

Abstract
Disruptions in FOXP2, a transcription factor, are the only known monogenic cause of speech and language impairment. We report clinical findings for two new individuals with a submicroscopic deletion of FOXP2: a boy with severe apraxia of speech and his currently moderately affected mother. A 1.57 Mb deletion on chromosome 7q31 was detected by array Comparative Genomic Hybridization (aCGH). In addition to FOXP2, the patients’ deletion involves two other genes, MDFIC and PPP1R3A, neither of which has been associated with speech or language disorders. Thus, findings for these two family members provide informative phenotypic information on FOXP2 haploinsufficiency. Evaluation by a clinical geneticist indicated no major congenital anomalies or dysmorphic features. Evaluations by a clinical psychologist and occupational therapist indicated cognitive-linguistic processing and sensorimotor control deficits, but did not support a diagnosis of autism spectrum disorder. Evaluation by clinical and research speech pathologists confirmed that both patients’ speech deficits met contemporary criteria for apraxia of speech. Notably, the patients were not able to laugh, cough, or sneeze spontaneously, replicating findings reported for two other FOXP2 cases and a potential diagnostic sign of nonsyndromic apraxia of speech. Speech severity findings for the boy were not consistent with the hypothesis that loss of maternal FOXP2 should be relatively benign. Better understanding of the behavioral phenotype of FOXP2 disruptions will aid identification of patients, toward an eventual understanding of the pathophysiology of syndromic and nonsyndromic apraxia of speech.
Keywords: aCGH, apraxia of speech, autism, dyspraxia, speech sound disorder
INTRODUCTION
Developmental speech-language disorders are a heterogeneous group of conditions that have high heritability, but few replicated genetic findings [Stromswold, 2008; Ramus and Fisher, 2009; Newbury and Monoco, 2010]. The first insights into the molecular basis of developmental speech-language disorders came from the discovery that a speech sound disorder termed Childhood Apraxia of Speech [CAS; ASHA 2007]. was caused by a heterozygous missense mutation in the FOXP2 gene co-segregating with nonword repetition task deficits in the multigenerational ‘KE’ family [Lai et al., 2001]. FOXP2 codes for a protein from the FOX-family of winged-helix/forkhead transcription factors. The FOXP2 protein is expressed widely in the fetal and adult brain, where it regulates the expression of other genes within and among cortical, basal ganglia, and cerebellar circuits [Vernes and Fisher, 2009]. The expression pattern is specific to defined neuronal subpopulations in these different structures (e.g., deep layers of the cortex, medium spiny neurons of the striatum, Purkinje cells in the cerebellum). Deficits in these regions during embryogenesis and/or postnatal development are risk factors for early and persistent speech-language disorder and associated cognitive, sensorimotor, and learning deficits.
Genetic manipulation of Foxp2 in different vertebrate species, including mice and songbirds, have documented the consequences of reduced levels for acquisition of conspecific vocal communication and associated sensorimotor behaviors [Fisher and Scharrf, 2009]. For example, mouse pups homozygous for null alleles have had severe motor delays whereas those heterozygous for inactivation mutations show inconsistent phenotypes [French et al., 2007; Groszer et al., 2008]. Mice that carry heterozygous mutations equivalent to those in affected members of the KE family have deficits in learning rapid motor sequences, as well as impaired synaptic plasticity in corticostriatal and cerebellar circuits [Groszer et al., 2008].. FOXP2 is also the continuing focus of a large number of studies and discussions on the evolutionary biology of speech-language in humans [Fisher and Marcus, 2006].
In addition to the KE study series [see overview in Ramus and Fisher, 2009] MacDermot et al. [2005] reported an inactivating FOXP2 mutation affecting a mother and sibship, Lai et al. [2001] and a two-paper series [Shriberg et al., 2006; Tomblin et al., 2009] reported cases of balanced chromosomal translocations with breakpoints in FOXP2, Feuk et al, [2006] reported 7q31 chromosomal disruptions involving relatively large, cytogenetically visible deletions, Zeesman et al. [2006)] reported a deletion of FOXP2 and 51 additional genes, and Lennon et al. [2007] reported a boy with 7q31.1–7q31.31 deletion All cases in these reports have been associated with the rare (<.01%), severe, and persistent speech sound disorder most recently termed CAS [previously termed Developmental Verbal Dyspraxia]. The patients in these studies also had a range of associated deficits in cognition, language, morphology, sensorimotor control, and psychosocial function [Watkins et al., 2002; Tomblin et al., 2009; Shriberg, 2010a]. Of notable interest for an eventual understanding of the sensorimotor pathobiology of FOXP2 interruptions, the mother of the boy described in Lai et al. (2000) reported that he was never able to laugh spontaneously or to sneeze and the patient in Zeesman et al. [2006] had frequent gagging episodes, and reportedly was also unable to spontaneously laugh, cough, or sneeze.
Notwithstanding the wide-ranging interest in FOXP2 catalyzed by the speech, cognitive, motor, and learning findings in affected KE family members, relatively few reports are available that characterize the behavioral phenotype of other patients with FOXP2 haploinsufficiency using standardized clinical instruments. The present report describes clinical assessment findings for two new individuals with a submicroscopic deletion of the FOXP2 gene referred to author LDS for possible participation in a genetic research project in pediatric motor speech disorders: a boy with severe apraxia of speech and his currently moderately affected mother.
CLINICAL REPORT
Patient 1
History
Patient 1, evaluated at 4 years 10 months of age, carried a diagnosis of CAS and Pervasive Developmental Disorder-Not Otherwise Specified (PDD-NOS). Patient 1 was born following an uncomplicated, full term pregnancy. His birth weight was 3.6kg. He was healthy following birth but was described as a messy bottle feeder with a strong suck. He drooled and had frequent gagging with soft foods. His early fine and gross motor development were normal. He walked independently at 14 months. His family first became concerned about his speech and language development at 14 months of age, because he had never babbled and had a paucity of vocalizations. He was able to cry and squeal but did not develop a laugh and rarely coughed or sneezed. He said his first word “mama” at 24 months and at 4 years of age was still not combining 2 words together. Due to frequent episodes of otitis media, he had bilateral myringotomy tube placement and hearing was normal.
Physical examination
Patient 1 was nonsyndromic in appearance with normal growth (Figure 1). His height was 114.5cm (99th percentile), weight was 21.2kg (90th percentile) and head circumference was 52cms (70th percentile). He had the appearance of mild telecanthus with an intercanthal distance of 2.8cm (50–75th percentile) and his palpebral fissure length was 2.4cm (5th percentile) bilaterally. He had slightly high arched eyebrows. His hair was thick and curly with a widow’s peak. The nasal tip had a swallow groove. His ears were normal in morphology and position. His philtrum was long but normally formed. Palate and uvula were intact and not high arched. The neurologic exam was normal for tone, strength, coordination, gait and reflexes.
Figure 1.
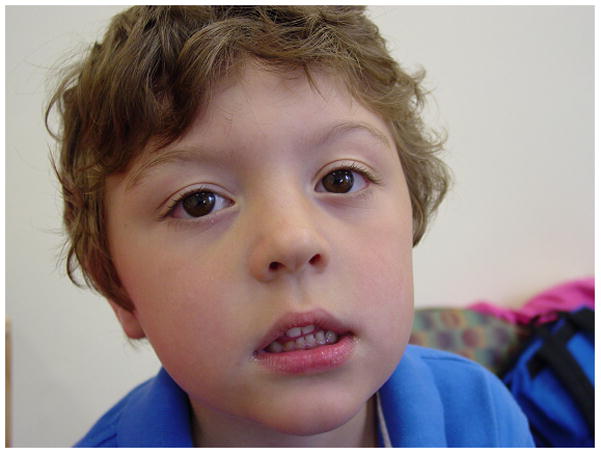
Patient 1 at 4 years, 10 months of age. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Speech
Patient 1’s speech was assessed using the Madison Speech Assessment Protocol (MSAP), a battery of tasks assessing his competence, precision, and stability of speech, prosody, and voice [Shriberg et al., 2010]. On the Goldman-Fristoe Test of Articulation-2 [GFTA-2; Goldman and Fristoe, 2000], he made 60 errors, resulting in a standard score of 47, placing him below the 1st percentile for his age and sex. Analyses of his conversational speech performance in other speech tasks indicated numerous consonant and vowel omissions, imprecise sound substitutions and distortions, restricted word shapes, unstable phoneme and word errors, articulatory groping, difficulty managing loudness and voicing, frequent production of grunts and single consonants for words, and poor intelligibility. He was unable to laugh spontaneously, producing squeals and shrieks instead. He also was unable to cough spontaneously, even when a piece of food became lodged in his throat. Hearing was normal. The combination of these speech, prosody, and voice signs is consistent with a diagnosis of severe CAS [ASHA, 2007].
Language
Patient 1’s language was assessed using the Clinical Evaluation of Language Fundamentals Preschool-2 [CELF Preschool-2; Wiig et al., 2004] and the conversational speech sample. On the CELF Preschool-2, his standard score on the combined receptive language subtests was 90, indicating performance within normal limits; his standard score on the combined expressive language subtests was 55, indicating severe expressive language impairment. Patient 1’s conversational language consisted primarily of single-word utterances reflecting a severely limited expressive vocabulary. He demonstrated a pronounced desire to communicate and extreme frustration at not being able to be understood verbally. In social situations, he sought out others with whom to interact, and attempted to get the attention of others by pulling on their hands, vocalizing, and putting his hands on people’s faces to turn them to look at something. Functionally, Patient 1 attempted to verbally communicate for a variety of purposes, including asking questions, directing activities, negating, and labeling.
Cognition
Patient 1’s intellectual functioning was assessed using the Wechsler Preschool and Primary Scale of Intelligence-III [Wechsler 2002]. IQ scores were calculated with a mean of 100 and a standard deviation of 15. He received a Verbal scale IQ of 77 (6th percentile), Performance scale IQ of 75 (5th percentile), and Processing Speed IQ of 68 (2nd percentile). He demonstrated a significant weakness in the Block Design subtest. These scores combined to yield a Full Scale IQ of 71 (3rd percentile). However, his standardized scores were viewed as underestimating his true cognitive potential due to the verbal language confound affecting many of the subtests. Patient 1 demonstrated a significant strength on the Picture Completion subtest, suggesting that his perceptual organization skills and alertness to visual details are well-developed.
Motor
Patient 1’s age equivalency on the Miller Function & Participation Scales [M-FUN: Miller, 2006] visual motor scale and the fine motor scale were both below the 4-year-old level. He had significant difficulty with a paper folding task. He was unable to organize the activity despite a practice session with washcloths and was unable to correctly orient the paper or crease it. He lacked good isolated finger coordination and was inconsistent in the use of a pincer grasp when placing items that he moved from his palm to fingers. He lacked the fine motor control expected for his age which is required to precisely use his fingers when signing. For gross motor testing on the M-FUN, Patient 1’s best effort was measured throughout the various tasks and his gross motor skills were found to be in the average range. He had difficulty with novel tasks: taping string onto a paper fish, folding paper, and winding string onto a pencil. These and other performance deficits were interpreted as evidence of difficulty with motor planning of both gross and fine motor tasks. Among deficits in other self-help skills, Patient 1 was not toilet trained and was not dressing independently at the time of the evaluation.
Psychological
All of Patient 1’s scores on the Autism Diagnostic Interview-Revised [ADI-R; Rutter et al., 2003] subdomains (Qualitative Abnormalities in Reciprocal Social Interactions = 16; Restricted, Repetitive, and Stereotyped Patterns of Behavior = 2, Abnormality of Development Evident Before 36 months = 5) were above the cut-off criterion for an autism spectrum disorder with the exception of Qualitative Abnormalities in Communication (score = 3). On the revised Autism Diagnostic Observation Schedule [ADOS; Lord et al., 2000], using the new algorithms [e.g., Gotham et al., 2007] for DSM-IV Autism Diagnosis, Module 1 (Some Words), Patient 1 obtained scores of 1 on the Communication domain, 4 on the Social Affect domain, and 4 on the Restricted and Repetitive Behaviors domain. The latter two domains summed created a Social Affect and Restricted and Repetitive Behaviors Total score of 8, which is at the cut-off for an autism spectrum disorder. Although Patient 1’s ADI-R and ADOS scores were above or at the cut-off criterion for an autism spectrum disorder, observations during the ADOS and throughout the examination were not consistent with autism spectrum disorder. He demonstrated several well-developed social relatedness and communication behaviors including using gestures, pointing, and coordinating vocalizations with eye contact to communicate with others. He also displayed pretend play, engaged in games that require turn-taking, and showed interest in and enjoyment from interacting with his family and the clinical psychologist
Patient 2
History
Patient 2, Patient 1’s mother, was evaluated at 24 years of age. She carried a diagnosis of apraxia of speech (i.e., persistent CAS), receptive and expressive language delay, PDD-NOS, and specific learning disabilities. Her gestation was complicated by a maternal bicornate uterus leading to a difficult vaginal delivery. APGAR scores were 4 at 1 minute and 7 at 5 minutes. Her birth weight at 36 weeks gestation was 2.7kg. In infancy, she frequently gagged on soft foods and liquids but was not considered to be a messy eater. Videofluoroscopy had revealed delayed swallowing. She also had excessive drooling and did not laugh or cough. Her early gross and fine motor milestones were on time. Her early verbal developmental course was similar to her son’s. She did not begin using single words until age 4. She began to combine words at age 7. She was able to use sign language effectively. She had frequent episodes of otitis media that required bilateral myringotomy tube placement at age 5 and normal hearing. She had left esotropia requiring corrective surgery.
Physical examination
Patient 2 is nonsyndromic in appearance. P2’s weight was 104.2kg, height 173.5cm (90–95th percentile), and head circumference 57.5cm (95th percentile). She had mild ptosis on the left. She wore glasses for history of left esotropia. Her intercanthal distance was 2.8cm (25–50th percentile) and her palpebral fissure length was 2.5cm bilaterally (25th percentile). She had thick naturally curly hair. Her face appeared slightly long with mild malar flatting. Her philtrum, palate and uvula were normally formed. Her ears had normal morphology and position. Her hands, fingers, feet, and toes appeared slightly broad. She had a right transverse palmar crease. The neurologic exam was normal for tone, strength, coordination, gait and reflexes.
Speech
Patient 2’s speech and language was assessed using formal and informal measures. On the GFTA-2 [Goldman and Fristoe, 2000] she had only one speech error, which when compared to the highest age normative data (20 years), yielded a standard score of 96. Although her ability to articulate correctly in single words was excellent, analysis of her conversational speech yielded an array of speech sound errors and an overall articulatory imprecision, including inconsistent lateralization and prolongation of/s/, context-based sound errors in multisyllabic words, mild nasal emissions on words with high-pressure consonants, lexical stress errors, and intermittent low pitch (glottal fry). On a nonsense syllable task requiring rapid alternating tongue-jaw motions, she had consonant and vowel imprecision, slow repetition rates, and arrhythmicity. Patient 2 reported that she often found the act of speaking “exhausting.” Patient 2’s speech signs are presently characteristic of mild to moderate, persistent CAS and dysarthria [ASHA, 2007]. Due to the significant normalization of speech, prosody, and voice signs of CAS over time [Shriberg, Potter et al., 2011], however, it is difficult to gauge the reliability of adjectives used to index possible differences in the severity of speech involvement in Patient 2 compared to Patient 1. The relative severity issue is of particular interest. Feuk et al. [2006] previously proposed, based on observing paternal FOXP2 deletions and cases of maternal uniparental disomy of chromosome 7, that FOXP2 may be subject to parent-of-origin effects. If their proposal is correct, then paternal deletions of FOXP2 will result in severe phenotypes, while maternal deletions should be relatively benign. Our data do not support the Feuk et al. hypothesis, since Patient 1, who appears to be more severely affected than Patient 2, inherited the deletion on the maternal chromosome..
Language
Seven subtests of the Clinical Evaluation of Language Fundamentals-4 [CELF-4; Semel et al., 2003] were administered to assess Patient 2’s receptive, expressive, and social language. The oldest available normative CELF age data (17 to 21 years) were used to derive scaled scores. She scored within one standard deviation of the mean on 2 of the 7 (29%) subtests (Word Definition, Word Classes), and from 1 to 3 standard deviations below the mean on 5 (71%) of the subtests (Understanding Spoken Paragraphs, Formulating Sentences, Recalling Sentences, Semantic Relationships, and Sentence Assembly). Six subtests from the Woodcock-Johnson III Tests of Achievement [Woodcock et al., 2001] were administered to assess Patient 2’s reading and oral and written language skills. Her grade-equivalency scores were Passage Comprehension: (grade) 8.9, Picture Vocabulary: 8.2, Letter-Word Identification: 8.0, Writing Samples: 7.6, Spelling: 5.4, and Word Attack: 2.9. Thus, in all domains assessed, Patient 2’s scaled scores and school grade equivalent data were consistent with severe language impairment.
Cognition
The Wechsler Adult Intelligence Scale-Third Edition [WAIS-III; Wechsler 1997] was administered to Patient 2 to allow for comparison with previous studies of adults with FOXP2 (see Discussion). Her full scale IQ score of 89 (23rd percentile) was in the average range, and, as discussed later, is similar to the scores reported for the affected adults in Tomblin et al. [2009] and Watkins et al. [2002]. Patient 2’s Performance IQ score of 92 (30th percentile) was higher than her Verbal IQ score of 87 (19th percentile). Her overall Adaptive Behavior Composite (Vineland Adaptive Behavior Scales, Second Edition, Parent/Caregiver Rating form) score was 54 (<1st percentile), considered to be in the low range. Thus, despite her average IQ score, her everyday living skills were severely impaired.
Motor
On the Beery-Buktenica Developmental Test of Visual Motor Integration, 5th edition [Beery and Beery, 2006], Patient 2 scored in the average range in visual motor skills (form copying), below average in visual perception (a form constancy task), and in the very low range in motor coordination (a pencil task drawing within boundaries). Her specific test scores were as follows (subtest, raw score, standard score, percentile, classification): Visual Motor Integration, 27, 92, 30, Average; Visual Perception, 26, 86, 18, Below Average; and, Motor Coordination, 21, 56, .4, Very Low. She demonstrated good finger dexterity skills and hand grip and pinch strength, but performed manipulative tasks at a slower rate than expected for her age (Nine Hole Peg Test: left 21 seconds, right 18 seconds, both between 1 and 2 SD below the mean). She lacked precise pencil control for fine drawing and as a result received a score below the expectations for her age. She also demonstrated some challenges with other types of visual perceptual skills including spatial relations, visual discrimination, figure-ground, and visual closure.
MATERIALS AND METHODS
Array Comparative Genomic Hybridization (aCGH)
aCGH analysis was performed using the Agilent 2×105K custom array (Agilent Technologies, Inc., Santa Clara, CA), designed for clinical testing by the International Standards for Cytogenomic Arrays Consortium [ISCA: https://www.iscaconsortium.org/]. This array contains more than 105,000 oligonucleotide probes and provides high density coverage for clinically relevant deletion/duplication syndromes and the telomere and pericentromeric regions, together with genome-wide coverage with an average probe spacing of ~35 kb. Preparation of test and control DNA, labeling and hybridization were performed following the manufacturer’s protocols.
RESULTS
aCGH Analyses
aCGH testing detected a copy number loss (deletion) on the long arm of chromosome 7, affecting the segment between the bands q31.1 and q31.2 (Figure 2). The deletion was 1.57 Mb in size, from position 112,946,520 to 114,520,576, based on the Human Genome March 2006 (hg18) assembly. Figure 3 is a screenshot from the UCSC genome browser showing the deleted region, which also includes MDFIC and PPP1R3A.
Figure 2.

Graphic representation of the data from the ISCA 105K oligo-aCGH analysis. A: Ideogram of chromosome 7, with the the symbol on the right side of the image showing the position of the deleted region. B: Chromosome 7 findings from aCGH analysis software (OneClick CGH, InfoQuant, London, UK). [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Figure 3.

A screenshot from the UCSC genome browser showing the deleted region at cytogenetic location 7q31.2q31.2, from 112,946,520 to 114,520,576 based on the Human Genome March 2006 (hg18) assembly. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
DISCUSSION
As reviewed, inactivating point mutations and chromosomal deletions affecting FOXP2 have been causally associated with autosomal dominant CAS. In our patients, aCGH testing identified a 1.57 Mb submicroscopic deletion at 7q31.2q31.2, which, in addition to FOXP2, affected only two other known genes: MDFIC and PPP1R3A. While abnormalities of the FOXP2 gene have been reported in patients with speech disorders, as reviewed previously, neither MDFIC nor PPP1R3A have been associated with any type of communicative disorder in humans. The MDFIC gene (MyoD family inhibitor domain containing protein) codes for a protein with transcriptional regulatory activity, involved in modulating the expression from both cellular and viral promoters [Kusano and Eizuru, 2010]. The product of PPP1R3A (protein phosphatase 1, regulatory-inhibitor) binds to muscle glycogen with high affinity, thereby enhancing dephosphorylation of glycogen-bound substrates for protein phosphatase-1 (PP1) [Tang et al., 1991]. PPP1R3A has been associated with severe insulin resistance [Online Mendelian Inheritance in Man, 2011; Savage et al., 2002]. Because these genes, to date, have not been associated with communicative disorders, the behavioral phenotype provided in this report is presumed to closely estimate the speech, cognitive-linguistic, sensorimotor, and learning effects of FOXP2 haploinsufficiency.
On possible between-patient differences in severity of speech involvement, copy number abnormalities other then the 7q31.2q31.2 deletion were not detected by aCGH in either individual. However, the 105K custom array used in this study has an estimated resolution of 150–200 kilobases, and could not detect very small, intragenic deletions and duplications. We therefore cannot exclude the possibility that the child has small copy number variants in addition to the 7q31.2q31.2 deletion, that may have contributed to the severity of the disorder, but were not detected by our analysis. Genetic differences other than copy number changes (single nucleotide polymorphisms, point mutations, etc.) could have also modified the phenotype in the proband and his mother.
On another substantive severity issue, it has previously been proposed, based on indirect evidence, that the maternal allele of FOXP2 is comparatively underexpressed, such that while loss of paternal FOXP2 yields severe speech problems, loss of maternal FOXP2 should be relatively benign [Feuk et al., 2006]. Our present data, as well as findings from other families [MacDermot et al., 2005; Shriberg et al., 2006; Tomblin et al., 2009] challenge this hypothesis, since they indicate similarly severe phenotypic effects for maternally inherited disruptions of FOXP2.
Extended speech, language, prosody, and voice analyses similar to those reported for cases B and T [Shriberg et al., 2006] and three siblings with CAS associated with an unbalanced 4q:16q translocation [Shriberg et al., 2008] will be reported elsewhere. The following discussion addresses cognitive-linguistic, affective, and motor findings for the present patients compared to findings for FOXP2 cases in prior reports.
The cognitive-linguistic phenotype of FOXP2 mutations and deletions has been a controversial issue in explanatory models of CAS [Shriberg et al., 1997; Tomblin et al., 2009]. A central question is whether processing deficits in cognition, language, and learning are core aspects of isolated FOXP2 haploinsufficiency, or whether such deficits are due to more extensive genomic rearrangements encompassing FOXP2. Figure 4 adds the WAIS-III subscale findings for the present patients to findings from the two patients with FOXP2 disruptions described in Shriberg et al. [2006] and Tomblin et al. [2009] and averaged data from the 13 members of the KE family with a FOXP2 mutation [Watkins et al., 2002]. As shown, the trend across the four cases and the family is for higher Performance IQs than Verbal IQs, which follows from their verbal trait deficit. Notably, P2, B, and T, have performance IQs within the normal range (above 85) compared to the mean IQ of the 13 affected members of KE, with P2 having the highest scores (P2 was approximately the same age as T when each was assessed). The data in Figure 4 for P2, who had both Performance and Verbal IQs within the normal range, indicate that lowered IQ is not an inevitable consequence of FOXP2 haploinsufficiency.
Figure 4.

WAIS-III Performance and Verbal IQ scores for Patient 2 in comparison to B and T [Tomblin et al., 2009] and the means and standard deviations for the 13 affected members of the KE family [Watkins et al., 2002].
Figure 5 is a profile of WAIS-III Verbal and Nonverbal scores for Patient 2, the TB family and mean standardized scores for the 13 affected members of the KE family. Scores above 7 (solid horizontal line) are within the normal range. The subtest data are arranged in descending order based on P2’s scores (filled circles). Two observations on these data are useful to underscore in the present context of FOXP2 phenotype characterization. First are the large individual differences in profiles across cases, as indicated by the crossing trend lines above nearly all 10 subtests. There is no one cognitive processing characteristic profile approximated by the three cases and the averaged family members. Second, P2’s profile indicates strength in 7 of the 10 subtests, with lower performance in Arithmetic, Digit Span, and Digit Symbol Coding tasks. As shown, weakness in Arithmetic and Digit Span were also reported for B and KE and weakness in Digit Symbol Coding was also reported for B, T, and KE. Thus, these data support prior findings indicating that core endophenotypic features of FOXP2 interruptions include constraints in the cognitive processing domains underlying performance on these WAIS III subtasks—attention, short term memory, and speed of processing.
Figure 5.

WAIS-III subtest scale scores (10 = average, 3 =SD) for Patient 2 in comparison with the scores for B and T [Tomblin et al., 2009] and the means for the 13 affected members of the KE family [Watkins et al., 2002]. Scores above the solid line are within the normal range.
In both of our patients concerns have been raised about autism spectrum disorder and in P2 there is a concern with executive function deficits associated with a history of difficulty in activities of daily living. Several association studies have linked the AUTS1 autism loci to the FOXP2 region of chromosome 7q31, but FOXP2 mutations have not been found to be causal [Li et al., 2005]. Moreover, other reported cases of focused FOXP2 disruptions (i.e., point mutations, balanced translocations, and small deletions) are also not consistent with diagnoses of autism spectrum disorder. Vernes et al. [2008] reported that FOXP2 down-regulates CNTNAP2, a cortically-expressed gene that is independently associated with speech delays in autistic children and with language deficits in families affected by specific language impairment. Although both patients in the present study carried a diagnosis of PDD-NOS, observations of the social and communication behaviors during formal assessment and planned and spontaneous activities during the three-day assessment period were not consistent with an autism spectrum disorder. Both patients initiated social interactions, used gestures, coordinated eye contact with vocalizations, and were socially engaged.
Although there were case history data indicating on time early gross and fine motor milestones, both patients have gross and fine motor control deficits, phenotypic features that are widely reported in the autism literature [Mostofsky et al., 2007; Dowell et al., 2009] and that currently are controversial relative to explanatory models for concomitant CAS in autism [Shriberg, 2010b; Shriberg, Paul, et al., 2011]. In particular, Patient 1’s motor planning difficulties were consistent with the transcoding (planning/programming) deficits that define all forms of apraxia (i.e., limb, ideational, speech). His inconsistent maintenance of focus on the fine motor tasks and inability to maintain correct movement patterns is consistent with both cognitive (Figure 5) and sensorimotor consequences of reduced FOXP2 expression in cortical, cerebellar, and especially basal ganglia regions and pathways. The lack or at least paucity of spontaneous laugh, cough, and sneeze replicates these sensorimotor deficits in the patients reported by Lai et al. [2000] and Zeesman et al. [2006] and noted anecdotally in case studies [e.g., Unique, 2011]. These neurodevelopmental sensorimotor deficits have not been observed in emerging studies of CAS in syndromic and nonsyndromic contexts [Shriberg, 2010c; Shriberg, Potter, et al., 2011].
After over two decades of programmatic studies of the KE family, FOXP2 remains the sole but rare cause of nonsyndromic CAS, itself a rare speech sound disorder. Lack of consensus on the core phenotypic signs of FOXP2 deficits continues to be a major constraint on genetic research and on clinical decision making. Findings reported for the present two family members add support to the view that FOXP2 haploinsufficiency can disrupt development in cognition, speech, language, and sensorimotor domains. Clinicians should be alert for these phenotypic features when considering molecular testing for FOXP2 interruptions in a patient with a valid diagnosis of CAS.
Acknowledgments
This work was supported by a grant from the National Institute on Deafness and Other Communicative Disorders (DC000496) to Lawrence D. Shriberg and a Core Grant from the National Institute of Child Health and Development (HD03352) to the Waisman Center. We thank the patients and their family and the following individuals for their contributions to this report: Jon Douglas, Craig Jackson, Heather Karlsson, Lynn Levin, Heather Lohmeier, Jane McSweeny, Malgorzata Nowaczyk, Edythe Strand, and ChristieTilkens.
References
- American Speech-Language-Hearing Association. Childhood apraxia of speech [Technical report] 2007 Available from www.asha.org/policy.
- Beery KE, Beery NA. The Beery-Buktenica Developmental Test of Visual Motor Integration Administration, Scoring, and Teaching Manual. 5. Bloomington, MN: Pearson; 2006. [Google Scholar]
- Dowell LR, Mahone EM, Mostofsky SH. Associations of postural knowledge and basic motor skill with dyspraxia in autism: Implication for abnormalities in distributed connectivity and motor learning. Neuropsychology. 2009;23:563–570. doi: 10.1037/a0015640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuk L, Kalervo A, Lipsanen-Nyman M, Skaug J, Nakabayashi K, Finueane B, Hartung D, Innes M, Kerem B, Nowaczyk MJ, Rivlin J, Roberts W, Senman L, Summers A, Szatmari P, Wong V, Vincent JB, Zeesman S, Osborne LR, Cardy JO, Kere J, Scherer SW, Hannula-Jouppi K. Absence of a paternally-inherited FOXP2 gene in developmental verbal dyspraxia. Am J Hum Genet. 2006;79:965–972. doi: 10.1086/508902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher SE. Molecular windows into speech and language disorders. Folia Phoniatr Logop. 2007;59:130–140. doi: 10.1159/000101771. [DOI] [PubMed] [Google Scholar]
- Fisher SE, Marcus GF. The eloquent ape: genes, brains and the evolution of language. Nature Rev Genet. 2006;7:9–20. doi: 10.1038/nrg1747. [DOI] [PubMed] [Google Scholar]
- Fisher SE, Scharrf C. FOXP2 as a molecular window into speech and language [Review article] Trends Genet. 2009;25:166–177. doi: 10.1016/j.tig.2009.03.002. [DOI] [PubMed] [Google Scholar]
- French CA, Groszer M, Preece C, Coupe A-M, Rajewsky K, Fisher SE. Generation of mice with a conditional Foxp2 null allele. Genesis. 2007;45:440–446. doi: 10.1002/dvg.20305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman R, Fristoe M. Goldman–Fristoe Test of Articulation. 2. Circle Pines, MN: American Guidance Service, Inc; 2000. [Google Scholar]
- Gothman K, Risi S, Pickles A, Lord C. The Autism Diagnostic Observation Schedule: Revised algorithms for improved diagnostic validity. J Autism Dev Disord. 2007;37:613–627. doi: 10.1007/s10803-006-0280-1. [DOI] [PubMed] [Google Scholar]
- Groszer M, Keays DA, Deacon RM, de Bono JP, Prasad-Mulcare S, Gaub S, Baum MG, French CA, Nicod J, Coventry JA, Enard W, Fray M, Brown SD, Nolan PM, Pääbo S, Channon KM, Costa RM, Eilers J, Ehret G, Rawlins JN, Fisher SE. Impaired synaptic plasticity and motor learning in mice with a point mutation implicated in human speech deficits. Curr Biol. 2008;18:354–362. doi: 10.1016/j.cub.2008.01.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst JA, Baraitser M, Auger E, Graham F, Norell S. An extended family with a dominantly inherited speech disorder. Dev Med Child Neurol. 1990;32:352–355. doi: 10.1111/j.1469-8749.1990.tb16948.x. [DOI] [PubMed] [Google Scholar]
- Lai CSL, Fisher SE, Hurst JA, Vargha-Khadem F, Monaco AP. A forkhead-domain gene is mutated in a severe speech and language disorder. Nature. 2001;413:519–523. doi: 10.1038/35097076. [DOI] [PubMed] [Google Scholar]
- Lennon PA, Cooper ML, Peiffer DA, Gunderson KL, Patel A, Peters S, Cheung SW, Bacino CA. Deletion of 7q31.1 supports involvement of FOXP2 in language impairment: clinical report and review. Am J Med Genet A. 2007;143A:791–798. doi: 10.1002/ajmg.a.31632. [DOI] [PubMed] [Google Scholar]
- Li H, Yamagata T, Mori M, Momoi MY. Absence of causative mutations and presence of autism-related allele in FOXP2 in Japanese autistic patients. Brain Dev. 2005;27:207–210. doi: 10.1016/j.braindev.2004.06.002. [DOI] [PubMed] [Google Scholar]
- Lord C, Rutter M, DeLavore P, Risi S. Autism diagnostic observation schedule-General. Los Angeles, CA: Western Psychological Services; 2000. [Google Scholar]
- MacDermot KD, Bonora E, Sykes N, Coupe AM, Lai CS, Vernes SC, Vargha-Khadem F, McKenzie F, Smith RL, Monaco AP, Fisher SE. Identification of FOXP2 runcation as a novel cause of developmental speech and language deficits. Am J Hum Genet. 2005;76:1074–80. doi: 10.1086/430841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller LJ. Miller Function & Participation Scales. San Antonio, TX: Pearson; 2006. [Google Scholar]
- Mostofsky SH, Burgess MP, Gidley Larson JC. Increased motor cortex white matter volume predicts motor impairment in autism. Brain. 2007;30:2117–2122. doi: 10.1093/brain/awm129. [DOI] [PubMed] [Google Scholar]
- Newbury DF, Monaco AP. Genetic advances in the study of speech and language disorders. Neuron. 2010;68:309–320. doi: 10.1016/j.neuron.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Online Mendelian Inheritance in Man, OMIM (TM) MIM Number 600917. Johns Hopkins University; Baltimore, MD: 2011. World Wide Web URL: http://www.ncbi.nlm.nih.gov/omim/ [Google Scholar]
- Ramus F, Fisher SE. Genetics of language. In: Gazzaniga MS, editor. The Cognitive Neurosciences IV. Cambridge: MIT Press; 2009. pp. 855–871. [Google Scholar]
- Rutter M, LeCouteur A, Lord C. Autism diagnostic interview-revised. Los Angeles, CA: Western Psychological Services; 2003. [Google Scholar]
- Savage DB, Agostini M, Barroso I, Gurnell M, Luan J, Meirhaeghe A, Harding A-H, Ihrke G, Rajanayagam O, Soos MA, George S, Berger D, Thomas EL, Bell JD, Meeran K, Ross RJ, Vidal-Puig A, Wareham NJ, O’Rahilly S, Chatterjee VK, Schafer AJ. Digenic inheritance of severe insulin resistance in a human pedigree. Nature Genet. 2002;31:379–384. doi: 10.1038/ng926. [DOI] [PubMed] [Google Scholar]
- Semel E, Wiig E, Secord W. Clinical evaluation of language fundamentals. 4. San Antonio, TX: Harcourt Assessment; 2003. [Google Scholar]
- Shriberg LD. A neurodevelopmental framework for research in Childhood Apraxia of Speech. In: Maassen B, van Lieshout P, editors. Speech Motor Control: New developments in basic and applied research. Oxford: Oxford University Press; 2010a. pp. 259–270. [Google Scholar]
- Shriberg LD. Apraxia of speech and nonverbal school-aged children with autism. Paper presented at the National Institutes of Health Workshop on Nonverbal School-Aged Children with Autism; Bethesda, MD. 2010b. [Google Scholar]
- Shriberg LD. Diagnostic marker research in childhood apraxia of speech. Paper presented at the Academy of Neurologic Communication Disorders and Sciences Scientific Meeting; Philadelphia, PA. 2010c. [Google Scholar]
- Shriberg LD, Aram DM, Kwiatkowski J. Developmental apraxia of speech: I. Descriptive perspectives. J Speech Lang Hear R. 1997;40:273–285. doi: 10.1044/jslhr.4002.273. [DOI] [PubMed] [Google Scholar]
- Shriberg LD, Ballard KJ, Tomblin JB, Duffy JR, Odell KH, Williams CA. Speech, prosody, and voice characteristics of a mother and daughter with a 7;13 translocation affecting FOXP2. J Speech Lang Hear R. 2006;49:500–525. doi: 10.1044/1092-4388(2006/038). [DOI] [PubMed] [Google Scholar]
- Shriberg LD, Fourakis M, Hall S, Karlsson HK, Lohmeier HL, McSweeny J, Potter NL, Scheer-Cohen AR, Strand EA, Tilkens CM, Wilson DL. Extensions to the Speech Disorders Classification System (SDCS) Clin Linguist Phonet. 2010;24:795–824. doi: 10.3109/02699206.2010.503006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shriberg LD, Jakielski KJ, El-Shanti H. Breakpoint localization using array-CGH in three siblings with an unbalanced 4q:16q translocation and Childhood Apraxia of Speech (CAS) Am J Hum Genet Part A. 2008;146A:2227–2233. doi: 10.1002/ajmg.a.32363. [DOI] [PubMed] [Google Scholar]
- Shriberg LD, Paul R, Black LM, van Santen JP. The hypothesis of apraxia of speech in children with Autism Spectrum Disorder. J Autism Dev Disord. 2011;41:405–426. doi: 10.1007/s10803-010-1117-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shriberg LD, Potter NL, Strand EA. Prevalence and phenotype of childhood apraxia of speech in youth with galactosemia. J Speech Lang Hear R. 2011;54:487–519. doi: 10.1044/1092-4388(2010/10-0068). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stromswold K. The genetics of speech and language impairments. N Engl J Med. 2008;359:2381–2383. doi: 10.1056/NEJMe0807813. [DOI] [PubMed] [Google Scholar]
- Tang PM, Bondor JA, Swiderek KM, DePaoli-Roach AA. Molecular cloning and expression of the regulatory (RG1) subunit of the glycogen-associated protein phosphatase. J Biol Chem. 1991;266:15782–15789. [PubMed] [Google Scholar]
- Tomblin JB, O’Brien M, Shriberg LD, Williams C, Murray J, Patil S, Bjork J, Anderson S, Ballard K. Language features in a mother and daughter of a chromosome 7;13 translocation involving FOXP2. J Speech Lang Hear R. 2009;52:1157–1174. doi: 10.1044/1092-4388(2009/07-0162). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unique. 7q deletions: between 7q21 & 7q32 [Brochure] Caterham, UK: Author; 2011. [Google Scholar]
- Vernes SC, Fisher SE. Unravelling neurogenetic networks implicated in developmental language disorders. Biochem Soc Trans. 2009;37:1263–1269. doi: 10.1042/BST0371263. [DOI] [PubMed] [Google Scholar]
- Vernes SC, Newbury DF, Abrahams BS, Winchester L, Nicod J, Groszer M, Alarcón M, Oliver PL, Davies KE, Geschwind DH, Monaco AP, Fisher SE. A functional genetic link between distinct developmental language disorders. N Engl J Med. 2008;359:2337–2345. doi: 10.1056/NEJMoa0802828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins KE, Dronkers NF, Vargha-Khadem F. Behavioural analysis of an inherited speech and language disorder: Comparison with acquired aphasia. Brain. 2002;125:452–464. doi: 10.1093/brain/awf058. [DOI] [PubMed] [Google Scholar]
- Wechsler D. Wechsler Adult Intelligence Scale. 3. San Antonio, TX: Harcourt Assessment; 1997. [Google Scholar]
- Wechsler D. Wechsler Primary and Preschool Scale of Intelligence. 3. San Antonio, TX: Harcourt Assessment; 2002. [Google Scholar]
- Wiig EH, Secord WA, Semel E. Clinical evaluation of language fundamentals-Preschool. 2. San Antonio, TX: The Psychological Corporation: Harcourt Assessment, Inc; 2004. [Google Scholar]
- Woodcock RW, McGrew K, Mather N. Woodcock-Johnson Tests of Cognitive Abilities and Tests of Achievement. 3. Rolling Meadows, IL: Riverside Publishing; 2001. [Google Scholar]
- Zeesman S, Nowaczyk MJ, Teshima I, Roberts W, Cardy JO, Brian J, Senman L, Feuk L, Osborne LR, Scherer SW. Speech and language impairment and oromotor dyspraxia due to deletion of 7q31 that involves FOXP2. Am J Med Genet A. 2006;140A:509–514. doi: 10.1002/ajmg.a.31110. [DOI] [PubMed] [Google Scholar]