clear. However, there are reports that they can down-regulate T cell mediated antigen-specific responses68, down-regulate TCRs/CD3-zeta chains69 and promote the development of Treg cells70,71. Other mechanisms of MDSCs immunosuppression include secretion of TGF-β72, up-regulation of cyclo-oxygenase 2 (COX-2) and prostaglandin E2 (PGE2)73 as well as negatively regulate NK cells by inhibiting effector functions72. These issues are discussed in recent reviews60,66,74.
Several in vivo studies of pancreatic cancer have shown increased numbers of MDSCs in the tumor microenvironment75–77. In one study of spontaneous pancreatic carcinoma, it was shown that not only are the MDSCs increased in frequency but they also have arginase activity and suppress T-cell responses76. Moreover, by evaluating the suppressive mechanisms from tumor inception throughout tumor development results suggests that the suppressive mechanism exists in early pre-malignant lesions and increase during tumor progression76. In another study, of mouse pancreatic cancer, the number of MDSCs inversely correlated with CD8+ T cells infiltrates and MDSCs were present in both the primary and metastatic lesions and not merely correlated with chronic inflammation77. It is easy to appreciate that therapeutic strategies designed to either inhibit MDSCs, their products or possibly promote their differentiation should be considered to treat tumor development and progression.
Most cells in the immune system have both pro-and anti-tumor activity. Macrophages can induce T cell recruitment and activation at the tumor site, as well as promote tumor cell growth, angiogenesis and immunosuppression. Tumor associated macrophages (TAMs) are derived from blood monocytes in response to tumor -derived signals such as macrophage-CSF (M-CSF), chemokine ligand 2 (CCL2), VEGF and Angiopoietin-278–83. TAMs are functionally divided into two subtypes M1 and M2. TAMs M1 are activated in response to IFN-γ or microbial products and are characterized by production of high IL-12, IL-23, toxic intermediates and pro-inflammatory cytokines including TNF-α. The M2 subset is induced by IL-4, IL-10, IL-13, glucocorticoids and immunoglobulin (Ig) complexes. They produce TGF-β and IL-10 and promote adaptive Th2 immunity, angiogenesis, tissue remodeling and repair83.
In PDAC, the role of macrophages is beginning to be explored. Macrophages are significantly more numerous in PDAC than in normal pancreatic tissue, and their accumulation does not correlate with chronic pancreatitis-like features in the surrounding tissue58. The TAM M2 subtype has been associated with a poor prognosis84. In an in vivo mouse model when large numbers of human monocytes were co-engrafted with human tumor cells, tumor growth was enhanced. However, when a low ratio of human monocytes were co-grafted with human tumor cells inhibition of tumor growth was observed85. This group has shown that repeated contact of monocytes with tumor cells leads to decreased production of cytotoxic molecules (TNF-α, reactive oxygen intermediates and IL-12) and increased production of immunosuppressive cytokine IL-1085,86. This suggests that there may be a maximum ratio of monocytes to tumor cells and a threshold of the molecules they produce that when exceeded no longer has anti-tumor effects. In an in vitro study by Baran et al. (2009), the production of TNF-α by TAMs lead to an increased number of pancreatic tumor cells as well as macrophage motility, ultimately inducing phenotypic tumor cell changes characteristic of epithelial-mesenchymal transition (EMT)87. These studies support the hypothesis that the increase in number of TAM and their products such as TNF-α in PDAC, may overcome a certain threshold and switch from an anti-tumor to a pro-tumor response, but further studies are needed to better understand the significance of the number and type of TAMs that play a role in PDAC.
The field of tumor immunology as applied to pancreatic cancer is in its infancy but several studies support the notion that immune cells are actively engaged in eliminating tumor cells and generating anti-tumor memory cells but that the response is either not robust enough to control tumor growth or potentially, too robust and causes damage that triggers immunosuppression and subsequent tumor growth. In a review by Sica et al. (2008) the role of M1/M2 macrophages in tumor development was hypothesized. The authors hypothesized that early in the course of tumor development, macrophages with an M1 phenotype (high IL-12, IL-23, toxic intermediates and TNF-α production) dominate and the production of pro-inflammatory cytokines and toxic intermediates support tumor formation. Once the tumor is established macrophages with the M2 phenotype (TGF-β, IL-10 production) dominate, thereby impairing the anti-tumor Th1 response and promoting tumor growth83. This supports the hypothesis of an early active Th1 response (production of anti-tumor T cells, NK cells, antibodies and cytokines) that becomes less effective as the disease progresses. This is accompanied by increased numbers of Treg cells and Th2 cytokine production. However, the time line of the immune response in PDAC is questionable. Clark et al. (2007) studied the immune response in pancreatic cancer from early disease to invasive cancer in a murine model of spontaneous PDAC. They found that early on there were few effector T cells and that the majority of infiltrating cells were macrophages, MDSCs and Treg cells77. Their findings suggest that from the inception of PDAC the immune system is suppressed and is never able to mount a robust anti-tumor response77,88. This might represent normal immune tolerance to self-tissue since most antigens on tumor cells are not recognized as foreign. Moreover, due to the vast heterogeneity seen in PDAC, both an early active Th1 response and suppression may occur. The balance between the two effects could be dependent upon the etiology of the disease as well as the immune system of the patient in question.
Dendritic cells (DCs) are responsible for the recognition of “danger”, i.e. pathogens as well as damaged tissue, activation of immunity and preservation of tolerance to self-antigens89. They constitute the critical link between the innate and adaptive immune systems since they can traffic from damaged/invaded tissue sites to regional lymph nodes and present antigen to T cells. Once this occurs the adaptive immune system is activated. DCs play a critical role in initiating the immune response against developing tumors. However, tumor progression and the influence of the tumor microenvironment can inhibit DC recruitment, differentiation, maturation and survival90. In a recent review by Ma et al. (2010) the mechanisms by which tumor cells regulate dendritic cells are thoroughly discussed. In brief, several factors are involved in the dendritic cell-tumor cell cross-talk including GM-CSF, VEGF, TGF-β, IL-10 and ROS90. Tumor cells can produce or express various metabolites or proteins that can prevent DCs from engulfing, recruiting, differentiating, migrating, activating, and cross-presenting antigens, thereby inhibiting a tumor-specific T cell response90. Furthermore, tumor cell death may either establish tumor-induced tolerance or enhance immune responses by exposing “cell death-associated patterns” that can ultimately induce a variety of innate immune responses90.
In PDAC, DCs are rare but when present, are located on the outside margin of the tumor91. A study evaluating the influence of circulating myeloid DCs (c-m-DCs), circulating lymphoid DCs (c-l-DCs) and DCs within the tumor on patient survival, it was found that a high percentage of c-m-DCs or high numbers of DCs in the tumor prolonged survival92. Other studies found that blood myeloid DCs in PDAC were only “partially mature” and the change in their expression of surface markers led to an impairment of their immunostimulatory function93. This change was also observed in patients with chronic pancreatitis suggesting that systemic inflammatory factors may play a role in this change93,94. In addition, it appears that preservation of mature blood DCs correlates with disease control and prolonged survival93,94. Several therapeutic strategies involve the vaccination of enhanced DC number and function in combination with other immune modulators and/or chemotherapy.
The future of immunotherapy will be dependent upon elucidating the roles of immune cell subtypes and their capacity to function or dysfunction at various stages during the development of pancreatic tumors.
Section III: Immune Interaction with Microenvironment
PDAC is a hypoxic environment with a dense stroma, which can comprise up to 90% of the tumor volume95–97. It is now understood that this dense stroma is derived from overgrowth of the extracellular matrix (ECM) and can “protect” and enhance tumor development by forming a barrier against both chemotherapeutic drugs as well as the immune system98. The production of fibroblast growth factor type 2 (FGF-2), epidermal growth factor (EGF) as well as the EGF receptor (EGFR)99, transforming growth factor alpha and beta type 1 (TGF-α100, TGF-β1), insulin-like growth factor-1 (IGF-1), platelet derived growth factor (PDGF) and VEGF by both tumor cells, immune cells and other stromal cells contribute to stromal production as well as tumor cell survival and growth99,101–105.
Pancreatic stellate cells (PSCs) are myofibroblast-like cells and considered to be the main pancreatic cancer-associated stromal fibroblasts. These cells are recognized to be the key players in the development of desmoplasia as recently reviewed by Duner et al. (2011)106. When PSCs are activated by stress, cytokines or growth factors they become ECM protein-producing fibroblasts. In addition to producing ECM protein, stellate cells secrete periostin107, as well as produce MMP-2/gelatinase-A, MMP-9/gelatinase-B108 and MMP-12109. Periostin is a protein that enhances the fibrogenic activity of PSCs while promoting endothelial cell growth and motility107,109. MMP-2, MMP-9 can break down components of the basement membrane and help promote angiogenesis, ultimately leading to local invasion and disease progression108,110,111. On the other hand, MMP-12 can induce the production of an anti-angiogenic molecule, endostatin. Endostatin is a cleavage product of type XVIII collagen and can inhibit the proliferation of endothelial cells and ultimately, angiogenesis112. PSCs are the main producers of VEGF in the tumor microenvironment107. In a study of tumor cell-stellate cell interactions in PDAC by Erkan et al. (2009), it was found that while tumor cells induce secretion of VEGF by PSCs, PSCs increase endostatin production of tumor cells109. Although this balance between pro- and anti-angiogenic effects probably involves a variety of factors in addition to VEGF and endostatin, it is useful in understanding the stimulatory and inhibitory forces at play in the PDAC microenvironment (Figure 1A). Pancreatic stellate cells provide a promising target for pancreatic cancer therapy. A recent study has shown that the specific up-regulation of hedgehog receptor smoothened (SMO) gene expression activates the sonic hedgehog signaling pathway in pancreatic cancer-associated stromal fibroblasts but not in normal pancreatic tissue113. This finding led to the development of SMO antagonists to target stromal fibroblasts in the tumor microenvironment. This is a challenging approach that relies heavily on interstitial fluid pressure for drug delivery. However, with the improved penetration of chemotherapeutic drugs into the tumor mass along with anti-angiogenic agents that help stabilize local vasculature SMO antagonists may find clinical utility for the treatment of PDAC.
Figure 1. A–B Interaction between PDAC and the Microenvironment.
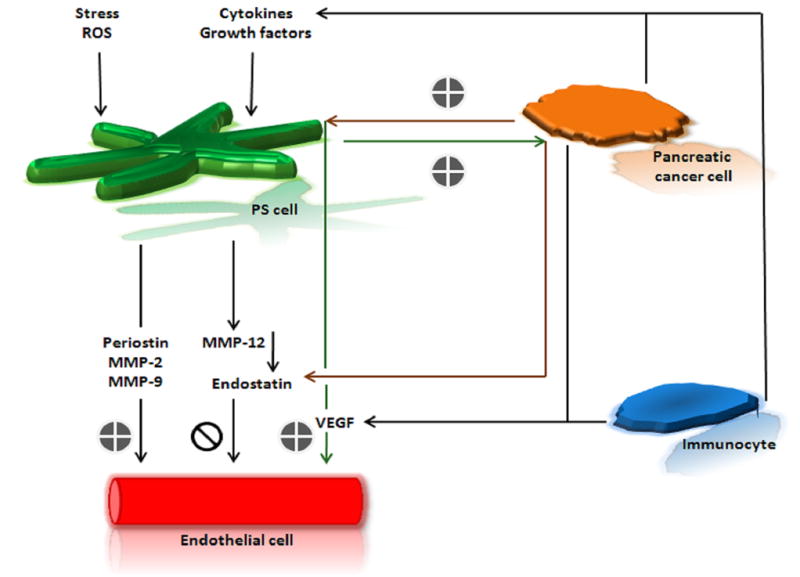
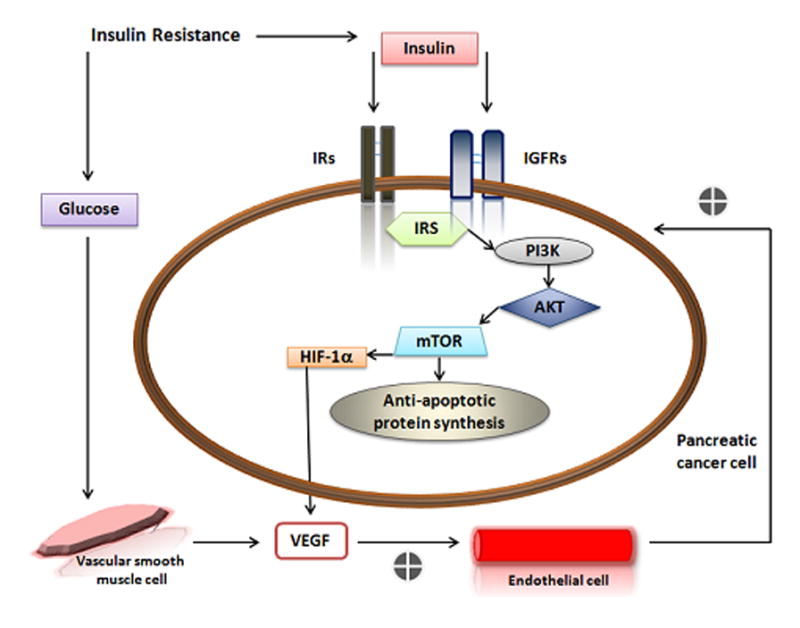
A. The pro- and anti-angiogenic function of Pancreatic Stelate (PS) cells. Immunocytes can release cytokines and growth factors that promote neo angiogenesis as well as activate PS cells. Upon activation by stress, ROS, cytokines and/or growth factors PS cells can secrete periostin to mediate endothelial cell adhesion and migration as well as secrete MMPs. MMPs can both promote neo angiogenesis through basal membrane destruction (MMP-2, MMP-9) or inhibit neo angiogenesis by triggering production of endostatin (MMP-12). Pancreatic cancer cells can also promote neo angiogenesis by stimulating PS cells to secrete VEGF or inhibit neo angiogenesis by increasing endostatin secretion. B. Relationship between insulin resistance and pancreatic cancer development and survival. Insulin resistance can lead to increased insulin and glucose in the blood. When the level of IGF-1 is low in the tumor microenviroment, IGFRs and IRs that can be overexpressed on cancer cells are free for insulin to bind and stimulate cancer cell growth. Furthermore, downstream signaling of the PI3K/Akt/mTOR pathway can sustain cellular survival through the synthesis of anti-apoptotic proteins. Under hypoxic conditions, insulin can mediate VEGF secretion from pancreatic cancer cells via expression of HIF-1α. Elevated levels of blood glucose may also stimulate VEGF.
AKT (protein kinase B); HIF-1 α (hypoxia inducible factor-1α); IGFR (insulin-like growth factor receptor); IR (insulin receptor) IRS (insulin receptor substrate); MMP (matrix metalloprotease); mTOR (mammalian target of rapamycin) PI3K (Phosphatidylinositol 3-kinases) ROS (reactive oxygen species) VEGF (vascular endothelial growth factor).
Endothelial cells line all blood vessels and without a constant blood supply tumors cannot enlarge beyond 1–2 mm and cannot grow at distal sites103. Hence, angiogenesis involves the growth of these cells. The factors that can promote angiogenesis are VEGF, FGF-2, TGF-β1 and PDGF102–105. The most potent of which is VEGF type A, a soluble growth factor commonly known as VEGF. Soluble VEGF binds to the VEGF tyrosine kinase receptors type 1, 2, 3 (VEGFR-1, 2, 3). VEGFR-2 is restricted to endothelial cells. VEGF/VEGFR2 complexes on endothelial cells can result in several downstream events that promote angiogenesis103. Neuropilin-1 (NRP-1) and neuropilin-2 (NRP-2) are co-receptors for VEGFRs on endothelial cells114. Recently, it was reported that NRP-1 and NRP-2 are expressed on tumor cells115 and their expression correlates with a more malignant phenotype116,117. In vivo, decreased expression of NRP-2 in PDAC slowed tumor growth. The inhibition of tumor growth was attributed to indirect effects on angiogenesis as opposed to anti-proliferative effects on tumor cells111, making NRP-2 a potential therapeutic target as recently reviewed by Muders et al. (2011)118,119. This study, as well as others, support the idea that the normal balance between pro- and anti-angiogenic effects can be lost in PDAC. Although PDAC is not considered a “vascular” tumor, it has areas of enhanced endothelial cell proliferation with significant correlations between blood vessel density and disease progression suggesting that anti-angiogenic targets might be attractive candidates for anti-tumor therapy. Moreover, although anti-angiogenic strategies that target VEGF alone have not yet shown efficacy for the treatment of pancreatic cancer, some anti-angiogenic molecules have been shown to reduce the immunosuppression associated with cancer120. In a review by Tartour et al. (2011) the link between angiogenesis and immunity is discussed120. In brief, anti-angiogenic molecules can decrease immunosuppressive cells (MDSC, Treg cells), immunosuppressive cytokines (IL-10 and TGF-β), as well as inhibitory molecules (PD-1)120. This suggests that anti-angiogenic therapy may be most beneficial when used in conjunction with immunotherapy.
Endocrine cells play a role in the development and progression of pancreatic cancer has not been well established. In vitro studies have demonstrated that insulin can enhance the growth of pancreatic tumor cells121. However, in patients with PDAC the association of hyperglycemia and hyperinsulinemia is questionable122. Although the link between endocrine disturbances such as diabetes and pancreatic cancer is still under debate122 it is hypothesized that increased proliferation and function of beta cells as a result of systemic insulin tolerance is involved in the progression of pancreatic cancer123. Patients with type II diabetes become unresponsive to insulin and the pancreas compensates by producing more insulin. Insulin can act as a tumor growth factor when tumor cells over express both insulin receptor substrates, 1 and 2, as is the case for pancreatic cancer124,125. Insulin can also act as a growth factor when circulating IGF-1 is low126 and the IGF-1 receptor is available to bind to insulin. Moreover, IGF-1R activation also leads to the activation of the mammalian target of rapamycin (mTOR) -mediated PI3K/Akt pathway, providing anti-apoptotic signals to the cell127. Thus the inhibitions of IGF-1/IGF-1R activity as well as mTOR are potential therapeutic targets for pancreatic cancer127. In addition, under hypoxic conditions insulin can also stimulate the expression of hypoxia inducible factor-1α (HIF-1α) in pancreatic cancer cells128,129 thereby promoting angiogenesis and further tumor development. Moreover, hyperglycemia can stimulate the expression of VEGF by human vascular smooth muscle cells130. This demonstrates a potential role for hyperglycemia in endothelial cell dysfunction130. Patients with type II diabetes can have impaired immune cell functions, particularly with regard of neutrophils and cytokines leading to an immunosuppressive state. Thus, endocrine cell dysfunction and its relationship to the development and progression of PDAC may be more closely related then is currently appreciated (Figure 1B).
Section IV: Immune Failure and Tumor Escape in PDAC
Tumor cell escape involves a complex network of dynamic interactions among cells of the tumor, the immune system and the stroma. Although we assume that the immune response in PDAC has the potential to eliminate tumor cells, during cancerogenesis, it is possible that the immune system is activated but fails to eliminate the tumor. For example:
The tumor antigens are not recognized as foreign or dangerous.
The activation of the response is either not rapid or robust enough to eradicate the tumor cells in early stages. In late stages, tumors grow too rapidly to be controlled by Tc cells.
Antigens, to which an immune response has been generated, signal cytokine overproduction that alter the immune response or actually suppress/kill cells of the immune system.
The immune system can also provide a “pro-tumor response”, whereby components of the immune response can stimulate the growth of tumor cells themselves or dampen (alter) the anti-tumor immune response.
The products of the immune system such as antibodies, activated T cells and cytokines might also have many collateral effects on normal tissues causing immune dysregulation as well as tissue damage.
Clearly, there are a variety of other mechanisms that might also be involved in the ability of tumor cells to evade or circumvent the immune system either during the activation phase or the effector phase of the immune response. This is not a new concept and it has been extensively reviewed131–133. However, the likelihood for several more mechanisms that have yet to be elucidated is high. A general view of tumor escape in pancreatic cancer is depicted in Figure 2.
Figure 2. Mechanisms of tumor escape in PDAC development and survival.

A. Pancreatic cancer cells avoid apoptosis induced by immune cells and/or induce apoptosis in immunocytes. cancer cells manipulate ‘extrinsic’ apoptotic pathways through up-regulation of apoptotic inducing ligands (FasL, TRAIL, RCAS1) or down-regulation of apoptotic receptors (Fas, TRAILR, RCAS1R); B. Pancreatic cancer cells avoid immune detection and the effector phase of the immune response. Cancerogenesis is a dynamic sum of multiple genomic and proteomic alterations with the final result of vast heterogeneity in expression of molecules responsible for immune regulation such as HLA, MICA/MICB, TAA or CRP; C. Pancreatic cancer cells promote suppression and/or alteration of immune response. Aberrant expression of immune co-stimulatory molecules (CD40, CD40L, CD70, B7 family molecules) and adhesion molecules (ICAM-1) as well as loss of molecules necessary for immune recognition (CD3-ζ) by cancer cells leads to disruption of the immune response allowing tumor progression and invasion; D. Pancreatic cancer cells and immunocytes secrete immunosuppressive factors (TGF-β, IL-10, MUC1, MUC5AC, IDO, Galectin-1, ROS) that can dampen the immune response in the tumor microenvironment.
B7-H1 (PD-L1, programmed death-1 ligand, PD-L1); B7-H3 (CD276, co-stimulatory molecule belonging to B7 family); B7-H4 (co-stimulatory molecule belonging to B7 family); CD3-ζ (T cell co-receptor-zeta chain); CD40 (tumor necrosis factor receptor superfamily member 5); CD40L (CD40-ligand, CD154); CD70 (tumor necrosis factor receptor superfamily member 7); CRP (complement regulatory protein); FasL (Fas ligand, CD95L); FasR (Fas receptor, CD95, Apo-1 tumor necrosis factor receptor superfamily member 6); ICAM-1 (inter-cellular adhesion molecule 1CD54); IDO (Indoleamine 2,3-Dioxygenase); IL-10 (interleukin 10); HLA (human leukocyte antigen); MICA/MICB (major histocompatibility complex class I chain-related genes A and B; MUC1 (mucin 1); MUC5AC (mucin 5AC); NKG2D (natural killer cell receptor); PD-1 (programmed death 1); RCAS1 (receptor-binding cancer antigen 1); RCAS1R (receptor-binding cancer antigen 1 receptor); ROS (reactive oxygen species); TAA (tumor-associated antigen); TAP (tumor-associated antigens); TGF- β (transforming growth factor beta); Th2 (T helper type 2 lymphocytes); TRAIL (tumor necrosis factor-related apoptosis-inducing ligand); TRAILR (tumor necrosis factor-related apoptosis-inducing ligand receptor).
Pancreatic tumor cells can “escape” immune surveillance by several mechanisms. They can avoid apoptosis, immune detection and the effector phase of the immune system, and kill tumor specific Tc cells. Moreover, pancreatic tumor cells can migrate to other tissues and promote immune suppression and dysregulation. This raises the question of whether “tumor escape” is a true failure of the immune system to recognize an altered normal cell and mount an anti-tumor response or whether the tumor cells silence and/or attack the immune system. Indeed both might occur.
Tumor Cells Avoid Undergoing Apoptosis and Induce Apoptosis in Other Cells
Apoptosis is natural programmed non-necrotic cell death. It plays a crucial role in maintaining homeostasis as well as immune-mediated cell killing. PDAC cells have developed several mechanisms to avoid undergoing apoptosis and or induce apoptosis in immune cells (Tc cells) and surrounding normal epithelial cells. Either mechanism could promote tumor progression. Samm et al. (2010) extensively reviewed the role of apoptosis in the pathology of pancreatic cancer demonstrating a correlation between disease occurrence with failures in apoptotic mechanisms134. This section will review the key apoptotic evasion strategies employed by PDAC cells.
Apoptosis can normally occur through ‘extrinsic’ or ‘intrinsic’ pathways. Death receptors (DRs), Fas (CD95, Apo-1), TNF-related apoptosis-inducing ligand receptor, (TRAILR, Apo-2) and TNF receptor, and their corresponding ligands FasL, TRAIL and TNF-α mediate the extrinsic pathway. Pro- and anti-apoptotic molecules of the mitochondria mediate the intrinsic pathway. Molecules involved in apoptotic pathways are ideal targets for killing tumor cells particularly if they are over-expressed. However in PDAC, tumor cells have developed mechanisms to down-regulate apoptotic receptors and/or up-regulate the apoptosis-inducing ligands as well as mutate regulatory apoptotic pathways.
The FAS System is comprised of Fas ligand (FasL) that when bound to Fas receptor (CD95) can induce apoptosis in cells that express functional Fas receptor135. Tumor escape can be achieved through down-regulation or loss of Fas, dysfunctional Fas signal transduction or expression of functional FasL136,137. In PDAC, Fas is expressed on the majority of established cell lines. However, most are resistant to Fas-ligand-mediated apoptosis137–139. This resistance can be attributed to Fas-associated phosphatase-1, an inhibitor of Fas signal transduction, that is over-expressed in Fas-resistant pancreatic cancer cell lines137,140. However, several pancreatic cancer cell lines and surgical specimens express functional FasL allowing these tumor cells to induce apoptosis in activated Tc cells137,141,142 as well as other FasR expressing cell types.
TRAIL can induce apoptosis in susceptible cells through interaction with membrane receptors DR4 and DR5143 and decoy receptors DcR1 and DcR2144. The TRAIL death receptor pathway is regulated by inhibitory proteins such as bcl-2-related proteins, bcl-2, bcl-xL and fas-like IL-1 converting enzyme (FLICE)-like inhibitor protein and stimulated by Bax145. Pancreatic cancer cell lines are heterogeneous in their expression of TRAIL, its receptors DR4, DR5, DcR1 and DcR2 as well as regulatory proteins Bax and bcl-2 and bcl-xL146. Therefore, some pancreatic cancer cell lines are susceptible to TRAIL-mediated apoptosis whereas others are completely resistant146. The cell lines that express TRAIL can induce apoptosis in tumor infiltrating lymphocytes (TILs) as well as other TRAIL-receptor expressing cell types. This provides TRAIL- expressing tumor cells with a way to escape the immune system. It also confers a growth advantage for an aggressive tumor cell by eliminating the less aggressive clones146,147. In addition, this promotes metastasis through the apoptosis of surrounding normal cells148. Initially, TRAIL-based therapy was postulated to be a good treatment option for PDAC considering that high TRAIL expression correlated with an increased apoptotic index149,150. Unfortunately, primary human tumors are often resistant to TRAIL-induced apoptosis despite the expression of TRAIL receptors, DR4 and DR5 as well as the mediators for the pathway151–155. Several proteins that can promote resistance to TRAIL-mediated killing in PDAC have been identified and these include: histone deacetylase 2 (HDAC2)154, STAT3156, CUX1157, cFLIP158–160, XIAP161–165, MCL1156,166, bclXL156,167,168 or surviving169, and SKP2155. Due to the heterogeneity in tumors from different patients immunotherapy targeted to TRAIL could be beneficial in some, but not all patients with pancreatic cancer.
RCAS1 (receptor-binding cancer antigen 1) was identified from cancer cells and can induce apoptosis in immune cells that express RCAS-1-receptor (RCAS1R)170. Although evidence for the role of RCAS1 in PDAC is limited, a few studies have found trends in tumor cell positivity and up-regulation of RCAS1, correlating to histopathologic grade and poor patient prognosis, respectively171–173. This suggests that up-regulation of RCAS1 may play an important role in PDAC progression by evading the immune system. However, further investigation is warranted. In addition, increased serum levels of RCAS1 correlated with tumor stage and when compared to CA19-9, RCAS1 showed greater fidelity as a diagnostic marker for pancreatic cancer. However, when used together, diagnostic efficiency was enhanced172.
Both pro- and anti-apoptotic molecules of the mitochondria mediate the intrinsic pathway of apoptosis. In PDAC, alterations of the Bcl-2 protein family regulated intra-mitochondrial signal transduction pathway have been reported134. Tumor cells promote their own survival, progression and metastasis by manipulating both ‘intrinsic’ and ‘extrinsic’ apoptotic pathways.
Tumor Cells Avoid Immune Detection and the Effector Phase of the Immune System
Normal cells undergo many alterations in the progression to adult cancer cells. These alterations can be advantageous to the tumor cell and lead to down-or up-regulation of various genes and their corresponding expression of molecules such as HLA, TAAs or complement regulatory proteins (CRPs). In addition, alterations can lead to expression of abnormal genes and proteins that can provide specific targets for therapy.
Dysregulation of HLA
For the immune system to initiate a response against a tumor, DCs must transport tumor antigens to the regional lymph nodes. Tumor antigens are processed with the help of transporter for antigen presentation (TAP), and presented to T cells by HLA class I and class II molecules on the surface of DCs. For the tumor cells to be killed by the resulting Tc cells, they must express the specific tumor antigens in class I molecules. In PDAC, tumor cells down-regulate or lose expression of HLA class I, its associated β2-microglobulin174 and TAP175. Therefore, some tumor cells no longer present antigen to immune cells and avoid immune detection as well as killing by Tc cells. Although this does render them sensitive to NK cell-mediated killing, NK cells are far less effective in eliminating tumor cells.
Tumor cells can express HLA class II molecules de novo175. This suggests that tumor cells are promoting a Th2/humoral immune response, by influencing the type of HLA molecule expressed on their cell surface. Ultimately, this can have detrimental effects on tumor cell killing by preventing cellular immunity, i.e. Tc cells, and promoting humoral immunity, i.e. Th cells. Interestingly however, it has been reported that HLA class I and TAP expression can be re-induced in PDAC cell lines in vitro by treatment with IFN-γ175 thus providing a possible means of altering the balance between cellular and humoral immunity by promoting Th1/cell-mediated immunity.
HLA related molecules, MHC class I chain-related genes A and B (MICA/MICB), are intestinal surface glycoproteins that can be up-regulated in response to stress or by epithelial tumors176. MICA/MICB are ligands for the NKG2D activating receptor found on NK and gamma delta T cells of the immune system. NK cells can recognize cells that either down-regulate MHC antigens or completely lose HLA class I molecules. In PDAC, cells express MIC and sera from PDAC patients contain elevated levels of soluble MIC (sMIC) that correlate to tumor stage and differentiation177. Moreover, sMIC in the sera of patients with PDAC can inhibit the cellular cytotoxicity of NK and gamma/delta T cells, thereby inhibiting the ability of the innate immune response to eliminate PDAC cells177.
Down-regulation of TAAs
Tumor cells can up-regulate normally expressed molecules as well as express abnormal self-molecules. These phenomena are some of the main factors driving the development of targeted immunotherapy. Unfortunately, after tumor cells “escape” from immune surveillance, they often become “resistant” to TAA-specific induced immune effector cells. In addition, as the more aggressive tumor cells differentiate, the expression of TAAs can mutate or decrease to a point of complete loss of expression from the surface of the remaining tumor cells.
Expression of CRPs
Complement inhibitors CD46 (membrane cofactor protein), CD55 (complement decay accelerating factor), and CD59 (protectin) can protect tumor cells from lysis by activated complement178–180. PDAC cell lines express high levels of these molecules on their surface (unpublished results, Pop, Vitetta et al.). This suggests that tumor cells are able to regulate complement- dependent effector functions. Hence anti-tumor antibodies made by the host or administered therapeutically (e.g., anti-CA19.9) would fail to kill the tumor cells by C’ mediated lysis181.
Tumor Cells Promote Immune Suppression and Immune Dysfunction Co-stimulatory Molecules
Interestingly, in tumor progression, tumor cells can also aberrantly express T cell co-stimulatory molecules. These molecules are typically limited to cells of the immune system and are involved in lymphocyte signaling pathways. The over-expression of these molecules by tumor cells can lead to either amplification or dampening of local immunity with devastating consequences for normal body physiology. For example, tumor cells that express inhibitory co-stimulatory molecules can suppress or eliminate specific anti-tumor immunocytes, thereby allowing the tumor to progress. On the other hand, tumor cells that express activating co-stimulatory molecules can enhance the immune response such that the inflammatory milieu causes damage to the surrounding normal tissue and results in further progression of tumor growth.
The B7 super family are co-stimulatory molecules expressed on antigen presenting cells that included; B7-1 (CD80) and B7-2 (CD86) and their receptors CD28 and cytotoxic T-lymphocyte antigen-4 (CTLA-4), as well as the B7-homolog molecules B7-H1, B7-DC (B7-DC), B7-H2, B7-H3 and B7-H4. In the immune response, T cells and antigen-MHC complexes determine specificity whereas the co-stimulatory molecules of the B7 family determine the magnitude and type of immune response. Therefore, the B7 ligands can provide an activating or inhibitory signal depending upon the receptor bound and the influence of the local environment182.
The B7/CD28 and B7/CTLA-4 systems are T cell co-stimulatory pathways that act on antigen-presenting cells and T cells. The B7/CD28 interaction promotes T and B cell activation, Th1/Th2 differentiation, cell migration, and homeostasis of CD25+CD4+ Treg cells183. In contrast, B7/CTLA-4 interactions downregulate T cell function and ongoing immune responses as well as help maintain peripheral tolerance183. By blocking co-stimulatory pathways, specific clones of activated T cells are turned off183. This pathway has been explored in developing strategies for immune intervention therapies (discussed in the therapy section).
B7-H1 (Programmed death-1 ligand, PD-L1; CD274) and B7-DC (PD-L2; CD273) are cell surface ligands for programmed death-1 (PD-1) receptor that is expressed on activated T cells, B cells and monocytes184–188. The expression of B7-H1 is induced by IFN-γ on several cells types189,190. The expression of B7-DC is limited to DCs and activated macrophages and induced by IL-4 and IL-13191. Both ligands can induce PD-1 to negatively regulate both cellular and humoral immune responses184,188,189,192. However, the interaction of B7-DC with PD-1193 also has stimulatory effects suggesting another receptor interaction or influence of other environmental factors189,194. B7-H1 inhibits anti-tumoral T-cell immunity by interacting with PD-1 on T cells resulting in tumor-specific T cell apoptosis or impaired cytotoxicity and cytokine production by activated T cells195–198. In addition, ligation of B7-H1 to T-cells can result in the preferential production of IL-10199,200. IL-10 is an immunosuppressive cytokine that can inhibit Th1 type immune responses201,202 by modulating antigen presenting cells (APCs) and DC function and promoting Treg cell responses203,204. In PDAC, the expression of both B7-H1 and IL-10 is up-regulated as compared to normal tissues. The expression of B7-H1 correlates with advanced tumor stage and poor prognosis and is inversely correlated with TILs188,205. This suggests that tumor cells are expressing B7-H1 to suppress the anti-tumor immune responses while promoting the production of the immunosuppressive cytokine IL-10. Furthermore, in an in vivo mouse model of pancreatic cancer, blocking the B7-H1, B7-DC206 or the PD-Ls/PD-1 pathways188 with MAbs can induce anti-tumor effects and promote infiltration of T cells into the tumor188,206. This is important because it identifies specific molecules in pathways that can be therapeutically targeted in order to restore the anti-tumor immune response. Moreover, the B7-DC blockade decreases IL-10 and FoxP3 levels whereas B7-H1 blockade increases IFN-γ and FoxP3 in the tumor site206. Thus further demonstrating the important role of each ligand not just as specific Tc cell inhibitors but also as general immunosupressors involving Treg cells. As we better understand the mechanisms by which tumor cells inhibit anti-tumor response, we can design more effective therapies.
Although no studies to date report the expression of B7-H2 (CD275) on pancreatic cancer cells it would not be surprising if it were expressed.
The role of B7-H3 (CD276) in anti-tumor immunity was recently reviewed by Loos et al. (2010) and will be briefly discussed in this section207. B7-H3 is expressed on several non-immune cell types throughout the body. However, its expression can be induced on activated DCs, monocytes, T cells and some tumor cell lines208–211. The role of B7-H3 in immune regulation is controversial due to the fact that both stimulatory and inhibitory immune functions have been reported and possibly attributed to two distinct receptors207–216. However, only one receptor has been identified, it is the triggering receptor expressed on myeloid cells (TREM)-like transcription 2 (TLT-2)217. The interaction of B7-H3 with TLT-2 on T cells enhances T cell activation, proliferation, cytokine production and cytotoxicity217. In PDAC, B7-H3 tumor- related expression has been reported to be significantly higher than in non-cancer tissue or normal pancreas218. Its expression correlated with lymph node metastasis and advanced pathologic stage218. In an in vivo mouse model of pancreatic cancer, B7-H3 blockade promoted CD8+ T cell infiltration into the tumor and induced substantial anti-tumor effects that were synergistic with gemcitabine218.
B7-H4 is expressed predominantly on human epithelial cells of the female genital tract, kidney, lung and pancreas with low/no expression on other cell types219,220. Although the receptor for B7-H4 is unknown, the expression of B7-H4 can be induced on monocytes, macrophages and myeloid DCs by both IL-6 and IL-10 and down-regulated by GM-CSF and IL-4221–223. Much remains to be elucidated concerning the role of B7-H4 in immune regulation. However, studies have shown that B7-H4 inhibits the proliferation of both CD4+ and CD8+ T cells as well as cytokine production by inducing cell cycle arrest224–226. B7-H4 is highly expressed on several human cancers219,220 and although data are limited, one study has investigated the expression of B7-H4 in PDAC. B7-H4 was expressed more often than p53, a potential marker for pancreatic cancer, and B7-H4 positive tumor cells were inversely correlated to tumor grade227. These findings suggest an early induction followed by loss of B7-H4 expression leading to a decrease in tumor-associated immunogenicity in higher-grade tumors227. The role of B7-H4 in normal pancreatic tissue has not been investigated. However B7-H4 interactions in normal pancreas may block T cell-mediated immunity where as in PDAC, this protection may be lost due to B7-H4 expression227.
CD40 is a membrane glycoprotein member of the TNF receptor family. It is expressed on several cell types including B lymphocytes, DCs and monocytes228–230. Its ligand, CD154 (CD40L), is expressed on the surface of T cells. The interaction of CD40+ B cells with CD154+ T cells induces B cell proliferation, immunoglobulin production, somatic hypermutation of B cell receptors and immunoglobulin class-switching231–234. The interaction of CD40+ DCs, and CD154+ T cells leads to up-regulation of co-stimulatory molecules (CD80, CD86) on APC cells to help T cell activation, proliferation and cytokine expression235. The normal expression and interaction of CD40 and CD154 by immune cells results in the proliferation of the immune response with the potential to ultimately affect anti-tumor immunity. In a recent study by Shoji et al. (2011) it was found that both CD40 and CD154 are expressed by PDAC cell lines and patient specimens and although the study did not directly evaluate TILs they found the frequency of CD154 expression on TILs to be low in their xenograft model236. These findings suggest that PDAC cells can potentially use CD40 and CD154 expression as an autocrine mechanism to promote tumor cell proliferation as well as potentially alter CD154 expression on TILs. This alteration may be explained by the ligation of CD40 on PDAC cells inducing the secretion of several types of pro and anti-inflammatory cytokines (IL-6, IL-10, IL-12) as found by Shoji et al. (2011). Moreover, studies on other malignant cell types support the secretion of cytokines following CD40 ligation237–239. The balance of pro-versus anti-tumor immune response can tip in favor of either response or remain in equilibrium based upon the expression of surface molecules by tumor cells, immune cells, stromal cells as well as the factors that they release. A good example of this was the finding that very high expression of CD154 in patient specimens correlated with a favorable prognosis236. On the one hand, PDAC cells promote their own growth with the expression of CD40-CD154 and immune cell suppression with secretion of IL-10. The ligation of CD40 on these tumor cells leads to the secretion of pro-inflammatory cytokines IL-6 and IL-12 that could ultimately result in an anti-tumor response. These seemingly contradictory findings within the same study best illustrate the complexity of the tumor-immune system interactions. Moreover, when pancreatic cancer patients were treated with an agonist CD40 antibody and gemcitabine chemotherapy tumor regression was observed240. When Beatty et al. (2011) evaluated this effect in a mouse model of PDAC they found that CD40-activated macrophages but not T cells nor gemcitabine infiltrated tumors and mediated tumor regression and depletion of tumor stromal cells240.
CD54 (inter-cellular adhesion molecule 1, ICAM-1) exists in both membrane-bound and soluble forms. The adhesion molecule CD54 is expressed on several different cell types241–243 and can be secreted as soluble CD54 (sCD54) by mononuclear cells, endothelial cells, keratinocytes, hepatocytes and some tumor cells244. The regulation of sCD54 is not well understood, but TNF-α, IFN-γ, and IL-1 can induce the expression of membrane-bound CD54 while, glucocorticoids are the most well known inhibitors241–243. CD54 binds to the β2 integrins lymphocyte function-associated antigen (LFA-1; CD11a/CD18) and macrophage 1 (Mac-1; CD11b/CD18) on leukocytes, as well as sialophorin (CD43) on leukocytes and platelets and soluble fibrinogen242,245,246. It functions predominantly as an adhesion molecule, but it can elicit a variety of effects including T cell and NK cell activation and leukocyte migration242,245,246. CD54 is associated with disease states characterized by local or systemic inflammation247,248 and although CD54 is not tumor specific and is expressed on many normal cells in humans, it can play a crucial role in the tumor microenvironment. It has been hypothesized that CD54 dictates the metastatic potential and lethality of many types of cancer cells249. The over-expression of CD54 at the leading edge of tumor invasion has been correlated with a poor patient prognosis250. Although no published studies have evaluated the role of CD54 in pancreatic cancer, our unpublished data derived from pancreatic cell lines suggest that its expression is down-regulated or lost on some PDAC cell lines.
CD70 (TNFSF7) ligand is a member of the TNF superfamily that interacts with CD27. The interaction of CD70 ligand with CD27 regulates long-term maintenance of T cell immunity as well as B-cell activation and immunoglobulin synthesis251–260. CD70 expression is normally limited to antigen-activated T and B lymphocytes254,261,262 and is found infrequently in a few other normal cell types263,264. However, aberrant expression of CD70 has been reported in several tumor types including pancreatic cancer cells265–269. Since CD70 expression has a limited normal distribution and aberrant cell surface expression in tumors, CD70 makes an attractive target for therapy269. In an in vivo model of human pancreatic cancer, mice were treated with an anti-CD70 drug conjugate (SGN-75) all 7 mice treated showed a delay in tumor growth with 2 of 7mice showing a complete and sustained regression269.
Loss of CD3-zeta Chain Expression
The T cell receptor (TCR)/CD3-zeta chain is a crucial component in the T cell signal transduction complex. Although it is important for the initial activation of Tc cells in the regional lymph node, and not in the effector function of Tc cells at the tumor site, it is important to note that specimens from PDAC patients have shown significant down-regulation or loss of TCRs/CD3-zeta chains on TILs137. The significance of this finding has yet to be determined but it is proposed that environmental factors such as ROS and arginase produced by macrophages in tumor sites can decrease the expression of TCRs on effector T cells such that they can no longer recognize the tumor antigens expressed in HLA class I molecules. Thus, the effector T cells in the tumor microenvironment might not recognize their target cells and hence, not kill them.
Production and Secretion of Immunosuppressive Factors
A more global mechanism of PDAC tumor escape is the production and secretion of immunosuppressive molecules. In addition to several preciously mentioned immunosuppressive molecules, PDAC cells can produce and secrete TGF-β, MUC1, MUC5AC, Indoleamine 2,3-Dioxygenase (IDO), Galectin-1 (Gal-1), and ROS.
The role of TGF-β270 in blocking the activation of lymphocytes and monocytes has been covered in other sections of this review. However, its role in tumor cells will be addressed here. In PDAC, TGF-β has been shown to up-regulate proteases such as MMP-2 and urokinase plasminogen activator (uPA)271, to down-regulate cell surface CD54272 and to stimulate the secretion of VEGF by tumor cells271. These effects can result in degradation of the extracellular matrix, thereby promoting tumor cell invasion and metastasis while providing an angiogenic stimulus to promote further development. Moreover, TGF-β2 has been shown to induce PDAC cells to express functional Foxp3, possibly allowing tumor cells to mimic the function of Treg cells273. This suggests another mechanism by which tumor cells can suppress anti-tumor responses, by functioning as suppressor cells themselves.
MUC1 is an epithelial cell membrane-bound glycoprotein that is approximately 80% carbohydrate274,275. It is associated with the progression of normal pancreatic ductal cells to infiltrating ductal carcinoma and has been shown to enhance the invasiveness of pancreatic cancer cells by inducing an epithelial to mesenchymal transition276–282. MUC1 is also an immunogen that elicits CD8+ T cell responses23 and induces the production of anti-MUC1 antibodies of both the IgM and IgG isotypes24,25. Increased serum levels of anti-MUC1 antibodies correlate with increased patient survival25. Tumor-derived mucin has been shown to profoundly affect the cytokine repertoire of monocyte-derived DCs, producing regulatory APCs (IL-10highIL-12low) that lead to a Th1 immune response283. Again, this supports the hypothesis that the tumor cells do elicit an anti-tumor immune response. However, it is either not robust enough or is quickly thwarted by other escape mechanisms employed by tumor cells. To better elucidate the immunosuppressive effects of MUC1, Tinder et al. (2008) compared pancreatic tumors that expressed MUC1 to those that lacked MUC1 expression in a mouse model of spontaneous PDAC284. The tumors derived from MUC1+ mice expressed higher levels of COX-2 and IDO compared with tumors from MUC1− mice especially during early stages of development. In addition, MUC1+ mice had an increased pro-inflammatory milieu with elevated levels of Treg cells and myeloid suppressor cells within the tumor and draining lymph nodes284. In subsequent in vivo studies, Besmer et al. (2011) showed that MUC1− mice have significantly slower tumor progression and rates of metastasis285. Moreover, from their in vitro studies it is suggested that MUC1 is necessary for MAPK activity and oncogenic signaling285. MUC1-mediated mechanisms can enhance the onset and progression of the disease, which in turn, regulate the immune response284. It is possible that early in disease MUC1 expression is recognized by the immune system and initially promotes a robust Th1 anti-tumor immune response. However, over time the progressive inflammation can evoke an immunosuppressive response established either by the efforts of the immune system to maintain balance or the attempts by the tumor cells to suppress the response.
MUC5AC, another glycoprotein from the mucin family, is over-expressed only by PDAC cells and not by normal pancreatic cells. A recent study revealed that by knocking down MUC5AC expression of wild-type MUC5AC positive pancreatic cell lines by siRNA, there was a decrease in tumorigenicity and tumor development286. This suggests that MUC5AC expression may play an important role in the development of PDAC as well as provide a potential tumor specific target for therapy.
The up-regulation of enzymes or proteins crucial for immune cell function can be an important mechanism by which tumor cells control the tumor environment and prevent tumor specific immune responses. IDO is an IFN-γ induced immune regulatory enzyme that catabolizes tryptophan287. IDO can create an immunosuppressive environment by deleting tryptophan, a crucial metabolite for T cells undergoing antigen-dependent activation, ultimately leading to T cell arrest, anergy or death288–291. In PDAC up-regulation of IDO in tumor cells is associated with an increased number of Treg cells289. Recently, another immunoregulatory molecule secreted by pancreatic tumor cells and activated pancreatic stellate cells, Gal-1 was identified292. Gal-1 is a carbohydrate- binding protein that is thought to be a regulator of T cell homeostasis, survival and inflammation293,294. Up-regulation of Gal-1 was suggested to induce apoptosis in T cells, activation of DCs, regulate immune cell trafficking and promote proliferation and invasion of tumor cells294,295. Thus PDAC cells can employ basic nutrient metabolism and secretion of immunosuppressive factors to locally regulate the immune response.
Although ROS is mainly implicated to promote cell death and is a key component of immune defense against invading microbes296–299 there is increasing evidence to suggest that it also plays a role in cell survival and signaling300–304. We previously reviewed ROS as products of neutrophils as well as macrophages. However, PDAC cells have been reported to produce ROS as well305. In a study by Vaquero et al. (2004), when PDAC cells were stimulated by IGF-I or FGF-2 they produced ROS which protected the cells from apoptosis305. It has been proposed that tumor cells require the presence of chemotactic molecules for growth, invasion and metastasis. Ultimately these “homing factors” may promote immune escape.
Strong expression of the chemokine receptor CXCR4 has been reported to correlate with advanced pancreatic cancer306. There is much more that remains to be uncovered about the role of CXCR4 and other chemotactic molecules in PDAC.
Unfortunately, tumor escape from normal immunity is not the main or only mechanism by which tumor cells survive. Genetic alterations and microenvironment factors that both promote tumor cell development and eliminate less robust tumor cells ultimately produce immortal cells with an infinite capacity for reproduction, if nutrients are available. In this regard, the precursors of cancer stem cells can evolve into new blood vessel progenitors307 and adult cancer cells, especially in hypoxic states, can induce and sustain new blood vessel formation that ensures nutrient supply for further tumor growth. Moreover, one of the most potent angiogenic factors, VEGF, is also an immunosuppressive molecule secreted by tumor cells308. Thus, as a part of the extraordinary innate and acquired abilities of tumor cells to defeat the host, they develop a variety of ‘escape’ mechanisms that can overcome almost any anti-tumor approach. However, the lessons learned from the various ways tumor cells continually adapt to ensure their survival should be used to develop rational multi-targeted immunotherapies.
Section V: A role for Immunotherapy in PDAC
The dismal prognosis of PDAC is due to late detection and limited treatment options. It is often diagnosed at an advanced stage of disease when the tumor is inoperable and frequently resistant to standard therapy. Clinical trials in patients with PDAC have focused on improving both surgery and radiation therapy as well as determining better drug-treatment combinations. Despite this, PDAC is almost uniformly fatal.
Several biological approaches have been studied for the treatment of pancreatic cancer, e.g. gene therapy, signal transduction modulators, anti-angiogenics, MMP inhibitors, oncolytic viral therapy, as well as immunotherapy. However, these have not improved patient survival as recently reviewed by Wong et al. (2008)309. It is important to note that clinical trials with targeted biologics are used in all patients with disease regardless of whether or not they express the target, making conclusions difficult and potentially erroneous. Several proposals have been made to address this issue by selecting patients with the appropriate targets on their tumors or in the tumor microenvironment310. Therefore, there is a need to establish the genetic and proteomic profile of the tumor cells in each patient as well as understand the key molecules involved in multi-drug resistance and use this information to successfully target the most effective agents to cells. This profile can be used to determine the treatment regimen as well as monitor responses to treatment. Ultimately, the hope is that by using this profile to screen study patients for the expression of the target in question in clinical trials with targeted therapeutics, we may actually begin to see clinically meaningful responses.
The rationale for immunotherapy is to augment a patient’s natural immune response to their pancreatic cancer or introduce components of an immune system to slow disease progression. The consequences of impaired immune function are significant. Tumor cells are capable of functioning as immunocytes with the ability to secrete immunosuppressive cytokines, ultimately impairing the immune system’s function to recognize TAAs and destroy tumor cells.
Currently, immunotherapy for PDAC is only available in clinical trials. There are several ongoing clinical trials to evaluate single-agent as well as combined cytotoxic therapy and combinations of targeted therapies (including MAbs)311. However, considering how refractory to conventional agents this disease is, clinical trials may offer the best treatment option as well as teach us how to define specific combinatorial therapy guidelines (e.g., dosing, timing, route of administration, adjuvant therapy, etc). Unfortunately, the benefits of immunotherapy have yet to meet initial expectations. Clinical studies have yielded undesirable results such as the stimulation of incorrect immune responses, cytokine storm, tumor progression and metastasis. From a mechanistic point of view, the failure of immunotherapy in treating pancreatic cancer is a consequence of the genomic instability inherent in cancer cells, which allow them to highjack immune defenses. Moreover, the potential existence of cancer stems cells may help explain tumor rescue and epithelial-to-mesenchymal transition (EMT), a phenomena believed to play a role in tumor progression and the ineffectiveness of current therapy. PDAC research should accelerate the understanding of the rescue mechanisms (tumorigenicity, invasiveness and resistance to therapy, angiogenesis, etc.) that tumors employ through the self-renewal “stem cell” subpopulations. Understanding these cell types can provide a new avenue for cancer-targeted therapeutics, as well as identify the patients with high-risk of unfavorable disease evolution or recurrence.
The future of immunotherapy stands on either identification of unique specific TAAs using proteomic analysis312 or finding avenues to “teach” the tumor cell to express a stable specific antigen which can be further targeted in an immunotherapy setting. The best-case scenario will be a simultaneous immuno-strategy, which will result in overcoming the tumor tolerance, stimulating the specific targeted anti-tumor response and shutting off the suppressor arm of immunity at the same time. This scenario relies on the development of novel agents to target specific biological processes not only unique to the tumor cells but also to their microenvironment. Novel agents include growth factor inhibitors, anti-angiogenic factors, MMP inhibitors as well as intracellular mediator or pathway inhibitors311,313. Moreover, to enhance the efficacy of pancreatic cancer immunotherapy we should also establish methods for earlier diagnosis with new and very specific molecular biomarkers312. This should facilitate the design of “early protective” measures such as vaccination (preventive immunotherapy).
The lessons learned from tumor escape mechanisms and failed clinical trials should represent the platform for the development of rational and successful combination therapy (e.g., cytotoxic and targeted/immunotherapy along with surgery) for patients with PDAC. However, one of the major challenges of combining immunotherapy with conventional chemotherapy is timing. Administering chemotherapy before immunotherapy eliminates the immunocytes that are activated with the latter. However, the two may work synergistically with one serving as the necessary adjuvant for a robust anti-tumor immune response. For example, an anti-tumor response may be facilitated by the upregulation of Fas, TAAs and cell chemosensitization314 or by cross-presentation of TAAs by DCs following tumor cytolysis. Ultimately, in order to improve old therapies and develop new ones, we must understand why our past attempts at treating PDAC have failed and apply that knowledge to our future approaches.
Acknowledgments
Supported in part by NIH CTSA Grant UL1 RR024982 and the Cancer Immunobiology Center
We thank Drs. Rolf Brekken, for his intellectual contribution in the discussion of pancreatic cancer, Michael McPhaul, for his editorial advise and Helen Mayo, Reference and Liaison Librarian at The University of Texas Southwestern Medical Center Dallas, for her assistance with finding references for this manuscript. Support for this article was received from the NIH CTSA Grant UL1 RR024982 and The Cancer Immunobiology Center.
References
- 1.Matzinger P, Kamala T. Tissue-based class control: the other side of tolerance. Nature reviews. Immunology. 2011;11:221–230. doi: 10.1038/nri2940. [DOI] [PubMed] [Google Scholar]
- 2.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331:1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 3.Emmrich J, Sparmann G, Hopt U, Lohr M, Liebe S. Typing of leukocytes in pancreatic tissue surrounding human pancreatic carcinoma. Ann N Y Acad Sci. 1999;880:171–174. doi: 10.1111/j.1749-6632.1999.tb09520.x. [DOI] [PubMed] [Google Scholar]
- 4.Fogar P, et al. Decreased total lymphocyte counts in pancreatic cancer: an index of adverse outcome. Pancreas. 2006;32:22–28. doi: 10.1097/01.mpa.0000188305.90290.50. 00006676-200601000-00004 [pii] [DOI] [PubMed] [Google Scholar]
- 5.Tassi E, et al. Carcinoembryonic antigen-specific but not antiviral CD4+ T cell immunity is impaired in pancreatic carcinoma patients. J Immunol. 2008;181:6595–6603. doi: 10.4049/jimmunol.181.9.6595. 181/9/6595 [pii] [DOI] [PubMed] [Google Scholar]
- 6.Bellone G, et al. Tumor-associated transforming growth factor-beta and interleukin-10 contribute to a systemic Th2 immune phenotype in pancreatic carcinoma patients. Am J Pathol. 1999;155:537–547. doi: 10.1016/s0002-9440(10)65149-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park H, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. ni1261 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fossiez F, et al. T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J Exp Med. 1996;183:2593–2603. doi: 10.1084/jem.183.6.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gnerlich JL, et al. Induction of Th17 cells in the tumor microenvironment improves survival in a murine model of pancreatic cancer. J Immunol. 185:4063–4071. doi: 10.4049/jimmunol.0902609. jimmunol.0902609 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamamoto M, et al. Enhancement of anti-tumor immunity by high levels of Th1 and Th17 with a combination of dendritic cell fusion hybrids and regulatory T cell depletion in pancreatic cancer. Oncol Rep. 2009;22:337–343. [PubMed] [Google Scholar]
- 11.Wilke CM, et al. Th17 cells in cancer: help or hindrance? Carcinogenesis. 2011;32:643–649. doi: 10.1093/carcin/bgr019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ikemoto T, et al. Clinical roles of increased populations of Foxp3+CD4+ T cells in peripheral blood from advanced pancreatic cancer patients. Pancreas. 2006;33:386–390. doi: 10.1097/01.mpa.0000240275.68279.13. 00006676-200611000-00013 [pii] [DOI] [PubMed] [Google Scholar]
- 13.Hiraoka N, Onozato K, Kosuge T, Hirohashi S. Prevalence of FOXP3+ regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clin Cancer Res. 2006;12:5423–5434. doi: 10.1158/1078-0432.CCR-06-0369. 12/18/5423 [pii] [DOI] [PubMed] [Google Scholar]
- 14.Yamagiwa S, Gray JD, Hashimoto S, Horwitz DA. A role for TGF-beta in the generation and expansion of CD4+CD25+ regulatory T cells from human peripheral blood. J Immunol. 2001;166:7282–7289. doi: 10.4049/jimmunol.166.12.7282. [DOI] [PubMed] [Google Scholar]
- 15.Baecher-Allan CM, Hafler DA. Functional analysis of highly defined, FACS-isolated populations of human regulatory CD4+CD25+ T cells. Clin Immunol. 2005;117:192. doi: 10.1016/j.clim.2005.08.008. discussion 193, S1521-6616(05)00287-1 [pii] [DOI] [PubMed] [Google Scholar]
- 16.Tan MC, et al. Disruption of CCR5-dependent homing of regulatory T cells inhibits tumor growth in a murine model of pancreatic cancer. J Immunol. 2009;182:1746–1755. doi: 10.4049/jimmunol.182.3.1746. 182/3/1746 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Viehl CT, et al. Depletion of CD4+CD25+ regulatory T cells promotes a tumor-specific immune response in pancreas cancer-bearing mice. Ann Surg Oncol. 2006;13:1252–1258. doi: 10.1245/s10434-006-9015-y. [DOI] [PubMed] [Google Scholar]
- 18.Moo-Young TA, et al. Tumor-derived TGF-beta mediates conversion of CD4+Foxp3+ regulatory T cells in a murine model of pancreas cancer. J Immunother. 2009;32:12–21. doi: 10.1097/CJI.0b013e318189f13c. 00002371-200901000-00002 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liyanage UK, et al. Increased prevalence of regulatory T cells (Treg) is induced by pancreas adenocarcinoma. J Immunother. 2006;29:416–424. doi: 10.1097/01.cji.0000205644.43735.4e. 00002371-200607000-00008 [pii] [DOI] [PubMed] [Google Scholar]
- 20.Lopez-Alemany R, et al. Inhibition of cell surface mediated plasminogen activation by a monoclonal antibody against alpha-Enolase. Am J Hematol. 2003;72:234–242. doi: 10.1002/ajh.10299. [DOI] [PubMed] [Google Scholar]
- 21.Nakatsura T, Senju S, Ito M, Nishimura Y, Itoh K. Cellular and humoral immune responses to a human pancreatic cancer antigen, coactosin-like protein, originally defined by the SEREX method. Eur J Immunol. 2002;32:826–836. doi: 10.1002/1521-4141(200203)32:3. <826::AID-IMMU826>3.0.CO;2-Y [pii] [DOI] [PubMed] [Google Scholar]
- 22.Johnston FM, et al. Circulating mesothelin protein and cellular antimesothelin immunity in patients with pancreatic cancer. Clin Cancer Res. 2009;15:6511–6518. doi: 10.1158/1078-0432.CCR-09-0565. 1078-0432.CCR-09-0565 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barnd DL, Lan MS, Metzgar RS, Finn OJ. Specific, major histocompatibility complex-unrestricted recognition of tumor-associated mucins by human cytotoxic T cells. Proc Natl Acad Sci U S A. 1989;86:7159–7163. doi: 10.1073/pnas.86.18.7159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kotera Y, Fontenot JD, Pecher G, Metzgar RS, Finn OJ. Humoral immunity against a tandem repeat epitope of human mucin MUC-1 in sera from breast, pancreatic, and colon cancer patients. Cancer Res. 1994;54:2856–2860. [PubMed] [Google Scholar]
- 25.Hamanaka Y, et al. Circulating anti-MUC1 IgG antibodies as a favorable prognostic factor for pancreatic cancer. Int J Cancer. 2003;103:97–100. doi: 10.1002/ijc.10801. [DOI] [PubMed] [Google Scholar]
- 26.Qin H, et al. CD4+ T-cell immunity to mutated ras protein in pancreatic and colon cancer patients. Cancer Res. 1995;55:2984–2987. [PubMed] [Google Scholar]
- 27.Shono Y, et al. Specific T-cell immunity against Ki-ras peptides in patients with pancreatic and colorectal cancers. Br J Cancer. 2003;88:530–536. doi: 10.1038/sj.bjc.6600697. 6600697 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kubuschok B, et al. Naturally occurring T-cell response against mutated p21 ras oncoprotein in pancreatic cancer. Clin Cancer Res. 2006;12:1365–1372. doi: 10.1158/1078-0432.CCR-05-1672. 12/4/1365 [pii] [DOI] [PubMed] [Google Scholar]
- 29.Imai K, et al. Identification of a novel tumor-associated antigen, cadherin 3/P-cadherin, as a possible target for immunotherapy of pancreatic, gastric, and colorectal cancers. Clin Cancer Res. 2008;14:6487–6495. doi: 10.1158/1078-0432.CCR-08-1086. 14/20/6487 [pii] [DOI] [PubMed] [Google Scholar]
- 30.Alters SE, Gadea JR, Philip R. Immunotherapy of cancer. Generation of CEA specific CTL using CEA peptide pulsed dendritic cells. Adv Exp Med Biol. 1997;417:519–524. [PubMed] [Google Scholar]
- 31.Peiper M, et al. The HER2/neu-derived peptide p654–662 is a tumor-associated antigen in human pancreatic cancer recognized by cytotoxic T lymphocytes. Eur J Immunol. 1997;27:1115–1123. doi: 10.1002/eji.1830270511. [DOI] [PubMed] [Google Scholar]
- 32.Argani P, et al. Discovery of new markers of cancer through serial analysis of gene expression: prostate stem cell antigen is overexpressed in pancreatic adenocarcinoma. Cancer Res. 2001;61:4320–4324. [PubMed] [Google Scholar]
- 33.Tanaka M, et al. Increased levels of IgG antibodies against peptides of the prostate stem cell antigen in the plasma of pancreatic cancer patients. Oncol Rep. 2007;18:161–166. [PubMed] [Google Scholar]
- 34.Marxsen J, et al. Detection of the anti-p53 antibody response in malignant and benign pancreatic disease. Br J Cancer. 1994;70:1031–1034. doi: 10.1038/bjc.1994.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Raedle J, et al. p53 autoantibodies in patients with pancreatitis and pancreatic carcinoma. Pancreas. 1996;13:241–246. doi: 10.1097/00006676-199610000-00005. [DOI] [PubMed] [Google Scholar]
- 36.Schmitz-Winnenthal FH, et al. High frequencies of functional tumor-reactive T cells in bone marrow and blood of pancreatic cancer patients. Cancer Res. 2005;65:10079–10087. doi: 10.1158/0008-5472.CAN-05-1098. 65/21/10079 [pii] [DOI] [PubMed] [Google Scholar]
- 37.Fukunaga A, et al. CD8+ tumor-infiltrating lymphocytes together with CD4+ tumor-infiltrating lymphocytes and dendritic cells improve the prognosis of patients with pancreatic adenocarcinoma. Pancreas. 2004;28:e26–31. doi: 10.1097/00006676-200401000-00023. 00006676-200401000-00023 [pii] [DOI] [PubMed] [Google Scholar]
- 38.Lund FE, Randall TD. Effector and regulatory B cells: modulators of CD4(+) T cell immunity. Nature reviews. Immunology. 2010;10:236–247. doi: 10.1038/nri2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Raulet DH, Guerra N. Oncogenic stress sensed by the immune system: role of natural killer cell receptors. Nat Rev Immunol. 2009;9:568–580. doi: 10.1038/nri2604. nri2604 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sutlu T, Alici E. Natural killer cell-based immunotherapy in cancer: current insights and future prospects. J Intern Med. 2009;266:154–181. doi: 10.1111/j.1365-2796.2009.02121.x. JIM2121 [pii] [DOI] [PubMed] [Google Scholar]
- 41.Perussia B. Lymphokine-activated killer cells, natural killer cells and cytokines. Curr Opin Immunol. 1991;3:49–55. doi: 10.1016/0952-7915(91)90076-d. [DOI] [PubMed] [Google Scholar]
- 42.Kitayama J, et al. Functional analysis of TCR gamma delta+ T cells in tumour-infiltrating lymphocytes (TIL) of human pancreatic cancer. Clin Exp Immunol. 1993;93:442–447. doi: 10.1111/j.1365-2249.1993.tb08198.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Degrate L, et al. Interleukin-2 immunotherapy action on innate immunity cells in peripheral blood and tumoral tissue of pancreatic adenocarcinoma patients. Langenbecks Arch Surg. 2009;394:115–121. doi: 10.1007/s00423-008-0393-4. [DOI] [PubMed] [Google Scholar]
- 44.Funa K, Nilsson B, Jacobsson G, Alm GV. Decreased natural killer cell activity and interferon production by leucocytes in patients with adenocarcinoma of the pancreas. Br J Cancer. 1984;50:231–233. doi: 10.1038/bjc.1984.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aparicio-Pages MN, Verspaget HW, Pena AS, Lamers CB. Natural killer cell activity in patients with adenocarcinoma in the upper gastrointestinal tract. J Clin Lab Immunol. 1991;35:27–32. [PubMed] [Google Scholar]
- 46.Frankel TL, et al. Identification and characterization of a tumor infiltrating CD56(+)/CD16 (−) NK cell subset with specificity for pancreatic and prostate cancer cell lines. Cancer Immunol Immunother. 59:1757–1769. doi: 10.1007/s00262-010-0897-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stangl S, Wortmann A, Guertler U, Multhoff G. Control of metastasized pancreatic carcinomas in SCID/beige mice with human IL-2/TKD-activated NK cells. J Immunol. 2006;176:6270–6276. doi: 10.4049/jimmunol.176.10.6270. 176/10/6270 [pii] [DOI] [PubMed] [Google Scholar]
- 48.Marincola FM, Da Pozzo LF, Drucker BJ, Holder WD., Jr Adoptive immunotherapy of human pancreatic cancer with lymphokine-activated killer cells and interleukin-2 in a nude mouse model. Surgery. 1990;108:919–929. 0039-6060(90)90291-9 [pii] [PubMed] [Google Scholar]
- 49.Wagner K, Schulz P, Scholz A, Wiedenmann B, Menrad A. The targeted immunocytokine L19-IL2 efficiently inhibits the growth of orthotopic pancreatic cancer. Clin Cancer Res. 2008;14:4951–4960. doi: 10.1158/1078-0432.CCR-08-0157. 14/15/4951 [pii] [DOI] [PubMed] [Google Scholar]
- 50.Houghton AM. The paradox of tumor-associated neutrophils: fueling tumor growth with cytotoxic substances. Cell Cycle. 9:1732–1737. doi: 10.4161/cc.9.9.11297. 11297 [pii] [DOI] [PubMed] [Google Scholar]
- 51.Aliustaoglu M, et al. The association of pre-treatment peripheral blood markers with survival in patients with pancreatic cancer. Hepatogastroenterology. 57:640–645. [PubMed] [Google Scholar]
- 52.An X, et al. Elevated neutrophil to lymphocyte ratio predicts survival in advanced pancreatic cancer. Biomarkers. 15:516–522. doi: 10.3109/1354750X.2010.491557. [DOI] [PubMed] [Google Scholar]
- 53.Ten Kate M, et al. Polymorphonuclear leukocytes increase the adhesion of circulating tumor cells to microvascular endothelium. Anticancer Res. 2007;27:17–22. [PubMed] [Google Scholar]
- 54.Bausch D, et al. Neutrophil granulocyte derived MMP-9 is a VEGF independent functional component of the angiogenic switch in pancreatic ductal adenocarcinoma. Angiogenesis. 2011 doi: 10.1007/s10456-011-9207-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nozawa H, Chiu C, Hanahan D. Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc Natl Acad Sci U S A. 2006;103:12493–12498. doi: 10.1073/pnas.0601807103. 0601807103 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Khazaie K, et al. The significant role of mast cells in cancer. Cancer and Metastasis Reviews. 2011;30:45–60. doi: 10.1007/s10555-011-9286-z. [DOI] [PubMed] [Google Scholar]
- 57.Boesiger, et al. Mast Cells Can Secrete Vascular Permeability Factor/Vascular Endothelial Cell Growth Factor and Exhibit Enhanced Release after Immunoglobulin E Aidependent Upregulation of Fc μ Receptor I Expression. The Journal of Experimental Medicine. 1998;188:1135–1145. doi: 10.1084/jem.188.6.1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Esposito I, et al. Inflammatory cells contribute to the generation of an angiogenic phenotype in pancreatic ductal adenocarcinoma. J Clin Pathol. 2004;57:630–636. doi: 10.1136/jcp.2003.014498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Strouch MJ, et al. Crosstalk between mast cells and pancreatic cancer cells contributes to pancreatic tumor progression. Clin Cancer Res. 2010;16:2257–2265. doi: 10.1158/1078-0432.CCR-09-1230. 1078-0432.CCR-09-1230 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. nri2506 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Almand B, et al. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol. 2001;166:678–689. doi: 10.4049/jimmunol.166.1.678. [DOI] [PubMed] [Google Scholar]
- 62.Bronte V, et al. Unopposed production of granulocyte-macrophage colony-stimulating factor by tumors inhibits CD8+ T cell responses by dysregulating antigen-presenting cell maturation. J Immunol. 1999;162:5728–5737. [PMC free article] [PubMed] [Google Scholar]
- 63.Melani C, Chiodoni C, Forni G, Colombo MP. Myeloid cell expansion elicited by the progression of spontaneous mammary carcinomas in c-erbB-2 transgenic BALB/c mice suppresses immune reactivity. Blood. 2003;102:2138–2145. doi: 10.1182/blood-2003-01-0190. 2003-01-0190 [pii] [DOI] [PubMed] [Google Scholar]
- 64.Kusmartsev S, et al. All-trans-retinoic acid eliminates immature myeloid cells from tumor-bearing mice and improves the effect of vaccination. Cancer Res. 2003;63:4441–4449. [PubMed] [Google Scholar]
- 65.Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. 2008;181:5791–5802. doi: 10.4049/jimmunol.181.8.5791. 181/8/5791 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Condamine T, Gabrilovich DI. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. 32:19–25. doi: 10.1016/j.it.2010.10.002. S1471-4906(10)00149-3 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Movahedi K, et al. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood. 2008;111:4233–4244. doi: 10.1182/blood-2007-07-099226. blood-2007-07-099226 [pii] [DOI] [PubMed] [Google Scholar]
- 68.Gabrilovich DI, Velders MP, Sotomayor EM, Kast WM. Mechanism of immune dysfunction in cancer mediated by immature Gr-1+ myeloid cells. J Immunol. 2001;166:5398–5406. doi: 10.4049/jimmunol.166.9.5398. [DOI] [PubMed] [Google Scholar]
- 69.Otsuji M, Kimura Y, Aoe T, Okamoto Y, Saito T. Oxidative stress by tumor-derived macrophages suppresses the expression of CD3 zeta chain of T-cell receptor complex and antigen-specific T-cell responses. Proc Natl Acad Sci U S A. 1996;93:13119–13124. doi: 10.1073/pnas.93.23.13119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huang B, et al. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006;66:1123–1131. doi: 10.1158/0008-5472.CAN-05-1299. 66/2/1123 [pii] [DOI] [PubMed] [Google Scholar]
- 71.Serafini P, Borrello I, Bronte V. Myeloid suppressor cells in cancer: recruitment, phenotype, properties, and mechanisms of immune suppression. Semin Cancer Biol. 2006;16:53–65. doi: 10.1016/j.semcancer.2005.07.005. S1044-579X(05)00058-1 [pii] [DOI] [PubMed] [Google Scholar]
- 72.Li H, Han Y, Guo Q, Zhang M, Cao X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J Immunol. 2009;182:240–249. doi: 10.4049/jimmunol.182.1.240. 182/1/240 [pii] [DOI] [PubMed] [Google Scholar]
- 73.Rodriguez PC, et al. Arginase I in myeloid suppressor cells is induced by COX-2 in lung carcinoma. J Exp Med. 2005;202:931–939. doi: 10.1084/jem.20050715. jem.20050715 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ostrand-Rosenberg S. Myeloid-derived suppressor cells: more mechanisms for inhibiting antitumor immunity. Cancer Immunol Immunother. 59:1593–1600. doi: 10.1007/s00262-010-0855-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tseng WW, et al. Development of an orthotopic model of invasive pancreatic cancer in an immunocompetent murine host. Clin Cancer Res. 16:3684–3695. doi: 10.1158/1078-0432.CCR-09-2384. 1078-0432.CCR-09-2384 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhao F, et al. Increase in frequency of myeloid-derived suppressor cells in mice with spontaneous pancreatic carcinoma. Immunology. 2009;128:141–149. doi: 10.1111/j.1365-2567.2009.03105.x. IMM3105 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Clark CE, et al. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007;67:9518–9527. doi: 10.1158/0008-5472.CAN-07-0175. 67/19/9518 [pii] [DOI] [PubMed] [Google Scholar]
- 78.Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006;66:605–612. doi: 10.1158/0008-5472.CAN-05-4005. 66/2/605 [pii] [DOI] [PubMed] [Google Scholar]
- 79.Mantovani A, Ming WJ, Balotta C, Abdeljalil B, Bottazzi B. Origin and regulation of tumor-associated macrophages: the role of tumor-derived chemotactic factor. Biochim Biophys Acta. 1986;865:59–67. doi: 10.1016/0304-419x(86)90013-2. [DOI] [PubMed] [Google Scholar]
- 80.Lewis JS, Landers RJ, Underwood JC, Harris AL, Lewis CE. Expression of vascular endothelial growth factor by macrophages is up-regulated in poorly vascularized areas of breast carcinomas. J Pathol. 2000;192:150–158. doi: 10.1002/1096-9896(2000)9999:9999. <::AID-PATH687>3.0.CO;2-G [pii] 10.1002/1096-9896(2000)9999:9999<::AID-PATH687>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 81.Sica A, Bronte V. Altered macrophage differentiation and immune dysfunction in tumor development. J Clin Invest. 2007;117:1155–1166. doi: 10.1172/JCI31422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Murdoch C, Tazzyman S, Webster S, Lewis CE. Expression of Tie-2 by human monocytes and their responses to angiopoietin-2. J Immunol. 2007;178:7405–7411. doi: 10.4049/jimmunol.178.11.7405. 178/11/7405 [pii] [DOI] [PubMed] [Google Scholar]
- 83.Sica A, et al. Macrophage polarization in tumour progression. Semin Cancer Biol. 2008;18:349–355. doi: 10.1016/j.semcancer.2008.03.004. S1044-579X(08)00023-0 [pii] [DOI] [PubMed] [Google Scholar]
- 84.Kurahara H, et al. Significance of M2-polarized tumor-associated macrophage in pancreatic cancer. The Journal of surgical research. 2011;167:e211–219. doi: 10.1016/j.jss.2009.05.026 . [DOI] [PubMed] [Google Scholar]
- 85.Mytar B, Baj-Krzyworzeka M, Majka M, Stankiewicz D, Zembala M. Human monocytes both enhance and inhibit the growth of human pancreatic cancer in SCID mice. Anticancer Res. 2008;28:187–192. [PubMed] [Google Scholar]
- 86.Mytar B, et al. Tumor cell-induced deactivation of human monocytes. Journal of Leukocyte Biology. 2003;74:1094–1101. doi: 10.1189/jlb.0403140. [DOI] [PubMed] [Google Scholar]
- 87.Baran B, et al. Blood monocytes stimulate migration of human pancreatic carcinoma cells in vitro: the role of tumour necrosis factor - alpha. Eur J Cell Biol. 2009;88:743–752. doi: 10.1016/j.ejcb.2009.08.002. S0171-9335(09)00291-X [pii] [DOI] [PubMed] [Google Scholar]
- 88.Clark CE, Beatty GL, Vonderheide RH. Immunosurveillance of pancreatic adenocarcinoma: insights from genetically engineered mouse models of cancer. Cancer Lett. 2009;279:1–7. doi: 10.1016/j.canlet.2008.09.037. S0304-3835(08)00799-4 [pii] [DOI] [PubMed] [Google Scholar]
- 89.Watowich SS, Liu YJ. Mechanisms regulating dendritic cell specification and development. Immunol Rev. 238:76–92. doi: 10.1111/j.1600-065X.2010.00949.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ma Y, Aymeric L, Locher C, Kroemer G, Zitvogel L. The dendritic cell-tumor crosstalk in cancer. Curr Opin Immunol. doi: 10.1016/j.coi.2010.09.008. S0952-7915(10)00135-4 [pii] [DOI] [PubMed] [Google Scholar]
- 91.Dallal RM, et al. Paucity of dendritic cells in pancreatic cancer. Surgery. 2002;131:135–138. doi: 10.1067/msy.2002.119937. S0039606002287929 [pii] [DOI] [PubMed] [Google Scholar]
- 92.Yamamoto T, et al. Circulating Myeloid Dendritic Cells as Prognostic Factors in Patients with Pancreatic Cancer Who Have Undergone Surgical Resection. J Surg Res. doi: 10.1016/j.jss.2010.09.027. S0022-4804(10)00778-X [pii] [DOI] [PubMed] [Google Scholar]
- 93.Tjomsland V, et al. Semi mature blood dendritic cells exist in patients with ductal pancreatic adenocarcinoma owing to inflammatory factors released from the tumor. PLoS One. 5:e13441. doi: 10.1371/journal.pone.0013441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tjomsland V, et al. Pancreatic adenocarcinoma exerts systemic effects on the peripheral blood myeloid and plasmacytoid dendritic cells: an indicator of disease severity? BMC Cancer. 10:87. doi: 10.1186/1471-2407-10-87. 1471-2407-10-87 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Neesse A, et al. Stromal biology and therapy in pancreatic cancer. Gut. doi: 10.1136/gut.2010.226092. gut.2010.226092 [pii] [DOI] [Google Scholar]
- 96.Reiser-Erkan C, et al. Hypoxia-inducible proto-oncogene Pim-1 is a prognostic marker in pancreatic ductal adenocarcinoma. Cancer Biol Ther. 2008;7:1352–1359. doi: 10.4161/cbt.7.9.6418. 6418 [pii] [DOI] [PubMed] [Google Scholar]
- 97.Couvelard A, et al. Expression of hypoxia-inducible factors is correlated with the presence of a fibrotic focus and angiogenesis in pancreatic ductal adenocarcinomas. Histopathology. 2005;46:668–676. doi: 10.1111/j.1365-2559.2005.02160.x. HIS2160 [pii] [DOI] [PubMed] [Google Scholar]
- 98.Bissell MJ, Radisky D. Putting tumours in context. Nat Rev Cancer. 2001;1:46–54. doi: 10.1038/35094059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Korc M. Pancreatic cancer-associated stroma production. Am J Surg. 2007;194:S84–86. doi: 10.1016/j.amjsurg.2007.05.004. S0002-9610(07)00348-0 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Korc M, et al. Overexpression of the epidermal growth factor receptor in human pancreatic cancer is associated with concomitant increases in the levels of epidermal growth factor and transforming growth factor alpha. J Clin Invest. 1992;90:1352–1360. doi: 10.1172/JCI116001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bachem MG, et al. Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology. 2005;128:907–921. doi: 10.1053/j.gastro.2004.12.036. S0016508504023315 [pii] [DOI] [PubMed] [Google Scholar]
- 102.Lohr M, et al. Transforming growth factor-beta1 induces desmoplasia in an experimental model of human pancreatic carcinoma. Cancer Res. 2001;61:550–555. [PubMed] [Google Scholar]
- 103.Vinay Kumar AKA, Fausto Nelson, Mitchell Richard N. Robbins Basic Pathology. 8. Saunders Elsevier; 2007. pp. 70–72.pp. 200–201. [Google Scholar]
- 104.Sipos B, et al. Vascular endothelial growth factor mediated angiogenic potential of pancreatic ductal carcinomas enhanced by hypoxia: an in vitro and in vivo study. Int J Cancer. 2002;102:592–600. doi: 10.1002/ijc.10753. [DOI] [PubMed] [Google Scholar]
- 105.Schniewind B, et al. Dissecting the role of TGF-beta type I receptor/ALK5 in pancreatic ductal adenocarcinoma: Smad activation is crucial for both the tumor suppressive and prometastatic function. Oncogene. 2007;26:4850–4862. doi: 10.1038/sj.onc.1210272. 1210272 [pii] [DOI] [PubMed] [Google Scholar]
- 106.Duner S, Lopatko Lindman J, Ansari D, Gundewar C, Andersson R. Pancreatic cancer: the role of pancreatic stellate cells in tumor progression. Pancreatology: official journal of the International Association of Pancreatology. 2010;10:673–681. doi: 10.1159/000320711. [DOI] [PubMed] [Google Scholar]
- 107.Erkan M, et al. Periostin creates a tumor-supportive microenvironment in the pancreas by sustaining fibrogenic stellate cell activity. Gastroenterology. 2007;132:1447–1464. doi: 10.1053/j.gastro.2007.01.031. S0016-5085(07)00166-7 [pii] [DOI] [PubMed] [Google Scholar]
- 108.Phillips PA, et al. Rat pancreatic stellate cells secrete matrix metalloproteinases: implications for extracellular matrix turnover. Gut. 2003;52:275–282. doi: 10.1136/gut.52.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Erkan M, et al. Cancer-stellate cell interactions perpetuate the hypoxia-fibrosis cycle in pancreatic ductal adenocarcinoma. Neoplasia. 2009;11:497–508. doi: 10.1593/neo.81618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Phillips PA, et al. Cell migration: a novel aspect of pancreatic stellate cell biology. Gut. 2003;52:677–682. doi: 10.1136/gut.52.5.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bergers G, et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol. 2000;2:737–744. doi: 10.1038/35036374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.O’Reilly MS, et al. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell. 1997;88:277–285. doi: 10.1016/s0092-8674(00)81848-6. S0092-8674(00)81848-6 [pii] [DOI] [PubMed] [Google Scholar]
- 113.Walter K, et al. Overexpression of smoothened activates the sonic hedgehog signaling pathway in pancreatic cancer-associated fibroblasts. Clinical cancer research: an official journal of the American Association for Cancer Research. 2010;16:1781–1789. doi: 10.1158/1078-0432.CCR-09-1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Soker S, Takashima S, Miao HQ, Neufeld G, Klagsbrun M. Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell. 1998;92:735–745. doi: 10.1016/s0092-8674(00)81402-6. S0092-8674(00)81402-6 [pii] [DOI] [PubMed] [Google Scholar]
- 115.Dallas NA, et al. Functional significance of vascular endothelial growth factor receptors on gastrointestinal cancer cells. Cancer Metastasis Rev. 2007;26:433–441. doi: 10.1007/s10555-007-9070-2. [DOI] [PubMed] [Google Scholar]
- 116.Wey JS, et al. Overexpression of neuropilin-1 promotes constitutive MAPK signalling and chemoresistance in pancreatic cancer cells. Br J Cancer. 2005;93:233–241. doi: 10.1038/sj.bjc.6602663. 6602663 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hansel DE, et al. Expression of neuropilin-1 in high-grade dysplasia, invasive cancer, and metastases of the human gastrointestinal tract. Am J Surg Pathol. 2004;28:347–356. doi: 10.1097/00000478-200403000-00007. [DOI] [PubMed] [Google Scholar]
- 118.Muders MH. Neuropilin and Neuropilin Associated Molecules as New Molecular Targets in Pancreatic Adenocarcinoma. Anticancer Agents Med Chem. 2011 doi: 10.2174/187152011795677481. [DOI] [PubMed] [Google Scholar]
- 119.Dallas NA, et al. Neuropilin-2-mediated tumor growth and angiogenesis in pancreatic adenocarcinoma. Clin Cancer Res. 2008;14:8052–8060. doi: 10.1158/1078-0432.CCR-08-1520. 14/24/8052 [pii] [DOI] [PubMed] [Google Scholar]
- 120.Tartour E, et al. Angiogenesis and immunity: a bidirectional link potentially relevant for the monitoring of antiangiogenic therapy and the development of novel therapeutic combination with immunotherapy. Cancer metastasis reviews. 2011;30:83–95. doi: 10.1007/s10555-011-9281-4. [DOI] [PubMed] [Google Scholar]
- 121.Ding XZ, Fehsenfeld DM, Murphy LO, Permert J, Adrian TE. Physiological concentrations of insulin augment pancreatic cancer cell proliferation and glucose utilization by activating MAP kinase, PI3 kinase and enhancing GLUT-1 expression. Pancreas. 2000;21:310–320. doi: 10.1097/00006676-200010000-00014. [DOI] [PubMed] [Google Scholar]
- 122.Kang SP, Saif MW. Clinical outcome of pancreatic cancer patients with diabetes mellitus: is diabetes a poor prognostic factor?. Highlights from the “2010 ASCO Annual Meeting”; Chicago IL USA . June 4–8, 2010; pp. 334–335. JOP. v11i04a09 [pii] [PubMed] [Google Scholar]
- 123.Schneider MB, et al. Prevention of pancreatic cancer induction in hamsters by metformin. Gastroenterology. 2001;120:1263–1270. doi: 10.1053/gast.2001.23258. S0016508501564938 [pii] [DOI] [PubMed] [Google Scholar]
- 124.Bergmann U, Funatomi H, Kornmann M, Beger HG, Korc M. Increased expression of insulin receptor substrate-1 in human pancreatic cancer. Biochem Biophys Res Commun. 1996;220:886–890. doi: 10.1006/bbrc.1996.0500. S0006-291X(96)90500-8 [pii] [DOI] [PubMed] [Google Scholar]
- 125.Kornmann M, et al. Enhanced expression of the insulin receptor substrate-2 docking protein in human pancreatic cancer. Cancer Res. 1998;58:4250–4254. [PubMed] [Google Scholar]
- 126.Teppala S, Shankar A. Association between serum IGF-1 and diabetes among U.S. adults. Diabetes Care. 2010;33:2257–2259. doi: 10.2337/dc10-0770. dc10-0770 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Rieder S, Michalski CW, Friess H, Kleeff J. Insulin-Like Growth Factor Signaling as a Therapeutic Target in Pancreatic Cancer. Anticancer Agents Med Chem. 2011 doi: 10.2174/187152011795677454. [DOI] [PubMed] [Google Scholar]
- 128.Treins C, Giorgetti-Peraldi S, Murdaca J, Van Obberghen E. Regulation of vascular endothelial growth factor expression by advanced glycation end products. J Biol Chem. 2001;276:43836–43841. doi: 10.1074/jbc.M106534200. M106534200 [pii] [DOI] [PubMed] [Google Scholar]
- 129.Wang F, et al. Hypoxia inducible factor-1 mediates effects of insulin on pancreatic cancer cells and disturbs host energy homeostasis. Am J Pathol. 2007;170:469–477. doi: 10.2353/ajpath.2007.060489. S0002-9440(10)60870-X [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Williams B, Gallacher B, Patel H, Orme C. Glucose-induced protein kinase C activation regulates vascular permeability factor mRNA expression and peptide production by human vascular smooth muscle cells in vitro. Diabetes. 1997;46:1497–1503. doi: 10.2337/diab.46.9.1497. [DOI] [PubMed] [Google Scholar]
- 131.Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991–998. doi: 10.1038/ni1102-991. ni1102-991 [pii] [DOI] [PubMed] [Google Scholar]
- 132.Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol. 2004;22:329–360. doi: 10.1146/annurev.immunol.22.012703.104803. [DOI] [PubMed] [Google Scholar]
- 133.Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annual Review of Immunology. 2011;29:235–271. doi: 10.1146/annurev-immunol-031210-101324. [DOI] [PubMed] [Google Scholar]
- 134.Samm N, et al. The Role of Apoptosis in the Pathology of Pancreatic Cancer. Cancers. 2010;3:1–16. doi: 10.3390/cancers3010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Nagata S, Golstein P. The Fas death factor. Science. 1995;267:1449–1456. doi: 10.1126/science.7533326. [DOI] [PubMed] [Google Scholar]
- 136.Strand S, et al. Lymphocyte apoptosis induced by CD95 (APO-1/Fas) ligand-expressing tumor cells--a mechanism of immune evasion? Nat Med. 1996;2:1361–1366. doi: 10.1038/nm1296-1361. [DOI] [PubMed] [Google Scholar]
- 137.Ungefroren H, et al. Immunological escape mechanisms in pancreatic carcinoma. Ann NY Acad Sci. 1999;880:243–251. doi: 10.1111/j.1749-6632.1999.tb09529.x. [DOI] [PubMed] [Google Scholar]
- 138.Ohta T, et al. Fas ligand expression in human pancreatic cancer. Oncol Rep. 2004;12:749–754. [PubMed] [Google Scholar]
- 139.von Bernstorff W, et al. Pancreatic cancer cells can evade immune surveillance via nonfunctional Fas (APO-1/CD95) receptors and aberrant expression of functional Fas ligand. Surgery. 1999;125:73–84. doi: 10.1067/msy.2099.93570. [DOI] [PubMed] [Google Scholar]
- 140.Sato T, Irie S, Kitada S, Reed JC. FAP-1: a protein tyrosine phosphatase that associates with Fas. Science. 1995;268:411–415. doi: 10.1126/science.7536343. [DOI] [PubMed] [Google Scholar]
- 141.Bellone G, et al. Production and pro-apoptotic activity of soluble CD95 ligand in pancreatic carcinoma. Clin Cancer Res. 2000;6:2448–2455. [PubMed] [Google Scholar]
- 142.Dhein J, Walczak H, Baumler C, Debatin KM, Krammer PH. Autocrine T-cell suicide mediated by APO-1/(Fas/CD95) Nature. 1995;373:438–441. doi: 10.1038/373438a0. [DOI] [PubMed] [Google Scholar]
- 143.Griffith TS, Lynch DH. TRAIL: a molecule with multiple receptors and control mechanisms. Curr Opin Immunol. 1998;10:559–563. doi: 10.1016/s0952-7915(98)80224-0. S0952-7915(98)80224-0 [pii] [DOI] [PubMed] [Google Scholar]
- 144.Pitti RM, et al. Genomic amplification of a decoy receptor for Fas ligand in lung and colon cancer. Nature. 1998;396:699–703. doi: 10.1038/25387. [DOI] [PubMed] [Google Scholar]
- 145.Scaffidi C, Kirchhoff S, Krammer PH, Peter ME. Apoptosis signaling in lymphocytes. Curr Opin Immunol. 1999;11:277–285. doi: 10.1016/s0952-7915(99)80045-4. imb308 [pii] [DOI] [PubMed] [Google Scholar]
- 146.Ibrahim SM, et al. Pancreatic adenocarcinoma cell lines show variable susceptibility to TRAIL-mediated cell death. Pancreas. 2001;23:72–79. doi: 10.1097/00006676-200107000-00011. [DOI] [PubMed] [Google Scholar]
- 147.Boni R, Wellmann A, Man YG, Hofbauer G, Brinkmann U. Expression of the proliferation and apoptosis-associated CAS protein in benign and malignant cutaneous melanocytic lesions. Am J Dermatopathol. 1999;21:125–128. doi: 10.1097/00000372-199904000-00003. [DOI] [PubMed] [Google Scholar]
- 148.Yoong KF, Afford SC, Randhawa S, Hubscher SG, Adams DH. Fas/Fas ligand interaction in human colorectal hepatic metastases: A mechanism of hepatocyte destruction to facilitate local tumor invasion. Am J Pathol. 1999;154:693–703. doi: 10.1016/S0002-9440(10)65316-3. S0002-9440(10)65316-3 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Sanlioglu AD, et al. High levels of endogenous tumor necrosis factor-related apoptosis-inducing ligand expression correlate with increased cell death in human pancreas. Pancreas. 2008;36:385–393. doi: 10.1097/MPA.0b013e318158a4e5. 00006676-200805000-00009 [pii] [DOI] [PubMed] [Google Scholar]
- 150.Sanlioglu AD, et al. High TRAIL death receptor 4 and decoy receptor 2 expression correlates with significant cell death in pancreatic ductal adenocarcinoma patients. Pancreas. 2009;38:154–160. doi: 10.1097/MPA.0b013e31818db9e3. 00006676-200903000-00009 [pii] [DOI] [PubMed] [Google Scholar]
- 151.Hamacher R, Schmid RM, Saur D, Schneider G. Apoptotic pathways in pancreatic ductal adenocarcinoma. Mol Cancer. 2008;7:64. doi: 10.1186/1476-4598-7-64. 1476-4598-7-64 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Fulda S. Apoptosis pathways and their therapeutic exploitation in pancreatic cancer. J Cell Mol Med. 2009;13:1221–1227. doi: 10.1111/j.1582-4934.2009.00748.x. JCMM748 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ozawa F, et al. Effects and expression of TRAIL and its apoptosis-promoting receptors in human pancreatic cancer. Cancer Lett. 2001;163:71–81. doi: 10.1016/s0304-3835(00)00660-1. S0304383500006601 [pii] [DOI] [PubMed] [Google Scholar]
- 154.Schuler S, et al. HDAC2 attenuates TRAIL-induced apoptosis of pancreatic cancer cells. Mol Cancer. 2010;9:80. doi: 10.1186/1476-4598-9-80. 1476-4598-9-80 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Schuler S, et al. SKP2 confers resistance of pancreatic cancer cells towards TRAIL-induced apoptosis. Int J Oncol. 2011;38:219–225. [PubMed] [Google Scholar]
- 156.Huang S, Sinicrope FA. Sorafenib inhibits STAT3 activation to enhance TRAIL-mediated apoptosis in human pancreatic cancer cells. Mol Cancer Ther. 2010;9:742–750. doi: 10.1158/1535-7163.MCT-09-1004. 1535-7163.MCT-09-1004 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ripka S, et al. CUX1: target of Akt signalling and mediator of resistance to apoptosis in pancreatic cancer. Gut. 2010;59:1101–1110. doi: 10.1136/gut.2009.189720. gut.2009.189720 [pii] [DOI] [PubMed] [Google Scholar]
- 158.Kauh J, et al. c-FLIP degradation mediates sensitization of pancreatic cancer cells to TRAIL-induced apoptosis by the histone deacetylase inhibitor LBH589. PLoS One. 2010;5:e10376. doi: 10.1371/journal.pone.0010376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Wang P, et al. Inhibition of RIP and c-FLIP enhances TRAIL-induced apoptosis in pancreatic cancer cells. Cell Signal. 2007;19:2237–2246. doi: 10.1016/j.cellsig.2007.06.001. S0898-6568(07)00176-3 [pii] [DOI] [PubMed] [Google Scholar]
- 160.Murtaza I, Saleem M, Adhami VM, Hafeez BB, Mukhtar H. Suppression of cFLIP by lupeol, a dietary triterpene, is sufficient to overcome resistance to TRAIL-mediated apoptosis in chemoresistant human pancreatic cancer cells. Cancer Res. 2009;69:1156–1165. doi: 10.1158/0008-5472.CAN-08-2917. 0008-5472.CAN-08-2917 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Vogler M, Durr K, Jovanovic M, Debatin KM, Fulda S. Regulation of TRAIL-induced apoptosis by XIAP in pancreatic carcinoma cells. Oncogene. 2007;26:248–257. doi: 10.1038/sj.onc.1209776. 1209776 [pii] [DOI] [PubMed] [Google Scholar]
- 162.Vogler M, et al. Targeting XIAP bypasses Bcl-2-mediated resistance to TRAIL and cooperates with TRAIL to suppress pancreatic cancer growth in vitro and in vivo. Cancer Res. 2008;68:7956–7965. doi: 10.1158/0008-5472.CAN-08-1296. 68/19/7956 [pii] [DOI] [PubMed] [Google Scholar]
- 163.Vogler M, et al. Small molecule XIAP inhibitors enhance TRAIL-induced apoptosis and antitumor activity in preclinical models of pancreatic carcinoma. Cancer Res. 2009;69:2425–2434. doi: 10.1158/0008-5472.CAN-08-2436. 0008-5472.CAN-08-2436 [pii] [DOI] [PubMed] [Google Scholar]
- 164.Dineen SP, et al. Smac mimetic increases chemotherapy response and improves survival in mice with pancreatic cancer. Cancer Res. 2010;70:2852–2861. doi: 10.1158/0008-5472.CAN-09-3892. 0008-5472.CAN-09-3892 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Mori T, et al. Effect of the XIAP inhibitor Embelin on TRAIL-induced apoptosis of pancreatic cancer cells. J Surg Res. 2007;142:281–286. doi: 10.1016/j.jss.2007.03.068. S0022-4804(07)00199-0 [pii] [DOI] [PubMed] [Google Scholar]
- 166.Huang S, Okumura K, Sinicrope FA. BH3 mimetic obatoclax enhances TRAIL-mediated apoptosis in human pancreatic cancer cells. Clin Cancer Res. 2009;15:150–159. doi: 10.1158/1078-0432.CCR-08-1575. 15/1/150 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Bai J, et al. Predominant Bcl-XL knockdown disables antiapoptotic mechanisms: tumor necrosis factor-related apoptosis-inducing ligand-based triple chemotherapy overcomes chemoresistance in pancreatic cancer cells in vitro. Cancer Res. 2005;65:2344–2352. doi: 10.1158/0008-5472.CAN-04-3502. 65/6/2344 [pii] [DOI] [PubMed] [Google Scholar]
- 168.Song JJ, An JY, Kwon YT, Lee YJ. Evidence for two modes of development of acquired tumor necrosis factor-related apoptosis-inducing ligand resistance. Involvement of Bcl-xL. J Biol Chem. 2007;282:319–328. doi: 10.1074/jbc.M608065200. M608065200 [pii] [DOI] [PubMed] [Google Scholar]
- 169.Retzer-Lidl M, Schmid RM, Schneider G. Inhibition of CDK4 impairs proliferation of pancreatic cancer cells and sensitizes towards TRAIL-induced apoptosis via downregulation of survivin. Int J Cancer. 2007;121:66–75. doi: 10.1002/ijc.22619. [DOI] [PubMed] [Google Scholar]
- 170.Nakashima M, Sonoda K, Watanabe T. Inhibition of cell growth and induction of apoptotic cell death by the human tumor-associated antigen RCAS1. Nat Med. 1999;5:938–942. doi: 10.1038/11383. [DOI] [PubMed] [Google Scholar]
- 171.Hiraoka K, et al. High expression of tumor-associated antigen RCAS1 in pancreatic ductal adenocarcinoma is an unfavorable prognostic marker. Int J Cancer. 2002;99:418–423. doi: 10.1002/ijc.10381. [DOI] [PubMed] [Google Scholar]
- 172.Ozkan H, Akar T, Koklu S, Coban S. Significance of serum receptor-binding cancer antigen (RCAS1) in pancreatic cancer and benign pancreatobiliary diseases. Pancreatology. 2006;6:268–272. doi: 10.1159/000092687. 000092687 [pii] [DOI] [PubMed] [Google Scholar]
- 173.Giaginis C, et al. Clinical significance of tumor-associated antigen RCAS1 expression in human pancreatic ductal adenocarcinoma. Dig Dis Sci. 2008;53:1728–1734. doi: 10.1007/s10620-007-0035-7. [DOI] [PubMed] [Google Scholar]
- 174.Imanishi T, et al. Correlation between expression of major histocompatibility complex class I and that of antigen presenting machineries in carcinoma cell lines of the pancreas, biliary tract and colon. Kobe J Med Sci. 2006;52:85–95. [PubMed] [Google Scholar]
- 175.Pandha H, Rigg A, John J, Lemoine N. Loss of expression of antigen-presenting molecules in human pancreatic cancer and pancreatic cancer cell lines. Clin Exp Immunol. 2007;148:127–135. doi: 10.1111/j.1365-2249.2006.03289.x. CEI3289 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Groh V, et al. Broad tumor-associated expression and recognition by tumor-derived gamma delta T cells of MICA and MICB. Proc Natl Acad Sci U S A. 1999;96:6879–6884. doi: 10.1073/pnas.96.12.6879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Marten A, von Lilienfeld-Toal M, Buchler MW, Schmidt J. Soluble MIC is elevated in the serum of patients with pancreatic carcinoma diminishing gammadelta T cell cytotoxicity. Int J Cancer. 2006;119:2359–2365. doi: 10.1002/ijc.22186. [DOI] [PubMed] [Google Scholar]
- 178.Harris CL, Abbott RJ, Smith RA, Morgan BP, Lea SM. Molecular dissection of interactions between components of the alternative pathway of complement and decay accelerating factor (CD55) J Biol Chem. 2005;280:2569–2578. doi: 10.1074/jbc.M410179200. M410179200 [pii] [DOI] [PubMed] [Google Scholar]
- 179.Purcell DF, Deacon NJ, Andrew SM, McKenzie IF. Human non-lineage antigen, CD46 (HuLy-m5): purification and partial sequencing demonstrates structural homology with complement-regulating glycoproteins. Immunogenetics. 1990;31:21–28. doi: 10.1007/BF00702485. [DOI] [PubMed] [Google Scholar]
- 180.Meri S, et al. Human protectin (CD59), an 18,000–20,000 MW complement lysis restricting factor, inhibits C5b-8 catalysed insertion of C9 into lipid bilayers. Immunology. 1990;71:1–9. [PMC free article] [PubMed] [Google Scholar]
- 181.Juhl H, et al. Frequent expression of complement resistance factors CD46, CD55, and CD59 on gastrointestinal cancer cells limits the therapeutic potential of monoclonal antibody 17-1A. J Surg Oncol. 1997;64:222–230. doi: 10.1002/(SICI)1096-9098(199703)64:3. <222::AID-JSO9>3.0.CO;2-C [pii] [DOI] [PubMed] [Google Scholar]
- 182.Collins M, Ling V, Carreno BM. The B7 family of immune-regulatory ligands. Genome Biol. 2005;6:223. doi: 10.1186/gb-2005-6-6-223. gb-2005-6-6-223 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Salomon BA, Bluestone JA. COMPLEXITIES OF CD28/B7: CTLA-4 COSTIMULATORY PATHWAYS IN AUTOIMMUNITY AND TRANSPLANTATION. Annual Review of Immunology. 2001;19:225–252. doi: 10.1146/annurev.immunol.19.1.225. [DOI] [PubMed] [Google Scholar]
- 184.Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu Rev Immunol. 2005;23:515–548. doi: 10.1146/annurev.immunol.23.021704.115611. [DOI] [PubMed] [Google Scholar]
- 185.Latchman Y, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. 2001;2:261–268. doi: 10.1038/85330. [DOI] [PubMed] [Google Scholar]
- 186.Freeman GJ, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027–1034. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Youngnak P, et al. Differential binding properties of B7-H1 and B7-DC to programmed death-1. Biochem Biophys Res Commun. 2003;307:672–677. doi: 10.1016/s0006-291x(03)01257-9. S0006291X03012579 [pii] [DOI] [PubMed] [Google Scholar]
- 188.Nomi T, et al. Clinical significance and therapeutic potential of the programmed death-1 ligand/programmed death-1 pathway in human pancreatic cancer. Clin Cancer Res. 2007;13:2151–2157. doi: 10.1158/1078-0432.CCR-06-2746. 13/7/2151 [pii] [DOI] [PubMed] [Google Scholar]
- 189.Ishiwata K, et al. Costimulator B7-DC attenuates strong Th2 responses induced by Nippostrongylus brasiliensis. J Immunol. 2010;184:2086–2094. doi: 10.4049/jimmunol.0804051. jimmunol.0804051 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Loke P, Allison JP. PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. Proc Natl Acad Sci U S A. 2003;100:5336–5341. doi: 10.1073/pnas.0931259100. 0931259100 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Matsumoto K, et al. B7-DC induced by IL-13 works as a feedback regulator in the effector phase of allergic asthma. Biochem Biophys Res Commun. 2008;365:170–175. doi: 10.1016/j.bbrc.2007.10.156. S0006-291X(07)02340-6 [pii] [DOI] [PubMed] [Google Scholar]
- 192.Carter L, et al. PD-1:PD-L inhibitory pathway affects both CD4(+) and CD8(+) T cells and is overcome by IL-2. Eur J Immunol. 2002;32:634–643. doi: 10.1002/1521-4141(200203)32:3<634::AID-IMMU634>3.0.CO;2-9. [pii] 10.1002/1521-4141(200203)32:3<634::AID-IMMU634>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 193.Brown JA, et al. Blockade of programmed death-1 ligands on dendritic cells enhances T cell activation and cytokine production. J Immunol. 2003;170:1257–1266. doi: 10.4049/jimmunol.170.3.1257. [DOI] [PubMed] [Google Scholar]
- 194.Ansari MJ, et al. The programmed death-1 (PD-1) pathway regulates autoimmune diabetes in nonobese diabetic (NOD) mice. J Exp Med. 2003;198:63–69. doi: 10.1084/jem.20022125. jem.20022125 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Dong H, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793–800. doi: 10.1038/nm730. nm730 [pii] [DOI] [PubMed] [Google Scholar]
- 196.Wintterle S, et al. Expression of the B7-related molecule B7-H1 by glioma cells: a potential mechanism of immune paralysis. Cancer Res. 2003;63:7462–7467. [PubMed] [Google Scholar]
- 197.Curiel TJ, et al. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat Med. 2003;9:562–567. doi: 10.1038/nm863. nm863 [pii] [DOI] [PubMed] [Google Scholar]
- 198.Iwai Y, et al. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A. 2002;99:12293–12297. doi: 10.1073/pnas.192461099. 192461099 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med. 1999;5:1365–1369. doi: 10.1038/70932. [DOI] [PubMed] [Google Scholar]
- 200.Tamura H, et al. B7-H1 costimulation preferentially enhances CD28-independent T-helper cell function. Blood. 2001;97:1809–1816. doi: 10.1182/blood.v97.6.1809. [DOI] [PubMed] [Google Scholar]
- 201.Moore KW, de Waal Malefyt R, Coffman RL, O’Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. 19/1/683 [pii] [DOI] [PubMed] [Google Scholar]
- 202.Akdis CA, Blaser K. Mechanisms of interleukin-10-mediated immune suppression. Immunology. 2001;103:131–136. doi: 10.1046/j.1365-2567.2001.01235.x. imm1235 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Vicari AP, Trinchieri G. Interleukin-10 in viral diseases and cancer: exiting the labyrinth? Immunol Rev. 2004;202:223–236. doi: 10.1111/j.0105–2896.2004.00216.x. IMR216 [pii] [DOI] [PubMed] [Google Scholar]
- 204.Erdman SE, et al. CD4(+)CD25(+) regulatory lymphocytes require interleukin 10 to interrupt colon carcinogenesis in mice. Cancer Res. 2003;63:6042–6050. [PubMed] [Google Scholar]
- 205.Geng L, et al. B7-H1 up-regulated expression in human pancreatic carcinoma tissue associates with tumor progression. J Cancer Res Clin Oncol. 2008;134:1021–1027. doi: 10.1007/s00432-008-0364-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Okudaira K, et al. Blockade of B7-H1 or B7-DC induces an anti-tumor effect in a mouse pancreatic cancer model. Int J Oncol. 2009;35:741–749. doi: 10.3892/ijo_00000387. [DOI] [PubMed] [Google Scholar]
- 207.Loos M, Hedderich DM, Friess H, Kleeff J. B7-h3 and its role in antitumor immunity. Clin Dev Immunol. 2010;2010:683875. doi: 10.1155/2010/683875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Chapoval AI, et al. B7-H3: a costimulatory molecule for T cell activation and IFN-gamma production. Nat Immunol. 2001;2:269–274. doi: 10.1038/85339. [DOI] [PubMed] [Google Scholar]
- 209.Sun M, et al. Characterization of mouse and human B7-H3 genes. J Immunol. 2002;168:6294–6297. doi: 10.4049/jimmunol.168.12.6294. [DOI] [PubMed] [Google Scholar]
- 210.Steinberger P, et al. Molecular characterization of human 4Ig-B7-H3, a member of the B7 family with four Ig-like domains. J Immunol. 2004;172:2352–2359. doi: 10.4049/jimmunol.172.4.2352. [DOI] [PubMed] [Google Scholar]
- 211.Zhang GB, et al. Characterization and application of two novel monoclonal antibodies against 2IgB7-H3: expression analysis of 2IgB7-H3 on dendritic cells and tumor cells. Tissue Antigens. 2005;66:83–92. doi: 10.1111/j.1399-0039.2005.00449.x. TAN449 [pii] [DOI] [PubMed] [Google Scholar]
- 212.Roth TJ, et al. B7-H3 ligand expression by prostate cancer: a novel marker of prognosis and potential target for therapy. Cancer Res. 2007;67:7893–7900. doi: 10.1158/0008-5472.CAN-07-1068. 0008-5472.CAN-07-1068 [pii] [DOI] [PubMed] [Google Scholar]
- 213.Suh WK, et al. The B7 family member B7-H3 preferentially down-regulates T helper type 1-mediated immune responses. Nat Immunol. 2003;4:899–906. doi: 10.1038/ni967. ni967 [pii] [DOI] [PubMed] [Google Scholar]
- 214.Castriconi R, et al. Identification of 4Ig-B7-H3 as a neuroblastoma-associated molecule that exerts a protective role from an NK cell-mediated lysis. Proc Natl Acad Sci U S A. 2004;101:12640–12645. doi: 10.1073/pnas.0405025101. 0405025101 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Prasad DV, et al. Murine B7-H3 is a negative regulator of T cells. J Immunol. 2004;173:2500–2506. doi: 10.4049/jimmunol.173.4.2500. 173/4/2500 [pii] [DOI] [PubMed] [Google Scholar]
- 216.Wang L, et al. B7-H3 promotes acute and chronic allograft rejection. Eur J Immunol. 2005;35:428–438. doi: 10.1002/eji.200425518. [DOI] [PubMed] [Google Scholar]
- 217.Hashiguchi M, et al. Triggering receptor expressed on myeloid cell-like transcript 2 (TLT-2) is a counter-receptor for B7-H3 and enhances T cell responses. Proc Natl Acad Sci U S A. 2008;105:10495–10500. doi: 10.1073/pnas.0802423105. 0802423105 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Yamato I, et al. Clinical importance of B7-H3 expression in human pancreatic cancer. Br J Cancer. 2009;101:1709–1716. doi: 10.1038/sj.bjc.6605375. 6605375 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Choi IH, et al. Genomic organization and expression analysis of B7-H4, an immune inhibitory molecule of the B7 family. J Immunol. 2003;171:4650–4654. doi: 10.4049/jimmunol.171.9.4650. [DOI] [PubMed] [Google Scholar]
- 220.Yi KH, Chen L. Fine tuning the immune response through B7-H3 and B7-H4. Immunol Rev. 2009;229:145–151. doi: 10.1111/j.1600-065X.2009.00768.x. IMR768 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Kryczek I, et al. Cutting edge: induction of B7-H4 on APCs through IL-10: novel suppressive mode for regulatory T cells. J Immunol. 2006;177:40–44. doi: 10.4049/jimmunol.177.1.40. 177/1/40 [pii] [DOI] [PubMed] [Google Scholar]
- 222.Kryczek I, et al. B7-H4 expression identifies a novel suppressive macrophage population in human ovarian carcinoma. J Exp Med. 2006;203:871–881. doi: 10.1084/jem.20050930. jem.20050930 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Kryczek I, et al. Relationship between B7-H4, regulatory T cells, and patient outcome in human ovarian carcinoma. Cancer Res. 2007;67:8900–8905. doi: 10.1158/0008-5472.CAN-07-1866. 67/18/8900 [pii] [DOI] [PubMed] [Google Scholar]
- 224.Prasad DV, Richards S, Mai XM, Dong C. B7S1, a novel B7 family member that negatively regulates T cell activation. Immunity. 2003;18:863–873. doi: 10.1016/s1074-7613(03)00147-x. S107476130300147X [pii] [DOI] [PubMed] [Google Scholar]
- 225.Sica GL, et al. B7-H4, a molecule of the B7 family, negatively regulates T cell immunity. Immunity. 2003;18:849–861. doi: 10.1016/s1074-7613(03)00152-3. S1074761303001523 [pii] [DOI] [PubMed] [Google Scholar]
- 226.Zang X, et al. B7x: a widely expressed B7 family member that inhibits T cell activation. Proc Natl Acad Sci U S A. 2003;100:10388–10392. doi: 10.1073/pnas.1434299100. 1434299100 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Awadallah NS, et al. Detection of B7-H4 and p53 in pancreatic cancer: potential role as a cytological diagnostic adjunct. Pancreas. 2008;36:200–206. doi: 10.1097/MPA.0b013e318150e4e0. 00006676-200803000-00016 [pii] [DOI] [PubMed] [Google Scholar]
- 228.Paulie S, et al. A p50 surface antigen restricted to human urinary bladder carcinomas and B lymphocytes. Cancer Immunol Immunother. 1985;20:23–28. doi: 10.1007/BF00199769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Alderson MR, et al. CD40 expression by human monocytes: regulation by cytokines and activation of monocytes by the ligand for CD40. J Exp Med. 1993;178:669–674. doi: 10.1084/jem.178.2.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Caux C, et al. Functional CD40 on B lymphocytes and dendritic cells. Res Immunol. 1994;145:235–239. doi: 10.1016/s0923-2494(94)80190-8. discussion 244–239. [DOI] [PubMed] [Google Scholar]
- 231.Banchereau J, et al. The CD40 antigen and its ligand. Annu Rev Immunol. 1994;12:881–922. doi: 10.1146/annurev.iy.12.040194.004313. [DOI] [PubMed] [Google Scholar]
- 232.Miyashita T, et al. Bidirectional regulation of human B cell responses by CD40-CD40 ligand interactions. J Immunol. 1997;158:4620–4633. [PubMed] [Google Scholar]
- 233.Merville P, et al. Bcl-2+ tonsillar plasma cells are rescued from apoptosis by bone marrow fibroblasts. J Exp Med. 1996;183:227–236. doi: 10.1084/jem.183.1.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Henriquez NV, Floettmann E, Salmon M, Rowe M, Rickinson AB. Differential responses to CD40 ligation among Burkitt lymphoma lines that are uniformly responsive to Epstein-Barr virus latent membrane protein 1. J Immunol. 1999;162:3298–3307. [PubMed] [Google Scholar]
- 235.Hunter TB, Alsarraj M, Gladue RP, Bedian V, Antonia SJ. An agonist antibody specific for CD40 induces dendritic cell maturation and promotes autologous anti-tumour T-cell responses in an in vitro mixed autologous tumour cell/lymph node cell model. Scand J Immunol. 2007;65:479–486. doi: 10.1111/j.1365-3083.2007.01927.x. SJI1927 [pii] [DOI] [PubMed] [Google Scholar]
- 236.Shoji Y, et al. The CD40-CD154 interaction would correlate with proliferation and immune escape in pancreatic ductal adenocarcinoma. J Surg Oncol. 2011;103:230–238. doi: 10.1002/jso.21812. [DOI] [PubMed] [Google Scholar]
- 237.Alexandroff AB, et al. Role for CD40-CD40 ligand interactions in the immune response to solid tumours. Mol Immunol. 2000;37:515–526. doi: 10.1016/s0161-5890(00)00079-1. S0161589000000791 [pii] [DOI] [PubMed] [Google Scholar]
- 238.Shorts L, et al. Stimulation through CD40 on mouse and human renal cell carcinomas triggers cytokine production, leukocyte recruitment, and antitumor responses that can be independent of host CD40 expression. J Immunol. 2006;176:6543–6552. doi: 10.4049/jimmunol.176.11.6543. 176/11/6543 [pii] [DOI] [PubMed] [Google Scholar]
- 239.Georgopoulos NT, et al. CD40-mediated death and cytokine secretion in colorectal cancer: a potential target for inflammatory tumour cell killing. Int J Cancer. 2007;121:1373–1381. doi: 10.1002/ijc.22846. [DOI] [PubMed] [Google Scholar]
- 240.Beatty GL, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331:1612–1616. doi: 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Roebuck KA, Finnegan A. Regulation of intercellular adhesion molecule-1 (CD54) gene expression. J Leukoc Biol. 1999;66:876–888. doi: 10.1002/jlb.66.6.876. [DOI] [PubMed] [Google Scholar]
- 242.Rothlein R, Dustin ML, Marlin SD, Springer TA. A human intercellular adhesion molecule (ICAM-1) distinct from LFA-1. J Immunol. 1986;137:1270–1274. [PubMed] [Google Scholar]
- 243.van de Stolpe A, van der Saag PT. Intercellular adhesion molecule-1. J Mol Med. 1996;74:13–33. doi: 10.1007/BF00202069. [DOI] [PubMed] [Google Scholar]
- 244.Witkowska AM, Borawska MH. Soluble intercellular adhesion molecule-1 (sICAM-1): an overview. Eur Cytokine Netw. 2004;15:91–98. [PubMed] [Google Scholar]
- 245.Boyd AW, Wawryk SO, Burns GF, Fecondo JV. Intercellular adhesion molecule 1 (ICAM-1) has a central role in cell-cell contact-mediated immune mechanisms. Proc Natl Acad Sci U S A. 1988;85:3095–3099. doi: 10.1073/pnas.85.9.3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Van Seventer GA, Shimizu Y, Horgan KJ, Shaw S. The LFA-1 ligand ICAM-1 provides an important costimulatory signal for T cell receptor-mediated activation of resting T cells. J Immunol. 1990;144:4579–4586. [PubMed] [Google Scholar]
- 247.Elsner J, et al. Synthesis and surface expression of ICAM-1 in polymorphonuclear neutrophilic leukocytes in normal subjects and during inflammatory disease. Immunobiology. 1995;193:456–464. doi: 10.1016/s0171-2985(11)80430-4. [DOI] [PubMed] [Google Scholar]
- 248.Miklossy J, et al. Role of ICAM-1 in persisting inflammation in Parkinson disease and MPTP monkeys. Exp Neurol. 2006;197:275–283. doi: 10.1016/j.expneurol.2005.10.034. S0014-4886(05)00373-0 [pii] [DOI] [PubMed] [Google Scholar]
- 249.Roland CL, Harken AH, Sarr MG, Barnett CC., Jr ICAM-1 expression determines malignant potential of cancer. Surgery. 2007;141:705–707. doi: 10.1016/j.surg.2007.01.016. S0039-6060(07)00104-3 [pii] [DOI] [PubMed] [Google Scholar]
- 250.Maruo Y, et al. ICAM-1 expression and the soluble ICAM-1 level for evaluating the metastatic potential of gastric cancer. Int J Cancer. 2002;100:486–490. doi: 10.1002/ijc.10514. [DOI] [PubMed] [Google Scholar]
- 251.Goodwin RG, et al. Molecular and biological characterization of a ligand for CD27 defines a new family of cytokines with homology to tumor necrosis factor. Cell. 1993;73:447–456. doi: 10.1016/0092-8674(93)90133-b. 0092-8674(93)90133-B [pii] [DOI] [PubMed] [Google Scholar]
- 252.Bowman MR, et al. The cloning of CD70 and its identification as the ligand for CD27. J Immunol. 1994;152:1756–1761. [PubMed] [Google Scholar]
- 253.Hintzen RQ, de Jong R, Lens SM, van Lier RA. CD27: marker and mediator of T-cell activation? Immunol Today. 1994;15:307–311. doi: 10.1016/0167-5699(94)90077-9. [DOI] [PubMed] [Google Scholar]
- 254.Hintzen RQ, et al. CD70 represents the human ligand for CD27. Int Immunol. 1994;6:477–480. doi: 10.1093/intimm/6.3.477. [DOI] [PubMed] [Google Scholar]
- 255.Jacquot S, Kobata T, Iwata S, Morimoto C, Schlossman SF. CD154/CD40 and CD70/CD27 interactions have different and sequential functions in T cell-dependent B cell responses: enhancement of plasma cell differentiation by CD27 signaling. J Immunol. 1997;159:2652–2657. [PubMed] [Google Scholar]
- 256.Agematsu K, et al. Generation of plasma cells from peripheral blood memory B cells: synergistic effect of interleukin-10 and CD27/CD70 interaction. Blood. 1998;91:173–180. [PubMed] [Google Scholar]
- 257.Hendriks J, et al. CD27 is required for generation and long-term maintenance of T cell immunity. Nat Immunol. 2000;1:433–440. doi: 10.1038/80877. [DOI] [PubMed] [Google Scholar]
- 258.Arens R, et al. Constitutive CD27/CD70 interaction induces expansion of effector-type T cells and results in IFNgamma-mediated B cell depletion. Immunity. 2001;15:801–812. doi: 10.1016/s1074-7613(01)00236-9. S1074-7613(01)00236-9 [pii] [DOI] [PubMed] [Google Scholar]
- 259.Tesselaar K, et al. Lethal T cell immunodeficiency induced by chronic costimulation via CD27-CD70 interactions. Nat Immunol. 2003;4:49–54. doi: 10.1038/ni869. ni869 [pii] [DOI] [PubMed] [Google Scholar]
- 260.Xiao Y, Hendriks J, Langerak P, Jacobs H, Borst J. CD27 is acquired by primed B cells at the centroblast stage and promotes germinal center formation. J Immunol. 2004;172:7432–7441. doi: 10.4049/jimmunol.172.12.7432. 172/12/7432 [pii] [DOI] [PubMed] [Google Scholar]
- 261.Hintzen RQ, et al. Engagement of CD27 with its ligand CD70 provides a second signal for T cell activation. J Immunol. 1995;154:2612–2623. [PubMed] [Google Scholar]
- 262.Lens SM, et al. Phenotype and function of human B cells expressing CD70 (CD27 ligand) Eur J Immunol. 1996;26:2964–2971. doi: 10.1002/eji.1830261223. [DOI] [PubMed] [Google Scholar]
- 263.Hishima T, et al. CD70 expression in thymic carcinoma. Am J Surg Pathol. 2000;24:742–746. doi: 10.1097/00000478-200005000-00014. [DOI] [PubMed] [Google Scholar]
- 264.Bullock TN, Yagita H. Induction of CD70 on dendritic cells through CD40 or TLR stimulation contributes to the development of CD8+ T cell responses in the absence of CD4+ T cells. J Immunol. 2005;174:710–717. doi: 10.4049/jimmunol.174.2.710. 174/2/710 [pii] [DOI] [PubMed] [Google Scholar]
- 265.Agathanggelou A, et al. Expression of immune regulatory molecules in Epstein-Barr virus-associated nasopharyngeal carcinomas with prominent lymphoid stroma. Evidence for a functional interaction between epithelial tumor cells and infiltrating lymphoid cells. Am J Pathol. 1995;147:1152–1160. [PMC free article] [PubMed] [Google Scholar]
- 266.Lens SM, et al. Aberrant expression and reverse signalling of CD70 on malignant B cells. Br J Haematol. 1999;106:491–503. doi: 10.1046/j.1365-2141.1999.01573.x. bjh1573 [pii] [DOI] [PubMed] [Google Scholar]
- 267.Diegmann J, et al. Identification of CD70 as a diagnostic biomarker for clear cell renal cell carcinoma by gene expression profiling, real-time RT-PCR and immunohistochemistry. Eur J Cancer. 2005;41:1794–1801. doi: 10.1016/j.ejca.2005.05.005. S0959-8049(05)00423-5 [pii] [DOI] [PubMed] [Google Scholar]
- 268.Law CL, et al. Lymphocyte activation antigen CD70 expressed by renal cell carcinoma is a potential therapeutic target for anti-CD70 antibody-drug conjugates. Cancer Res. 2006;66:2328–2337. doi: 10.1158/0008-5472.CAN-05-2883. 66/4/2328 [pii] [DOI] [PubMed] [Google Scholar]
- 269.Ryan MC, et al. Targeting pancreatic and ovarian carcinomas using the auristatin-based anti-CD70 antibody-drug conjugate SGN-75. Br J Cancer. 2010;103:676–684. doi: 10.1038/sj.bjc.6605816. 6605816 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Sulitzeanu D. Immunosuppressive factors in human cancer. Adv Cancer Res. 1993;60:247–267. doi: 10.1016/s0065-230x(08)60827-1. [DOI] [PubMed] [Google Scholar]
- 271.Teraoka H, et al. TGF-beta1 promotes liver metastasis of pancreatic cancer by modulating the capacity of cellular invasion. Int J Oncol. 2001;19:709–715. doi: 10.3892/ijo.19.4.709. [DOI] [PubMed] [Google Scholar]
- 272.Sawada T, et al. TGF-beta1 down-regulates ICAM-1 expression and enhances liver metastasis of pancreatic cancer. Adv Med Sci. 2006;51:60–65. [PubMed] [Google Scholar]
- 273.Hinz S, et al. Foxp3 expression in pancreatic carcinoma cells as a novel mechanism of immune evasion in cancer. Cancer Res. 2007;67:8344–8350. doi: 10.1158/0008-5472.CAN-06-3304. 67/17/8344 [pii] [DOI] [PubMed] [Google Scholar]
- 274.Gum JR., Jr Mucin genes the proteins they encode: structure diversity and regulation. Am J Respir Cell Mol Biol. 1992;7:557–564. doi: 10.1165/ajrcmb/7.6.557. [DOI] [PubMed] [Google Scholar]
- 275.Moniaux N, Escande F, Porchet N, Aubert JP, Batra SK. Structural organization and classification of the human mucin genes. Front Biosci. 2001;6:D1192–1206. doi: 10.2741/moniaux. [DOI] [PubMed] [Google Scholar]
- 276.Roy LD, et al. MUC1 enhances invasiveness of pancreatic cancer cells by inducing epithelial to mesenchymal transition. Oncogene. 2011;30:1449–1459. doi: 10.1038/onc.2010.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Balague C, et al. Altered expression of MUC2, MUC4, and MUC5 mucin genes in pancreas tissues and cancer cell lines. Gastroenterology. 1994;106:1054–1061. doi: 10.1016/0016-5085(94)90767-6. S0016508594001022 [pii] [DOI] [PubMed] [Google Scholar]
- 278.Balague C, Audie JP, Porchet N, Real FX. In situ hybridization shows distinct patterns of mucin gene expression in normal, benign, and malignant pancreas tissues. Gastroenterology. 1995;109:953–964. doi: 10.1016/0016-5085(95)90406-9. S0016508595003064 [pii] [DOI] [PubMed] [Google Scholar]
- 279.Terada T, Ohta T, Sasaki M, Nakanuma Y, Kim YS. Expression of MUC apomucins in normal pancreas and pancreatic tumours. J Pathol. 1996;180:160–165. doi: 10.1002/(SICI)1096-9896(199610)180:2<160::AID-PATH625>3.0.CO;2-A. 10.1002/(SICI)1096-9896(199610)180:2<160::AID-PATH625>3.0.CO;2-A [pii] [DOI] [PubMed] [Google Scholar]
- 280.Yonezawa S, et al. Differential mucin gene expression in human pancreatic and colon cancer cells. Biochem J. 1991;276 (Pt 3):599–605. doi: 10.1042/bj2760599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Park HU, et al. Aberrant expression of MUC3 and MUC4 membrane-associated mucins and sialyl Le(x) antigen in pancreatic intraepithelial neoplasia. Pancreas. 2003;26:e48–54. doi: 10.1097/00006676-200304000-00022. 00006676-200304000-00022 [pii] [DOI] [PubMed] [Google Scholar]
- 282.Nagata K, et al. Mucin expression profile in pancreatic cancer and the precursor lesions. J Hepatobiliary Pancreat Surg. 2007;14:243–254. doi: 10.1007/s00534-006-1169-2. [DOI] [PubMed] [Google Scholar]
- 283.Monti P, et al. Tumor-derived MUC1 mucins interact with differentiating monocytes and induce IL-10highIL-12low regulatory dendritic cell. J Immunol. 2004;172:7341–7349. doi: 10.4049/jimmunol.172.12.7341. 172/12/7341 [pii] [DOI] [PubMed] [Google Scholar]
- 284.Tinder TL, et al. MUC1 enhances tumor progression and contributes toward immunosuppression in a mouse model of spontaneous pancreatic adenocarcinoma. J Immunol. 2008;181:3116–3125. doi: 10.4049/jimmunol.181.5.3116. 181/5/3116 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Besmer DM, et al. Pancreatic Ductal Adenocarcinoma (PDA) mice lacking Mucin 1 have a profound defect in tumor growth and metastasis. Cancer research. 2011 doi: 10.1158/0008-5472.CAN-10-4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Hoshi H, et al. Tumor-associated MUC5AC stimulates in vivo tumorigenicity of human pancreatic cancer. Int J Oncol. 2011;38:619–627. doi: 10.3892/ijo.2011.911. [DOI] [PubMed] [Google Scholar]
- 287.Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Invest. 2007;117:1147–1154. doi: 10.1172/JCI31178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Fallarino F, et al. The combined effects of tryptophan starvation and tryptophan catabolites down-regulate T cell receptor zeta-chain and induce a regulatory phenotype in naive T cells. J Immunol. 2006;176:6752–6761. doi: 10.4049/jimmunol.176.11.6752. 176/11/6752 [pii] [DOI] [PubMed] [Google Scholar]
- 289.Witkiewicz A, et al. Expression of indoleamine 2,3-dioxygenase in metastatic pancreatic ductal adenocarcinoma recruits regulatory T cells to avoid immune detection. J Am Coll Surg. 2008;206:849–854. doi: 10.1016/j.jamcollsurg.2007.12.014. discussion 854-846 S1072-7515(07)01974-6 [pii] [DOI] [PubMed] [Google Scholar]
- 290.Munn DH, et al. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J Exp Med. 1999;189:1363–1372. doi: 10.1084/jem.189.9.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Fallarino F, et al. T cell apoptosis by tryptophan catabolism. Cell Death Differ. 2002;9:1069–1077. doi: 10.1038/sj.cdd.4401073. [DOI] [PubMed] [Google Scholar]
- 292.Chung JC, Oh MJ, Choi SH, Bae CD. Proteomic analysis to identify biomarker proteins in pancreatic ductal adenocarcinoma. ANZ J Surg. 2008;78:245–251. doi: 10.1111/j.1445-2197.2008.04429.x. ANS4429 [pii] [DOI] [PubMed] [Google Scholar]
- 293.Camby I, Le Mercier M, Lefranc F, Kiss R. Galectin-1: a small protein with major functions. Glycobiology. 2006;16:137R–157R. doi: 10.1093/glycob/cwl025. [DOI] [PubMed] [Google Scholar]
- 294.Xue X, et al. Galectin-1 secreted by activated stellate cells in pancreatic ductal adenocarcinoma stroma promotes proliferation and invasion of pancreatic cancer cells: an in vitro study on the microenvironment of pancreatic ductal adenocarcinoma. Pancreas. 2011;40:832–839. doi: 10.1097/MPA.0b013e318217945e. [DOI] [PubMed] [Google Scholar]
- 295.Cooper D, et al. Multiple functional targets of the immunoregulatory activity of galectin-1: Control of immune cell trafficking, dendritic cell physiology, and T-cell fate. Methods Enzymol. 2010;480:199–244. doi: 10.1016/S0076-6879(10)80011-4. S0076-6879(10)80011-4 [pii] [DOI] [PubMed] [Google Scholar]
- 296.Simon HU, Haj-Yehia A, Levi-Schaffer F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis. 2000;5:415–418. doi: 10.1023/a:1009616228304. [DOI] [PubMed] [Google Scholar]
- 297.Kannan K, Jain SK. Oxidative stress and apoptosis. Pathophysiology. 2000;7:153–163. doi: 10.1016/s0928-4680(00)00053-5. S0928468000000535 [pii] [DOI] [PubMed] [Google Scholar]
- 298.Davis W, Jr, Ronai Z, Tew KD. Cellular thiols reactive oxygen species in drug-induced apoptosis. J Pharmacol Exp Ther. 2001;296:1–6. [PubMed] [Google Scholar]
- 299.Babior BM, Lambeth JD, Nauseef W. The neutrophil NADPH oxidase. Arch Biochem Biophys. 2002;397:342–344. doi: 10.1006/abbi.2001.2642. S0003986101926426 [pii] [DOI] [PubMed] [Google Scholar]
- 300.Clement MV, Stamenkovic I. Superoxide anion is a natural inhibitor of FAS-mediated cell death. EMBO J. 1996;15:216–225. [PMC free article] [PubMed] [Google Scholar]
- 301.Lin KI, Pasinelli P, Brown RH, Hardwick JM, Ratan RR. Decreased intracellular superoxide levels activate Sindbis virus-induced apoptosis. J Biol Chem. 1999;274:13650–13655. doi: 10.1074/jbc.274.19.13650. [DOI] [PubMed] [Google Scholar]
- 302.Brar SS, et al. NOX5 NAD(P)H oxidase regulates growth and apoptosis in DU 145 prostate cancer cells. Am J Physiol Cell Physiol. 2003;285:C353–369. doi: 10.1152/ajpcell.00525.2002. 00525.2002 [pii] [DOI] [PubMed] [Google Scholar]
- 303.Sauer H, Wartenberg M, Hescheler J. Reactive oxygen species as intracellular messengers during cell growth and differentiation. Cell Physiol Biochem. 2001;11:173–186. doi: 10.1159/000047804. cpb11173 [pii] [DOI] [PubMed] [Google Scholar]
- 304.Chiarugi P, et al. Reactive oxygen species as essential mediators of cell adhesion: the oxidative inhibition of a FAK tyrosine phosphatase is required for cell adhesion. J Cell Biol. 2003;161:933–944. doi: 10.1083/jcb.200211118. jcb.200211118 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Vaquero EC, Edderkaoui M, Pandol SJ, Gukovsky I, Gukovskaya AS. Reactive oxygen species produced by NAD(P)H oxidase inhibit apoptosis in pancreatic cancer cells. J Biol Chem. 2004;279:34643–34654. doi: 10.1074/jbc.M400078200. M400078200 [pii] [DOI] [PubMed] [Google Scholar]
- 306.Wehler T, et al. Strong expression of chemokine receptor CXCR4 by pancreatic cancer correlates with advanced disease. Oncol Rep. 2006;16:1159–1164. [PubMed] [Google Scholar]
- 307.Shen R, et al. Precancerous stem cells can serve as tumor vasculogenic progenitors. PLoS One. 2008;3:e1652. doi: 10.1371/journal.pone.0001652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Gabrilovich DI, et al. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat Med. 1996;2:1096–1103. doi: 10.1038/nm1096-1096. [DOI] [PubMed] [Google Scholar]
- 309.Wong HH, Lemoine NR. Biological approaches to therapy of pancreatic cancer. Pancreatology. 2008;8:431–461. doi: 10.1159/000151536. 000151536 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Andre F, Delaloge S, Soria JC. Biology-Driven Phase II Trials: What Is the Optimal Model for Molecular Selection? Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2011;29:1236–1238. doi: 10.1200/JCO.2010.31.6877. [DOI] [PubMed] [Google Scholar]
- 311.Almhanna K, Philip PA. Defining New Paradigms for the Treatment of Pancreatic Cancer. Curr Treat Options Oncol. 2011 doi: 10.1007/s11864-011-0150-8. [DOI] [PubMed] [Google Scholar]
- 312.Chung JC, Oh MJ, Choi SH, Bae CD. Proteomic analysis to identify biomarker proteins in pancreatic ductal adenocarcinoma. ANZ journal of surgery. 2008;78:245–251. doi: 10.1111/j.1445-2197.2008.04429.x. [DOI] [PubMed] [Google Scholar]
- 313.O’Reilly EEA. Recent Findings in Pancreatic Cancer:Illuminating Emerging Treatment Strategies. 2010 http://cme.medscape.com/viewarticle/728727.
- 314.Dauer M, et al. Chemosensitization of pancreatic carcinoma cells to enhance T cell-mediated cytotoxicity induced by tumor lysate-pulsed dendritic cells. Journal of immunotherapy. 2005;28:332–342. doi: 10.1097/01.cji.0000164038.41104.f5. [DOI] [PubMed] [Google Scholar]