

Abstract
One of the most important and challenging problems in biomedicine and genomics is how to identify the disease genes. In this study, we developed a computational method to identify colorectal cancer-related genes based on (i) the gene expression profiles, and (ii) the shortest path analysis of functional protein association networks. The former has been used to select differentially expressed genes as disease genes for quite a long time, while the latter has been widely used to study the mechanism of diseases. With the existing protein-protein interaction data from STRING (Search Tool for the Retrieval of Interacting Genes), a weighted functional protein association network was constructed. By means of the mRMR (Maximum Relevance Minimum Redundancy) approach, six genes were identified that can distinguish the colorectal tumors and normal adjacent colonic tissues from their gene expression profiles. Meanwhile, according to the shortest path approach, we further found an additional 35 genes, of which some have been reported to be relevant to colorectal cancer and some are very likely to be relevant to it. Interestingly, the genes we identified from both the gene expression profiles and the functional protein association network have more cancer genes than the genes identified from the gene expression profiles alone. Besides, these genes also had greater functional similarity with the reported colorectal cancer genes than the genes identified from the gene expression profiles alone. All these indicate that our method as presented in this paper is quite promising. The method may become a useful tool, or at least plays a complementary role to the existing method, for identifying colorectal cancer genes. It has not escaped our notice that the method can be applied to identify the genes of other diseases as well.
Introduction
Colorectal cancer (CRC) is one of the most common malignancies in the western countries and a major cause of cancer-related death. Early detection of CRC could reduce the morbidity and improve the prognosis. Therefore, it is of great importance to identify cancer-related genes that could be used as biomarker for early diagnosis.
Recently, with the development of high-throughput biotechnologies, a large amount of biological data has been generated, such as yeast two-hybrid systems, protein complex and gene expression profiles, etc. These data are useful resources for deducing and understanding gene functions [1], [2], [3], [4], [5], [6], [7], [8]. So far the protein-protein interaction (PPI) data has been widely used for gene function prediction with the assumption that interacting proteins share the same or have similar functions and hence may be involved in the same pathway. This “guilty by association” rule was first proposed by Nabieva et al. [9] and can also be used to identify cancer related genes.
STRING is an online database resource which is an abbreviation for Search Tool for the Retrieval of Interacting Genes [10]. It provides both experimental as well as predicted interaction information with a confidence score. Algorithms based on PPI suggest that proteins with short distances to each other in the network are more likely to share the common biological functions [11], [12], [13], [14], and that interactive neighbors are more likely to have identical biological function than non-interactive ones [15], [16]. This is because the query protein and its interactive proteins may form a protein complex to perform a particular function or involved in a same pathway.
Although the successful application of the high-throughput data for gene function perdition and identification of novel genes associated with cancers, the errors in the high-throughput data have not been well solved yet. In this paper, we proposed a new method for identifying CRC related genes by integrating gene expression profile and a weighted functional protein association network constructed with PPI data from STRING. This method can make up the defect of only using high-throughput data. Meanwhile, the mRMR (maximum relevance minimum redundancy) algorithm [17] was utilized to identify six promising candidate genes distinguishing tumor and the normal colorectal samples. The Dijkstra's algorithm [18] was used to construct the shortest paths between each pair of the six genes. Moreover, additional 35 genes on these shortest paths were also identified and analyzed. For such 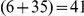 gene thus identified, it was observed that they contained more cancer genes than the genes identified from the gene expression profiles alone. Furthermore, the 41 genes also had greater functional similarity with the reported CRC genes than the genes identified from gene expression profiles alone. It is anticipated that some of the 41 genes thus identified might belong to novel CRC related genes.
gene thus identified, it was observed that they contained more cancer genes than the genes identified from the gene expression profiles alone. Furthermore, the 41 genes also had greater functional similarity with the reported CRC genes than the genes identified from gene expression profiles alone. It is anticipated that some of the 41 genes thus identified might belong to novel CRC related genes.
Materials and Methods
Dataset
We used the gene expression data from the colorectal cancer study of Hinoue et al. [19]. The gene expression profiling of 26 colorectal tumors and matched histologically normal adjacent colonic tissue samples were retrieved from NCBI Gene Expression Omnibus (GEO) with the accession number of GSE25070. The gene expression profile was obtained using the Illumina Ref-8 whole-genome expression BeadChip with 24526 probes corresponding to 18491 genes. Signal intensity was log2 transformed and then normalized with RSN (Robust Spline Normalization) method.
Tissue sample representation
Based on the above, the representation of a tissue sample can be formulated as a 24526-D (dimensional vector), as given by
| (1) |
where 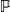 represents the tissue sample,
represents the tissue sample, 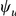 the value of it's
the value of it's 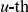 probe, and
probe, and 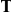 the transpose matrix (cf. Eq.6 of [20]).
the transpose matrix (cf. Eq.6 of [20]).
Cancer related gene list and two colorectal cancer related gene lists
We compiled three gene lists from public databases and published works to compare with the 41 candidate genes we identified. These three genes lists included one cancer related gene list and two colorectal cancer related gene lists.
742 cancer-related genes were derived from three sources. First, we obtained 457 cancer-related genes from the Cancer Gene Census of the Sanger Centre. Secondly, we retrieved cancer-related genes from the Atlas of Genetics and Cytogenetic in Oncology [21]. The third part was collected from the Human Protein Reference Database [22]. See Supporting Information S1.
The first colorectal cancer related gene list was retrieved from the study of Sabates-Bellver and coworkers [23]. They compared the transcriptomes of 32 adenomas with normal mucosa from the same individuals and identified 438 genes with markedly altered expression in colorectal adenomas compared with normal mucosa with Affymetrix U133 Plus 2.0 array. See Supporting Information S1.
The second colorectal cancer related gene list was retrieved form a recent work of Nagaraj et al. [24]. They proposed a Boolean based systems biology approach with guilt-by-association algorithm to identify novel cancer-associated genes. We compiled all the 134 novel CRC related genes identified in this study. See Supporting Information S1.
PPI data from STRING
The initial weighted PPI network was retrieved from STRING (version 9.0) [10] (http://string.embl.de/), which is a large database of known and predicted protein interactions. Proteins in the interaction network were represented with nodes, while the interaction between any two proteins therein was represented with an edge. These interactions contain direct (physical) and indirect (functional) interactions, derived from numerous sources such as experimental repositories, computational prediction methods. In the network, each edge is marked with a score to quantify the interaction confidence, i.e., the likelihood that an interaction may occur.
The mRMR (maximum relevance minimum redundancy) method
To find the genes that can distinguish colorectal tumors and normal adjacent tissues, we used the mRMR method, which was originally developed by Peng et al. [17] for analyzing the microarray data. The mRMR method could rank genes according to their relevance to the class of samples concerned, and meanwhile also could take the redundancy of genes into account. Those genes, which have the best trade-off between the maximum relevance to the sample class and the minimum redundancy, were considered as “good” biomarkers.
Both the relevance and redundancy were quantified by the following mutual information (MI):
| (2) |
where  and
and 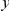 are vectors,
are vectors, 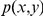 is their joint probabilistic density, and
is their joint probabilistic density, and 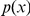 and
and 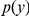 are the marginal probabilistic densities.
are the marginal probabilistic densities.
To quantify both the relevance and redundancy, let us define 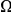 as the whole gene set,
as the whole gene set, 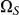 as the already-selected gene set containing
as the already-selected gene set containing  genes and
genes and 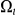 as the to-be-selected gene set containing
as the to-be-selected gene set containing  genes. The relevance
genes. The relevance 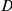 between the gene
between the gene 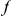 in
in 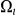 and the target
and the target  can be calculated by:
can be calculated by:
| (3) |
The redundancy 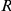 between the gene
between the gene 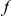 in
in 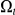 and all the genes in
and all the genes in 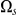 can be calculated by:
can be calculated by:
| (4) |
In order to obtain the gene 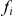 in
in 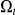 with the maximum relevance and minimum redundancy, let us combine Eq.3 and Eq.4, as can be formulated as follows:
with the maximum relevance and minimum redundancy, let us combine Eq.3 and Eq.4, as can be formulated as follows:
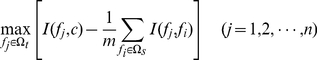 |
(5) |
Given a gene set with 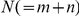 genes, the mRMR operation for the gene evaluation will continue
genes, the mRMR operation for the gene evaluation will continue 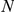 rounds. After these evaluations, the mRMR method will generate a gene set
rounds. After these evaluations, the mRMR method will generate a gene set 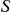 as formulated by
as formulated by
| (6) |
where the index 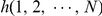 indicates which round the gene is selected. The smaller the index
indicates which round the gene is selected. The smaller the index 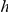 is, the earlier the gene satisfied Eq.5 and the better the gene is.
is, the earlier the gene satisfied Eq.5 and the better the gene is.
Prediction engine
In this study, the Nearest Neighbor Algorithm (NNA) [25], [26], which has been widely used in bioinformatics and computational biology [3], [27], [28], [29], [30], [31], [32], [33], [34], was adopted to predict the class of colorectal tissue samples. The “nearness” was calculated according to the following equation
| (7) |
where 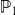 and
and 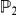 are two vectors representing two tissue samples,
are two vectors representing two tissue samples, 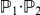 is their dot product,
is their dot product, 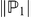 and
and 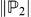 are their moduluses. The smaller the
are their moduluses. The smaller the 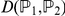 , the more similar the two samples are [35]. For an intuitive illustration of how NNA works, see Fig.5 of [20].
, the more similar the two samples are [35]. For an intuitive illustration of how NNA works, see Fig.5 of [20].
Performance validation
The following three cross-validation methods are often used in statistics for validating a statistical prediction method: independent dataset test, subsampling test, and jackknife test [36]. However, among the three validation methods, the jackknife test is the least arbitrary due to the following facts. (i) For the independent dataset test, although all the samples used to test the predictor are outside the training dataset used to train the prediction engine so as to exclude the “memory” effect or bias, the way of how to select the independent samples to test the predictor could be quite arbitrary unless the number of independent samples is sufficiently large. This kind of arbitrariness might lead to completely opposite conclusions. For instance, the conclusion that a predictor yielded a higher success rate than the other predictor for a given independent testing dataset might become just opposite when tested by another independent testing dataset [36]. (ii) For the subsampling test, the concrete procedure usually used in literatures is the 5-fold, 7-fold or 10-fold cross-validation. The problem with this kind of subsampling test is that the number of possible selections in dividing a benchmark dataset is extremely large even for a very simple and small dataset, as elucidated in [37] and demonstrated by Eqs.28–30 in [20]. Therefore, in any actual subsampling cross-validation tests, only a very tiny fraction of the possible selections are taken into account. Since different selections will always result in different outcomes even for a same benchmark dataset and a same predictor, the subsampling test cannot avoid the arbitrariness either. A test method unable to yield a unique outcome cannot be regarded as a good one. (iii) In the jackknife test, all the samples in the benchmark dataset will be singled out one-by-one and tested by the predictor trained by the remaining samples. During the process of jackknifing, both the training dataset and testing dataset are actually open, and each sample will be in turn moved between the two. The jackknife test can exclude the “memory” effect. Also, the arbitrariness problem as mentioned above for the independent dataset test and subsampling test can be avoided because the outcome obtained by the jackknife test is always unique for a given benchmark dataset. Accordingly, the jackknife test has been widely and increasingly used to inspect the quality of various predictors (see, e.g., [30], [31], [32], [38], [39], [40], [41], [42], [43], [44], [45], [46]). Accordingly, in this study the jackknife test was also used to examine the quality of the current prediction method.
The prediction accuracy was formulated by
| (8) |
where TP represents the true positive; TN, the true negative; FP, the false positive; and FN, the false negative.
Incremental feature selection (IFS)
Based on the ranked genes according to their importance after mRMR evaluation, we used the Incremental Feature Selection (IFS) (see, e.g., [1], [47]) to determine the optimal number of genes as biomarkers. During the IFS procedure, genes in the ranked gene set are added one by one from higher to lower rank. A new gene set is composed when one gene is added. Thus 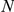 gene sets would be composed when given
gene sets would be composed when given 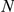 ranked genes. The
ranked genes. The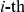 gene set is
gene set is
| (9) |
For each of the N gene sets, an NNA predictor was constructed and examined using the jackknife test to the benchmark dataset. By doing so we obtained an IFS table with one column for the index i and another column for the prediction accuracy. Thus, we could obtain the optimal gene set (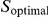 ), with which the predictor would yield the best prediction accuracy.
), with which the predictor would yield the best prediction accuracy.
Graph approach and shortest paths tracing
Graphs are a useful vehicle for studying complex biological systems because they can provide intuitive insights and the overall structure property, as demonstrated by various studies on a series of important biological topics (see, e.g., [48], [49], [50], [51], [52], [53], [54], [55], [56], [57], [58]). In this study, we first constructed a graph G(V, E) with the PPI data from STRING. In the graph, an edge was assigned for each pair of genes if they were in interaction with each other. The weight of edge E in graph G was derived from the confidence score according to the equation 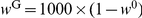 , where
, where 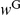 is the weight in graph G while
is the weight in graph G while 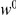 is the confidence score between two proteins concerned. Thus, we get a functional protein association network with edge weight. Dijkstra's algorithm [18] was used to find the shortest path from each of the six genes to all the other five genes in the graph. Then we picked out all the genes existing in the shortest paths and rank these genes according to their betweenness.
is the confidence score between two proteins concerned. Thus, we get a functional protein association network with edge weight. Dijkstra's algorithm [18] was used to find the shortest path from each of the six genes to all the other five genes in the graph. Then we picked out all the genes existing in the shortest paths and rank these genes according to their betweenness.
KEGG enrichment analysis
Functional annotation tool of DAVID [59] was used for KEGG pathway enrichment analysis. The enrichment p-value was corrected to control family-wide false discovery rate under certain rate (e.g., ≤0.05) with Benjamin multiple testing correction method [60]. All the genes on the BeadChip were selected as background during the enrichment analysis.
Results
mRMR results
The expression profile was retrieved from GEO with the accession number of GSE25070, which contained 52 samples and 24,526 probes and was transformed to a CSV file with 52 rows and 24526 columns as the input of mRMR. Each probe represented a feature and the 26 tumor samples belonged to class 1 while the paired26 paired normal samples belonged to class 2. After running the mRMR software, we obtained two tables (see Supporting Information S2), of which one was called MaxRel table that ranked the probes according to their relevance to the class of samples, and the other called mRMR feature table that listed the probes with the maximum relevance and minimum redundancy to the class of samples.
Six candidate genes identified by NNA and IFS
On the basis of the outputs of mRMR, we constructed 1000 feature subsets according to Eq.9. As described in the Materials and Methods section, we tested the predictor with one feature, two features, three features, etc., and the IFS result can be found in Supporting Information S3. Shown in Fig. 1 is the IFS curve plotted based on the data of Supporting Information S3. In the IFS curve, the X-axis is the number of probes used for classification, and the Y-axis is the prediction accuracies of the nearest neighbor algorithm evaluated by the jackknife test. The maximum accuracy was 1 when 6 features were included. The optimal probe set included 6 probes corresponding to 6 different genes, which were GUCA2B, PI16, CDH3, SPIB, BEST2, and HMGCLL1 ( Table 1 ).
Figure 1. IFS curve for the colorectal tumors and matched normal adjacent tissue samples classification.

In the IFS curve, the X-axis is for the number of probes used for classification, and the Y-axis for the prediction accuracies by the nearest neighbor algorithm (NNA) evaluated by the jackknife (Leave-One-Out) cross-validation test. The peak accuracy was 1 with six probes. The top 6 probes in the mRMR probe list formed the optimal discriminative probe set.
Table 1. mRMR top six genes.
| order | Probe name | Symbol | EntrezID | Protein ID |
| 1 | ILMN_1735578 | GUCA2B | 2981 | ENSP00000361662 |
| 2 | ILMN_1766264 | PI16 | 221476 | ENSP00000362778 |
| 3 | ILMN_1704294 | CDH3 | 1001 | ENSP00000264012 |
| 4 | ILMN_2143314 | SPIB | 6689 | ENSP00000270632 |
| 5 | ILMN_1755796 | BEST2 | 54831 | ENSP00000042931 |
| 6 | ILMN_2339192 | HMGCLL1 | 54511 | ENSP00000381654 |
Shortest paths genes
Meanwhile, we constructed an undirected graph with the PPI data from STRING. Then we picked out two genes from the six genes identified with the mRMR method as described above, and found out the shortest path between these two genes with the Dijkstra's algorithm. We obtained a total of 15 shortest paths with lowest cost (Supporting Information S4). Shown in Fig. 2 are the 15 shortest paths between the six candidate genes, where the interaction confidence was labeled on the edge for each of the interaction gene pairs. There were a total of 35 genes on the shortest paths and we ranked these genes according to their betweenness ( Table 2 ). Among these 35 genes, AR has the largest betweenness of 7, meaning that there are 7 shortest paths going through this gene. Accordingly, AR may play an important role in connecting the six candidate genes and hence may be related to CRC. Such a conclusion is fully consistent with the fact that AR protein was found in normal colorectal mucosa as well as in most CRC [61], [62], implying that the AR receptor is responsible for the mitogenic effects of the hormone as will be further discussed later.
Figure 2. 15 shortest paths between the six genes identified with mRMR method.

The 15 shortest paths between the six candidate genes were identified with Dijkstra's algorithm based on the PPI data from STRING. Yellow roundrect represents the top six candidate genes identified by the mRMR method. Red round represents the 35 genes existing within the range of the shortest paths. Numbers on edges represent the edge weights to quantify the interaction confidence. The smaller the number is, the stronger the interaction between two nodes is. See the text in the Section of “Graph approach and shortest paths tracing” for the quantitative relation of the edge weight with the confidence score between two proteins concerned.
Table 2. Shortest paths genes.
| order | Protein id | symbol | betweenness | P-value |
| 1 | ENSP00000363822 | AR | 7 | 0* |
| 2 | ENSP00000269305 | TP53 | 6 | 0.3442 |
| 3 | ENSP00000230354 | TBP | 5 | 0.0066* |
| 4 | ENSP00000250003 | MYOD1 | 5 | 0.0006* |
| 5 | ENSP00000263253 | EP300 | 5 | 0.0598 |
| 6 | ENSP00000287936 | HMGCR | 5 | 0* |
| 7 | ENSP00000314151 | KLK3 | 5 | 0* |
| 8 | ENSP00000344456 | CTNNB1 | 5 | 0.0984 |
| 9 | ENSP00000344741 | INSIG1 | 5 | 0* |
| 10 | ENSP00000349508 | CHD4 | 5 | 0* |
| 11 | ENSP00000351363 | MSMB | 5 | 0* |
| 12 | ENSP00000354620 | FOXJ3 | 5 | 0* |
| 13 | ENSP00000362649 | HDAC1 | 5 | 0.0108* |
| 14 | ENSP00000396219 | MEF2C | 5 | 0* |
| 15 | ENSP00000417884 | TRIM27 | 5 | 0* |
| 16 | ENSP00000342470 | NR1H3 | 4 | 0.005* |
| 17 | ENSP00000354476 | SREBF2 | 4 | 0.0038* |
| 18 | ENSP00000363868 | ABCA1 | 4 | 0.0098* |
| 19 | ENSP00000361066 | NCOA3 | 3 | 0.0038* |
| 20 | ENSP00000419692 | RXRA | 3 | 0.0098* |
| 21 | ENSP00000324806 | GSK3B | 2 | 0.1016 |
| 22 | ENSP00000399968 | NCOA2 | 2 | 0.0308* |
| 23 | ENSP00000206249 | ESR1 | 1 | 0.1968 |
| 24 | ENSP00000254227 | NR0B2 | 1 | 0.0346* |
| 25 | ENSP00000262367 | CREBBP | 1 | 0.0754 |
| 26 | ENSP00000265565 | SCAP | 1 | 0.0088* |
| 27 | ENSP00000268712 | NCOR1 | 1 | 0.0176* |
| 28 | ENSP00000297146 | GPR85 | 1 | 0.0104* |
| 29 | ENSP00000304895 | IRS1 | 1 | 0.0976 |
| 30 | ENSP00000329357 | SP1 | 1 | 0.1242 |
| 31 | ENSP00000348069 | SREBF1 | 1 | 0.023* |
| 32 | ENSP00000348551 | NCOR2 | 1 | 0.0162* |
| 33 | ENSP00000348827 | THRB | 1 | 0.0082* |
| 34 | ENSP00000348986 | INS-IGF2 | 1 | 0.0898 |
| 35 | ENSP00000353483 | MAPK8 | 1 | 0.1194 |
: P-value<0.05, significant.
To test whether our 35 shortest path genes were hubs in the background network or not, we ran a permutation to count the occurrence time of our 35 shortest path genes in the shortest paths between 6 random selected genes when they has greater betweenness than that in our study. We repeated this process 5000 times, and the p-value was calculated as the proportion of occurrence time of the 35 genes in 5000 permutation. For detail, please see Table 2 . There were 10 shortest path genes whose p-values were not significant. TP53 was a star molecular involved in numerous biological processes and nearly related to all kinds of cancers [63]. Therefore, it is nothing surprising that TP53 appeared many times in shortest path between 6 randomly picked genes. For EP300, it has been reported that this gene can acetylate TP53 and associated with lots of tumors [64]. CTNNB1 and GSK3B belong to the Wnt signaling pathway, the role of which in caners has been well documented [65]. For the remaining insignificant 6 genes, their betweennesses in our study were all one ( Table 2 ), and hence the number of occurrences for these genes in random shortest paths is prone to be greater than one. Most of these insignificant 6 shortest path genes fall behind in Table 2 according to their betweennesses, suggesting that they might not be important. Besides these 10 genes, the remaining 25 shortest path genes in our study were identified to be significant.
MaxRel table gene KEGG enrichment
Using the functional annotation tool of DAVID, the KEGG pathway enrichment analysis was carried out for the genes corresponding to the 1000 probes listed in the MaxRel. The enrichment results showed that these genes were significantly enriched in the energy metabolism pathways, including fatty acid metabolism, pentose and glucuronate interconversions, as well as starch and sucrose metabolism ( Table 3 ). These results suggested that metabolism of nutrients may play critical role in the tumorigenesis of CRC.
Table 3. MaxRel table genes KEGG enrichment.
| Term | KEGG ID | Counta | Percentageb | P-value | Benjamini Adjusted P-Value |
| Fatty acid metabolism | 00071 | 11 | 1.2 | 8.4E-5 | 1.5E-2 |
| Pentose and glucuronate interconversions | 00040 | 7 | 0.8 | 3.0E-4 | 2.7E-2 |
| Starch and sucrose metabolism | 00500 | 10 | 1.1 | 6.6E-4 | 3.8E-2 |
The number of genes belonging to a certain pathway.
The percentage of genes belonging to a certain pathway account for all the genes underwent KEGG pathway analysis.
Six candidate genes and shortest paths genes of KEGG enrichment
The KEGG pathway enrichment analysis was also performed on the 41 genes including the top six genes in the mRMR list and 35 genes in the shortest paths between these six genes with the functional annotation tool of DAVID. The enrichment result thus obtained showed that these genes were significantly enriched in the canonic cancer related pathways, such as prostate cancer, pathways in cancer, Wnt signaling pathway, cell cycle, colorectal cancer, thyroid cancer, and so on. It is instructive to note that among these pathways, some have been proved to be relevant to colorectal cancer including Wnt signaling pathway, cell cycle, colorectal cancer and insulin signaling pathway ( Table 4 ).
Table 4. mRMR top six genes and shortest path genes KEGG enrichment.
| Term | KEGG ID | Counta | Percentageb | P-value | Benjamini Adjusted P-Value |
| Prostate cancer | 05215 | 8 | 19.5 | 3.80E-08 | 2.40E-06 |
| Pathways in cancer | 05200 | 10 | 24.4 | 2.60E-06 | 8.00E-05 |
| Wnt signaling pathway | 04310 | 6 | 14.6 | 3.00E-04 | 6.30E-03 |
| Huntington's disease | 05016 | 6 | 14.6 | 6.70E-04 | 1.10E-02 |
| Notch signaling pathway | 04330 | 4 | 9.8 | 8.80E-04 | 1.10E-02 |
| Cell cycle | 04110 | 5 | 12.2 | 1.50E-03 | 1.60E-02 |
| Insulin signaling pathway | 04910 | 5 | 12.2 | 2.00E-03 | 1.80E-02 |
| Colorectal cancer | 05210 | 4 | 9.8 | 4.70E-03 | 3.60E-02 |
| Thyroid cancer | 05216 | 3 | 7.3 | 6.20E-03 | 4.20E-02 |
| Melanogenesis | 04916 | 4 | 9.8 | 7.40E-03 | 4.60E-02 |
The number of genes belonging to a certain pathway.
The percentage of genes belonging to a certain pathway account for all the genes underwent KEGG pathway analysis.
Overlap with cancer related gene list and two CRC related gene lists
We compiled 742 cancer-related genes from the following three different sources: Cancer Gene Census from the Sanger Centre, Atlas of Genetics and Cytogenetic in Oncology [21], and Human Protein Reference Database [22]. It was observed that 8 out of the 41 genes identified by us were proven to be cancer-related genes. Also, it was indicated by the Fisher's exact test that these 41 genes were significantly related to cancer (p-value = 0.0001908). See Supporting Information S5.
Moreover, we collected 438 genes that were differentially expressed between colorectal adenomas and normal mucosa from previous study [23]. Interestingly, the aforementioned 41 candidate genes identified by us had an overlap of 4 genes with the 438 genes, and the overlap was quite significant (p-value = 0.01057, Fisher's exact test). See Supporting Information S5.
Recently, the Boolean based systems biology approach was employed to identify 134 novel CRC related genes [24], of which three were identified by us in this study and the overlap was significant (p-value = 0.002017, Fisher's exact test). See Supporting Information S5.
Discussion
KEGG enrichment of MaxRel genes
The genes corresponding to the 1000 probes listed in the MaxRel table were significantly enriched in the energy metabolism pathways, including fatty acid metabolism, pentose and glucuronate interconversions, as well as starch and sucrose metabolism. It has been shown that diet has an important effect on the CRC development. Our finding is quite consistent with the fact that genetic polymorphisms influencing the metabolism of nutrients play an important role in the etiology of CRC and colorectal adenomatous polyps [62].
Multiple lines of evidences have indicated the implication or involvement of fat in the etiology of CRC [66]. The crucial role of fatty acids in numerous biological processes suggests that alteration in fatty acid metabolizing genes contributes to colon carcinogenesis [67]. It has been shown that starch and sucrose metabolism and pentose and glucuronateinterconversions were closely related to cancers. Christensen et al. [68] demonstrated that starch and sucrose metabolism and pentose and glucuronateinterconversions pathway were hypomethylated in isocitrate dehydrogenase mutant tumors. In addition, these two metabolic pathways were found to be significantly related to the risk of developing estrogen receptor-negative breast cancer [69].
A recent CRC disease-specific transcriptome research showed that starch and sucrose metabolism was one of the 7 common pathway significant differentially regulated using two different microarray platforms including Affymetrix HGU133 Plus2.0 array and the CRC disease specific array. Besides, fatty acid metabolism was identified as significantly differentially regulated pathway using colorectal disease specific array [70].
Six candidate genes identified by mRMR, NNA and IFS
In this study, we have identified the following six genes: GUCA2B, PI16, CDH3, SPIB, BEST2, and HMGCLL1. Below, let us briefly discuss their relationships with colorectal cancer.
GUCA2B (uroguanylin) is an endogenous activator of the guanylate cyclase-2C receptor found to be down regulated 8-fold in adenoma, and its expression is detected in blood and urine [71].Therefore, GUCA2B could be regarded as a non-invasive biomarker for the early detection of CRC. In addition, the radio labeled uroguanylin analogs have been used for detection of CRC in vivo [72].
PI16 (Peptidase inhibitor 16) is detected within the testis, prostate, small intestine, colon, and ovary with immunohistochemical analyses [73]. Decrease of PI16 level was detected in prostate cancer [73] and gastric cancer [74]. Our result also showed that the expression of PI16 in colorectal adenocarcinoma was significant decreased compared with the adjacent non-tumor colorectal tissue, which was consistent with the result of the research in prostate cancer and gastric cancer. Since PI16 is not well characterized and so far there is no report whatsoever about PI16 in colorectal cancer etiology, our result implied that PI16 may become a promising biomarker for colorectal cancer early diagnosis.
CDH3 is a classical cadherin, the demethylation of which is frequently detected in the advanced CRC which was associated with the overexpression of CDH3 [75]. Besides CRC, CDH3 was also overexpressed in the majority of pancreatic cancer and gastric cancer, but not in their noncancerous counterparts or in normal tissues. Thus CDH3 was regarded as a novel tumor-associated antigen useful for immunotherapy and early diagnosis of gastric cancer and CRC [76].
SPIB is a transcription factor of the E-twenty-six (ETS) family, which is known to act as positive or negative regulators of gene expression. SPIB is an adenoma condition-specific down regulated gene and its expression underwent a striking decrease in CRC tissues indicating that SPIB may serve as potential markers of CRC invasiveness and metastasis [77].
BEST2 (also known as VMD2L1) encodes a protein of the bestrophin family. Both RT-PCR analyses and X-gal staining revealed tissue-restricted BEST2 and VMD2L2 abundantly expressed in colon [78], [79]. It has been show that BEST2 mediates bicarbonate transport by goblet cells in mouse colon [80]. Straub et al. [81] identified BEST2 as one of the methylation markers for early detection and prognosis of CRC. Therefore, BEST2 was expected to become a therapy target for CRC with demethylation agent.
HMGCLL1 has been show to be related to various cancers, such as pancreatic cancers [82], glioblastoma multiforme [83], breast and colorectal cancers [84]. HMGCLL1 is one of the genes containing somatic mutations in pancreatic cancer [82]. Though mutation in HMGCLL1 has been reported to be involved in these cancers, the specific mechanisms underlying remain to be elucidated.
Shortest path genes
We totally identified 35 shortest paths genes. As we can see from Table 2 , some shortest path genes such as TP53, EP300, CTNNB1 and GSK3B were not significant for CRC due to their universality in numerous cancers. However, these genes have been well documented to be relevant to CRC, and also their role in CRC has been well characterized [85]. Besides these genes, most of the other shortest genes listed in Table 2 were quite specific to CRC (p-value<0.05). Below, let us focus on the specific genes with the large betweenness values and discuss the relationship of such genes with CRC.
AR (androgen receptor) is a ligand dependent transcription factor, which is involved in the control of cellular proliferation and differentiation [86]. Several studies have provided supporting evidences for its involvement of sex steroid hormones (estrogens and androgens) in the etiology and progression of CRC [87]. AR protein has been shown to be expressed in normal colorectal mucosa and in most colorectal cancer [61], [62], supporting that CRC expressing the AR receptor may respond to mitogenic effects of the hormone. Moreover, somatic reductions of the androgen receptor CAG repeat occur frequently, through a pathway different from microsatellite instability and early during colon carcinogenesis. Apparent growth selection of cells harboring shortened AR alleles suggests that androgens contribute to colon carcinogenesis in a yet unknown way [61].
TBP (the TATA-binding protein) is a key eukaryotic transcription factor used by all three cellular RNA polymerases. Compared to normal colon epithelium, TBP expression is elevated in the case of human colon carcinomas. Both Ras-dependent and Ras-independent mechanisms mediate the increases of TBP expression in colon carcinoma cell lines. Thus, TBP may be a crucial component in dysregulated signaling for causing tumors [88].
MYOD1 promoter methylation occurs in various malignancies including CRC. MYOD1 promoter methylation was detectable in tumor and normal colorectal samples, but was significantly higher in tumor than in normal mucosa. Patients without MYOD1 hypermethylation showed significantly longer survival than those with hypermethylation. Therefore, MYOD1 hypermethylation plays an important role in CRC and may be a novel prognostic factor [89].
HMGCR (3-hydroxy-3-methylglutaryl coenzyme A reductase) is an enzyme that catalyzes the rate-limiting step of cholesterol biosynthesis. HMGCR alternative splicing of exon 13 is not only a biomarker, but also a determinant of statin efficacy, which is a class of cholesterol-lowering drugs that inhibit HMGCR. HMGCR was used not only for the treatment of hypercholesterolemia, but also as a chemopreventive agent for CRC [90]. A genetic test of HMGCR was utilized to determine in which patients cholesterol-lowering statin drugs might have the most benefit in reducing the risk of CRC. A recent research has found a genetic variant may affect the way of how statins control both colorectal cancer and cardiovascular disease risk [91].
KLK3 (also known as prostate-specific antigen, PSA) is a kallikrein-like serine protease that is a widely used biomarker for prostate cancer [92]. In addition to prostate cancer, breast, colon, ovarian, liver and kidney tumors can also produce KLK3 [93]. Recently, several other members of KLK family like KLK7 have shown promise as potential biomarkers for various cancers including colon cancer [94], [95], [96]. Thus, with the progress of research, KLK3 may become a biomarker for CRC as well.
CHD (Chromodomain helicase DNA-binding protein) is a regulator of the chromatin remodeling process. CHD4 expression was detected in gastric cancers and CRCs by immunohistochemistry. It has been reported that loss of CHD4 expression was observed in 56.4% of the gastric cancers and 55.7% of the CRCs. In addition, Frameshift mutation and loss of expression of CHD genes are common in gastric cancers and CRCs with MSI-H. These alterations might contribute to cancer pathogenesis by deregulating CHD-mediated chromatin remodeling [97].
MSMB encodesβ-microsemino protein, which is a proposed biomarker for prostate cancer [98]. Genome-wide association studies (GWAS) have identified a variant, rs10993994, on chromosome 10q11 which is associated with prostate cancer risk. So far, there is no report about MSMB in CRC etiology. However, the expression of MSMB was detected in colon epithelial cells by immunohistochemistry [99]. Thus, it may be a potential biomarker for colorectal cancer diagnosis although it is remained to be verified.
FOXJ3 is a member of Human Forkhead-box (FOX) gene family. It has been shown that genetic and epigenetic changes of FOX family genes as well as alterations occurring in target genes of FOX transcription factors family could lead to human disease including carcinogenesis [100]. Recently, Niittymaki et al. [101] identified a SNP, rs2761880, locates in the binding site of FOXJ3 in CRC. It has been proposed that many of the predisposition loci for CRC are involved in control of gene expression by targeting transcription factor binding sites. In addition, oligonucleotide microarray analysis of distinct gene expression patterns in CRC tissues harboring BRAF and K-ras mutations has shown that FOXJ3 was identified by PAM (Prediction analysis of microarrays) and the jackknife (or leave-one-out) cross validation as candidate to distinguish the mutant groups [102].
HDAC1 (Histone deacetylase 1) is involved in tumorigenesis through their regulation of cell proliferation, differentiation and survival. In cancer cells, HDAC1 represses the expression of tumor suppress genes such as p21/WAF1/CIP1 and Bax, leading to aberrant cell proliferation and cell viability [103]. HDAC1 and HDAC3 are overexpressed in colon cancer cells and in primary colon cancer, and siRNA (small interfering RNA) mediated silencing of HDAC1 and HDAC3 in colon cancer cells induced apoptosis [104].
MEF2C (myocyte enhancer factor 2C) is a member of the MEF2 family of transcription factors. Recently, MEF2C was identified as a potential oncogenic transcription factor associated with CRC [24]. Besides, it has been shown that MEF2C was hypermethylated. Also, it was indicated by the significantly down-regulated in colon cancer that MEF2C may play a role in CRC etiology [105].
NR1H3 is a transcription factor involved in lipid homeostasis and inflammation. Recent evidences indicated that miRNAs can bind to the 3′untranslatedregions (UTRs) of mRNAs and regulates their translation. Genetic polymorphisms can locate in miRNA binding sites. Thus, miRNA regulation may be influenced by polymorphisms on the 3′UTRs. NR1H3 was identified as a candidate gene that harboring polymorphic in miRNA target sites which was associated with risk of sporadic CRC [106]. The specific relationship between NR1H3 and CRC remains to be further elucidated.
Overlap between selected genes and known cancer genes as well as known CRC related genes
Statistic test showed that the overlap between the 41 genes identified in our study and the 742 cancer-related genes we compiled was quite significant (p-value = 0.0001908). The KEGG analysis result of such 41 genes also implied that they were significantly enriched in cancer-related pathways (p-value = 8.00E-05). Taken together, it indicated that the 41 genes identified by us were closely associated with cancer. In addition, the overlaps of such 41 candidate genes with the previous (p-value = 0.01057) and recent (p-value = 0.002017) reported CRC biomarkers were significant. This suggested that the 41 candidate genes have the potential to be used as biomarkers for CRC diagnosis.
In addition, we compared the 41 genes identified by us with the top 41 genes in mRMR feature list and the top 41 differentially expressed genes identified by the traditional t-test method of R language [107]. See the Supporting Information S6 for such three sets of 41 genes. As can be seen from there, the 41 genes identified by us contain 8 cancer genes, which is more than 4 (p-value = 0.03965, proportion test) and 2 (p-value = 4.923e-05, proportion test) cancer genes than those contained in the 41 genes identified by mRMR and the 41 genes identified by the t-test, respectively ( Table 5 ).
Table 5. The overlap between 41 genes identified from three different methods and 742 cancer genes.
| Overlap with 742 Cancer genes | p-value | |
| Our 41 genes | 8 | |
| Top 41 mRMR genes | 4 | 0.03965 |
| Top 41 t-test genes | 2 | 4.923e-05 |
Functional similarity between selected genes and known CRC related genes
In this study, five gene sets were defined. The first gene set is our 41 selected genes. The second gene set is the top 41 mRMR genes. The third gene set is the top 41 t-test genes that have the smallest t-test p values. The second and third gene sets were from gene expression profiles alone. Our 41 gene were selected based on both gene expression profiles and protein interaction network. The fourth gene set is the 742 cancer genes mentioned above. The fifth gene set is the combined known CRC related genes of 742 cancer related genes, 438 genes from Sabates-Bellver's study [23]and 134 colorectal cancer related genes from Nagaraj's study [24]. These five gene sets can be found in the Supporting Information S6.
To compare the functional similarity between our selected genes and the known CRC related genes, we constructed their functional profiles using the −log10 of the hypergeometric test p value on Gene Ontology (GO) terms [1], [5], [108]. Then we calculated the Pearson correlation coefficient of their functional profiles [1], [109]. The functional similarities of the functional profiles for the five gene sets were shown in Table 6 . Our 41 genes had greater functional similarity with the cancer genes and the known CRC genes than the genes identified from gene expression profiles alone: top 41 mRMR genes and top 41 t-test genes. This suggests that the genes selected by our method are more reliable than the genes identified from the gene expression profiles alone. Combining the gene expression profiles and protein interaction network together can improve the identification of disease genes.
Table 6. The functional similarity between our 41 genes and known colorectal cancer genes.
| Cancer genes | Colorectal cancer genes | |
| Our 41 genes | 0.606068* | 0.491953* |
| Top 41 mRMR genes | 0.163112* | 0.244468* |
| Top 41 t-test genes | 0.203573* | 0.269548* |
Pearson correlation coefficient of functional profiles.
The reason why our method can generate more reliable results is because that the shortest pathway approach integrated here is based on all the information of genes from database, text mining, etc. that is quite stable and can avoid the false positives. In contrast to this, the method based on the gene expression data can cause lots of false positives. It is anticipated that our method may become a useful tool, or at least play a complementary role to the existing method, for identifying colorectal cancer genes.
It is instructive to point out that our method may have some limitations. This is because some hub genes that may simultaneously interact with lots of other genes can also occur in our shortest path and the randomly selected shortest paths, such as TP53 and EP300. Nevertheless, our method can provide a p-value to evaluate the significance that can be used to distinguish the hubs in the network background.
Conclusion
We proposed a novel method to identify cancer related genes. We applied this method on CRC and identified 41 genes which had the most potential to be biomarker for CRC early diagnose. Statistic test and KEGG analysis showed that the 41 candidate genes identified in our study are not only closely related to cancer but also have great potential to become biomarker for CRC diagnosis. In addition, the 41 candidate genes contain more cancer genes than the genes identified from gene expression profiles alone, and functional similarity analysis revealed that our genes had greater functional similarity with the reported CRC genes than the genes identified from gene expression profiles alone. We believe that our method may be helpful (or at least play a stimulative role) for predicting novel cancer related genes, and that it might have the potential applicability for the cancer research.
Supporting Information
The cancer-related gene list and the two colorectal cancer-related gene lists.
(XLS)
The MaxRel features table and mRMR features table.
(XLS)
Feature numbers and the first order accuracy which the IFS curve plot was based on.
(XLS)
The 15 shortest paths with the lowest cost presented with protein and gene, respectively.
(DOC)
The overlap between the 41 candidate genes and the three other datasets and the corresponding Fisher's exact test.
(DOC)
Five gene sets. First gene set is our 41 selected genes. The second gene set is the top 41 mRMR genes. The third gene set is the top 41 t-test genes that have the smallest t-test p values. The second and third gene sets were from gene expression profiles alone. Our 41 gene were selected based on both gene expression profiles and protein interaction network. The fourth gene set is the 742 cancer genes. The fifth gene set is the combined known colorectal cancer related genes.
(XLS)
Acknowledgments
The authors are very much indebted to the Editor and Reviewers for their constructive comments, which were very helpful for strengthening the presentation of this paper.
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: This work was supported by grants from the National Basic Research Program of China (2011CB510102, 2011CB510101, 2011CB910200 and 2010CB912702), the Natural Science Foundation of China (90913009), the Chinese Academy of Sciences (KSCX2-EW-R-04), a National High Tech Program Grant 2012AA022802 and the Innovation Program of Shanghai Municipal Education Commission (12ZZ087). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Huang T, Chen L, Cai Y-D, Chou K-C. Classification and analysis of regulatory pathways using graph property, biochemical and physicochemical property, and functional property. PLoS One. 2011;6:e25297. doi: 10.1371/journal.pone.0025297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang T, Cui W, Hu L, Feng K, Li YX, et al. Prediction of pharmacological and xenobiotic responses to drugs based on time course gene expression profiles. PLoS One. 2009;4:e8126. doi: 10.1371/journal.pone.0008126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cai YD, Huang T, Feng KY, Hu L, Xie L. A Unified 35-Gene Signature for both Subtype Classification and Survival Prediction in Diffuse Large B-Cell Lymphomas. PLoS One. 2010;5 doi: 10.1371/journal.pone.0012726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang T, Cai Y-D, Chen L, Hu L, Kong X-Y, et al. Selection of reprogramming factors of induced pluripotent stem cells based on the protein interaction network and functional profiles. Protein & Peptide Letters. 2011 doi: 10.2174/092986612798472884. [DOI] [PubMed] [Google Scholar]
- 5.Huang T, Shi XH, Wang P, He Z, Feng KY, et al. Analysis and prediction of the metabolic stability of proteins based on their sequential features, subcellular locations and interaction networks. PLoS One. 2010;5:e10972. doi: 10.1371/journal.pone.0010972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen L, Cai Y-D, Shi X-H, Huang T. Analysis of metabolic pathway using hybrid properties. Protein & Peptide Letters. 2011 doi: 10.2174/092986612798472857. [DOI] [PubMed] [Google Scholar]
- 7.Chen L, Cai Y-D, Huang T, Zeng W-M. Prediction of metabolic pathway using graph property, chemical functional group and chemical structure. Current Bioinformatics 2011 [Google Scholar]
- 8.Liu Q, Tan Y, Huang T, Ding G, Tu Z, et al. TF-centered downstream gene set enrichment analysis: Inference of causal regulators by integrating TF-DNA interactions and protein post-translational modifications information. BMC Bioinformatics. 2010;11:S5. doi: 10.1186/1471-2105-11-S11-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nabieva E, Jim K, Agarwal A, Chazelle B, Singh M. Whole-proteome prediction of protein function via graph-theoretic analysis of interaction maps. Bioinformatics. 2005;21(Suppl 1):i302–310. doi: 10.1093/bioinformatics/bti1054. [DOI] [PubMed] [Google Scholar]
- 10.Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39:D561–568. doi: 10.1093/nar/gkq973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharan R, Ulitsky I, Shamir R. Network-based prediction of protein function. Mol Syst Biol. 2007;3:88. doi: 10.1038/msb4100129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bogdanov P, Singh AK. Molecular function prediction using neighborhood features. IEEE/ACM Trans Comput Biol Bioinform. 2010;7:208–217. doi: 10.1109/TCBB.2009.81. [DOI] [PubMed] [Google Scholar]
- 13.Kourmpetis YA, van Dijk AD, Bink MC, van Ham RC, ter Braak CJ. Bayesian Markov Random Field analysis for protein function prediction based on network data. PLoS One. 2010;5:e9293. doi: 10.1371/journal.pone.0009293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ng KL, Ciou JS, Huang CH. Prediction of protein functions based on function-function correlation relations. Comput Biol Med. 2010;40:300–305. doi: 10.1016/j.compbiomed.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 15.Karaoz U, Murali TM, Letovsky S, Zheng Y, Ding C, et al. Whole-genome annotation by using evidence integration in functional-linkage networks. Proc Natl Acad Sci U S A. 2004;101:2888–2893. doi: 10.1073/pnas.0307326101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Letovsky S, Kasif S. Predicting protein function from protein/protein interaction data: a probabilistic approach. Bioinformatics. 2003;19(Suppl 1):i197–204. doi: 10.1093/bioinformatics/btg1026. [DOI] [PubMed] [Google Scholar]
- 17.Peng H, Long F, Ding C. Feature selection based on mutual information: criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans Pattern Anal Mach Intell. 2005;27:1226–1238. doi: 10.1109/TPAMI.2005.159. [DOI] [PubMed] [Google Scholar]
- 18.Dijkstra E. A Note on Two Problems in Connection with Graphs. Numerische Mathematik. 1959;1:269–271. [Google Scholar]
- 19.Hinoue T, Weisenberger DJ, Lange CP, Shen H, Byun HM, et al. Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res. 2011 doi: 10.1101/gr.117523.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chou KC. Some remarks on protein attribute prediction and pseudo amino acid composition (50th Anniversary Year Review). Journal of Theoretical Biology. 2011;273:236–247. doi: 10.1016/j.jtbi.2010.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huret JL, Dessen P, Bernheim A. Atlas of Genetics and Cytogenetics in Oncology and Haematology, year 2003. Nucleic Acids Res. 2003;31:272–274. doi: 10.1093/nar/gkg126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Keshava Prasad TS, Goel R, Kandasamy K, Keerthikumar S, Kumar S, et al. Human Protein Reference Database–2009 update. Nucleic Acids Res. 2009;37:D767–772. doi: 10.1093/nar/gkn892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sabates-Bellver J, Van der Flier LG, de Palo M, Cattaneo E, Maake C, et al. Transcriptome profile of human colorectal adenomas. Mol Cancer Res. 2007;5:1263–1275. doi: 10.1158/1541-7786.MCR-07-0267. [DOI] [PubMed] [Google Scholar]
- 24.Nagaraj SH, Reverter A. A Boolean-based systems biology approach to predict novel genes associated with cancer: Application to colorectal cancer. BMC Syst Biol. 2011;5:35. doi: 10.1186/1752-0509-5-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Friedman JH, Baskett F, Shustek LJ. An algorithm for finding nearest neighbors. IEEE Transaction on Information Theory. 1975;C-24:1000–1006. [Google Scholar]
- 26.Denoeux T. A k-nearest neighbor classification rule based on Dempster-Shafer theory. IEEE Transactions on Systems, Man and Cybernetics. 1995;25:804–813. [Google Scholar]
- 27.Hu L, Huang T, Shi X, Lu WC, Cai YD, et al. Predicting functions of proteins in mouse based on weighted protein-protein interaction network and protein hybrid properties. PLoS ONE. 2011;6:e14556. doi: 10.1371/journal.pone.0014556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu L, Zheng L, Wang Z, Li B, Liu L. Using pseudo amino Acid composition to predict protease families by incorporating a series of protein biological features. Protein and Peptide Letters. 2011;18:552–558. doi: 10.2174/092986611795222795. [DOI] [PubMed] [Google Scholar]
- 29.Yang XY, Shi XH, Meng X, Li XL, Lin K, et al. Classification of transcription factors using protein primary structure. Protein & Peptide Letters. 2010;17:899–908. doi: 10.2174/092986610791306670. [DOI] [PubMed] [Google Scholar]
- 30.Huang T, Chen L, Cai YD, Chou KC. Classification and analysis of regulatory pathways using graph property, biochemical and physicochemical property, and functional property. PLoS ONE. 2011;6:e25297. doi: 10.1371/journal.pone.0025297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hu LL, Huang T, Cai YD, Chou KC. Prediction of Body Fluids where Proteins are Secreted into Based on Protein Interaction Network. PLoS One. 2011;6:e22989. doi: 10.1371/journal.pone.0022989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang T, Niu S, Xu Z, Huang Y, Kong X, et al. Predicting Transcriptional Activity of Multiple Site p53 Mutants Based on Hybrid Properties. PLoS ONE. 2011;6:e22940. doi: 10.1371/journal.pone.0022940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang T, Shi XH, Wang P, He Z, Feng KY, et al. Analysis and prediction of the metabolic stability of proteins based on their sequential features, subcellular locations and interaction networks. PLoS ONE. 2010;5:e10972. doi: 10.1371/journal.pone.0010972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cai Y, Huang T, Hu L, Shi X, Xie L, et al. Prediction of lysine ubiquitination with mRMR feature selection and analysis. Amino Acids. 2011 doi: 10.1007/s00726-011-0835-0. [DOI] [PubMed] [Google Scholar]
- 35.Chou KC, Shen HB. Review: Recent progresses in protein subcellular location prediction. Analytical Biochemistry. 2007;370:1–16. doi: 10.1016/j.ab.2007.07.006. [DOI] [PubMed] [Google Scholar]
- 36.Chou KC, Zhang CT. Review: Prediction of protein structural classes. Critical Reviews in Biochemistry and Molecular Biology. 1995;30:275–349. doi: 10.3109/10409239509083488. [DOI] [PubMed] [Google Scholar]
- 37.Chou KC, Shen HB. Cell-PLoc: A package of Web servers for predicting subcellular localization of proteins in various organisms (updated version: Cell-PLoc 2.0: An improved package of web-servers for predicting subcellular localization of proteins in various organisms, Natural Science, 2010, 2, 1090–1103). Nature Protocols. 2008;3:153–162. doi: 10.1038/nprot.2007.494. [DOI] [PubMed] [Google Scholar]
- 38.Esmaeili M, Mohabatkar H, Mohsenzadeh S. Using the concept of Chou's pseudo amino acid composition for risk type prediction of human papillomaviruses. Journal of Theoretical Biology. 2010;263:203–209. doi: 10.1016/j.jtbi.2009.11.016. [DOI] [PubMed] [Google Scholar]
- 39.Georgiou DN, Karakasidis TE, Nieto JJ, Torres A. Use of fuzzy clustering technique and matrices to classify amino acids and its impact to Chou's pseudo amino acid composition. Journal of Theoretical Biology. 2009;257:17–26. doi: 10.1016/j.jtbi.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 40.Gu Q, Ding YS, Zhang TL. Prediction of G-Protein-Coupled Receptor Classes in Low Homology Using Chou's Pseudo Amino Acid Composition with Approximate Entropy and Hydrophobicity Patterns. Protein & Peptide Letters. 2010;17:559–567. doi: 10.2174/092986610791112693. [DOI] [PubMed] [Google Scholar]
- 41.Chou KC, Wu ZC, Xiao X. iLoc-Hum: Using accumulation-label scale to predict subcellular locations of human proteins with both single and multiple sites. Molecular Biosystems. 2012;8:629–641. doi: 10.1039/c1mb05420a. [DOI] [PubMed] [Google Scholar]
- 42.Mohabatkar H. Prediction of cyclin proteins using Chou's pseudo amino acid composition. Protein & Peptide Letters. 2010;17:1207–1214. doi: 10.2174/092986610792231564. [DOI] [PubMed] [Google Scholar]
- 43.Xiao X, Wu ZC, Chou KC. A multi-label classifier for predicting the subcellular localization of gram-negative bacterial proteins with both single and multiple sites. PLoS ONE. 2011;6:e20592. doi: 10.1371/journal.pone.0020592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu ZC, Xiao X, Chou KC. iLoc-Plant: a multi-label classifier for predicting the subcellular localization of plant proteins with both single and multiple sites. Molecular Bio Systems. 2011;7:3287–3297. doi: 10.1039/c1mb05232b. [DOI] [PubMed] [Google Scholar]
- 45.Mohabatkar H, Mohammad Beigi M, Esmaeili A. Prediction of GABA(A) receptor proteins using the concept of Chou's pseudo-amino acid composition and support vector machine. Journal of Theoretical Biology. 2011;281:18–23. doi: 10.1016/j.jtbi.2011.04.017. [DOI] [PubMed] [Google Scholar]
- 46.Chou KC, Wu ZC, Xiao X. iLoc-Euk: A Multi-Label Classifier for Predicting the Subcellular Localization of Singleplex and Multiplex Eukaryotic Proteins. PLoS One. 2011;6:e18258. doi: 10.1371/journal.pone.0018258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang T, Cui W, He ZS, Hu L, Liu F, et al. Functional association between influenza A (H1N1) virus and human. Biochem Biophys Res Commun. 2009;390:1111–1113. doi: 10.1016/j.bbrc.2009.08.131. [DOI] [PubMed] [Google Scholar]
- 48.Chou KC, Forsen S. Graphical rules for enzyme-catalyzed rate laws. Biochemical Journal. 1980;187:829–835. doi: 10.1042/bj1870829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou GP, Deng MH. An extension of Chou's graphic rules for deriving enzyme kinetic equations to systems involving parallel reaction pathways. Biochemical Journal. 1984;222:169–176. doi: 10.1042/bj2220169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chou KC. Graphic rules in steady and non-steady enzyme kinetics. Journal of Biological Chemistry. 1989;264:12074–12079. [PubMed] [Google Scholar]
- 51.Chou KC. Review: Applications of graph theory to enzyme kinetics and protein folding kinetics. Steady and non-steady state systems. Biophysical Chemistry. 1990;35:1–24. doi: 10.1016/0301-4622(90)80056-d. [DOI] [PubMed] [Google Scholar]
- 52.Althaus IW, Gonzales AJ, Chou JJ, Diebel MR, Chou KC, et al. The quinoline U-78036 is a potent inhibitor of HIV-1 reverse transcriptase. Journal of Biological Chemistry. 1993;268:14875–14880. [PubMed] [Google Scholar]
- 53.Chou KC, Kezdy FJ, Reusser F. Review: Steady-state inhibition kinetics of processive nucleic acid polymerases and nucleases. Analytical Biochemistry. 1994;221:217–230. doi: 10.1006/abio.1994.1405. [DOI] [PubMed] [Google Scholar]
- 54.Andraos J. Kinetic plasticity and the determination of product ratios for kinetic schemes leading to multiple products without rate laws: new methods based on directed graphs. Canadian Journal of Chemistry. 2008;86:342–357. [Google Scholar]
- 55.Chou KC. Graphic rule for drug metabolism systems. Current Drug Metabolism. 2010;11:369–378. doi: 10.2174/138920010791514261. [DOI] [PubMed] [Google Scholar]
- 56.Zhou GP. The disposition of the LZCC protein residues in wenxiang diagram provides new insights into the protein-protein interaction mechanism. Journal of Theoretical Biology. 2011;284:142–148. doi: 10.1016/j.jtbi.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chou KC, Lin WZ, Xiao X. Wenxiang: a web-server for drawing wenxiang diagrams. Natural Science. 2011;3:862–865. [Google Scholar]
- 58.Zhou GP. The Structural Determinations of the Leucine Zipper Coiled-Coil Domains of the cGMP-Dependent Protein Kinase I alpha and its Interaction with the Myosin Binding Subunit of the Myosin Light Chains Phosphase. Proteins & Peptide Letters. 2011;18:966–978. doi: 10.2174/0929866511107010966. [DOI] [PubMed] [Google Scholar]
- 59.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 60.Benjamini Y, Yekutieli D. The Control of the False Discovery Rate in Multiple Testing under Dependency. The Annals of Statistics. 2001;29:1165–1188. [Google Scholar]
- 61.Catalano MG, Pfeffer U, Raineri M, Ferro P, Curto A, et al. Altered expression of androgen-receptor isoforms in human colon-cancer tissues. Int J Cancer. 2000;86:325–330. doi: 10.1002/(sici)1097-0215(20000501)86:3<325::aid-ijc4>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 62.Castagnetta L, Traina A, Campisi I, Calabro M, Maratta A, et al. Androgen receptor status in nontumoral and malignant human colorectal tissues. Ann N Y Acad Sci. 2002;963:322–325. doi: 10.1111/j.1749-6632.2002.tb04124.x. [DOI] [PubMed] [Google Scholar]
- 63.Menendez D, Inga A, Resnick MA. The expanding universe of p53 targets. Nat Rev Cancer. 2009;9:724–737. doi: 10.1038/nrc2730. [DOI] [PubMed] [Google Scholar]
- 64.Gayther SA, Batley SJ, Linger L, Bannister A, Thorpe K, et al. Mutations truncating the EP300 acetylase in human cancers. Nat Genet. 2000;24:300–303. doi: 10.1038/73536. [DOI] [PubMed] [Google Scholar]
- 65.Polakis P. Wnt signaling and cancer. Genes Dev. 2000;14:1837–1851. [PubMed] [Google Scholar]
- 66.Howe GR, Aronson KJ, Benito E, Castelleto R, Cornee J, et al. The relationship between dietary fat intake and risk of colorectal cancer: evidence from the combined analysis of 13 case-control studies. Cancer Causes Control. 1997;8:215–228. doi: 10.1023/a:1018476414781. [DOI] [PubMed] [Google Scholar]
- 67.Hoeft B, Linseisen J, Beckmann L, Muller-Decker K, Canzian F, et al. Polymorphisms in fatty-acid-metabolism-related genes are associated with colorectal cancer risk. Carcinogenesis. 2010;31:466–472. doi: 10.1093/carcin/bgp325. [DOI] [PubMed] [Google Scholar]
- 68.Christensen BC, Smith AA, Zheng S, Koestler DC, Houseman EA, et al. DNA methylation, isocitrate dehydrogenase mutation, and survival in glioma. J Natl Cancer Inst. 2011;103:143–153. doi: 10.1093/jnci/djq497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li J, Humphreys K, Darabi H, Rosin G, Hannelius U, et al. A genome-wide association scan on estrogen receptor-negative breast cancer. Breast Cancer Res. 2010;12:R93. doi: 10.1186/bcr2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Allen WL, Jithesh PV, Oliver GR, Proutski I, Longley DB, et al. The colorectal cancer disease-specific transcriptome may facilitate the discovery of more biologically and clinically relevant information. BMC Cancer. 2010;10:687. doi: 10.1186/1471-2407-10-687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tsukahara H, Sekine K, Uchiyama M, Miura M, Nakazato M, et al. Uroguanylin level in umbilical cord blood. Pediatr Int. 2001;43:267–269. doi: 10.1046/j.1442-200x.2001.01393.x. [DOI] [PubMed] [Google Scholar]
- 72.Liu D, Overbey D, Watkinson LD, Daibes-Figueroa S, Hoffman TJ, et al. In vivo imaging of human colorectal cancer using radiolabeled analogs of the uroguanylin peptide hormone. Anticancer Res. 2009;29:3777–3783. [PubMed] [Google Scholar]
- 73.Reeves JR, Xuan JW, Arfanis K, Morin C, Garde SV, et al. Identification, purification and characterization of a novel human blood protein with binding affinity for prostate secretory protein of 94 amino acids. Biochem J. 2005;385:105–114. doi: 10.1042/BJ20040290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cui J, Chen Y, Chou WC, Sun L, Chen L, et al. An integrated transcriptomic and computational analysis for biomarker identification in gastric cancer. Nucleic Acids Res. 2011;39:1197–1207. doi: 10.1093/nar/gkq960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hibi K, Goto T, Mizukami H, Kitamura YH, Sakuraba K, et al. Demethylation of the CDH3 gene is frequently detected in advanced colorectal cancer. Anticancer Res. 2009;29:2215–2217. [PubMed] [Google Scholar]
- 76.Imai K, Hirata S, Irie A, Senju S, Ikuta Y, et al. Identification of a novel tumor-associated antigen, cadherin 3/P-cadherin, as a possible target for immunotherapy of pancreatic, gastric, and colorectal cancers. Clin Cancer Res. 2008;14:6487–6495. doi: 10.1158/1078-0432.CCR-08-1086. [DOI] [PubMed] [Google Scholar]
- 77.Deves C, Renck D, Garicochea B, da Silva VD, Giulianni Lopes T, et al. Analysis of select members of the E26 (ETS) transcription factors family in colorectal cancer. Virchows Arch. 2011;458:421–430. doi: 10.1007/s00428-011-1053-6. [DOI] [PubMed] [Google Scholar]
- 78.Stohr H, Marquardt A, Nanda I, Schmid M, Weber BH. Three novel human VMD2-like genes are members of the evolutionary highly conserved RFP-TM family. Eur J Hum Genet. 2002;10:281–284. doi: 10.1038/sj.ejhg.5200796. [DOI] [PubMed] [Google Scholar]
- 79.Bakall B, McLaughlin P, Stanton JB, Zhang Y, Hartzell HC, et al. Bestrophin-2 is involved in the generation of intraocular pressure. Invest Ophthalmol Vis Sci. 2008;49:1563–1570. doi: 10.1167/iovs.07-1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yu K, Lujan R, Marmorstein A, Gabriel S, Hartzell HC. Bestrophin-2 mediates bicarbonate transport by goblet cells in mouse colon. J Clin Invest. 2010;120:1722–1735. doi: 10.1172/JCI41129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.van Criekinge W, Meijer G, Straub J, De Carvalho BPM. Methylation markers for early detection and prognosis of colon cancers. 2007. International Application No: PCT/US2007/013803.
- 82.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 85.Morikawa T, Kuchiba A, Yamauchi M, Meyerhardt JA, Shima K, et al. Association of CTNNB1 (beta-catenin) alterations, body mass index, and physical activity with survival in patients with colorectal cancer. JAMA. 2011;305:1685–1694. doi: 10.1001/jama.2011.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.MacLean HE, Warne GL, Zajac JD. Localization of functional domains in the androgen receptor. J Steroid Biochem Mol Biol. 1997;62:233–242. doi: 10.1016/s0960-0760(97)00049-6. [DOI] [PubMed] [Google Scholar]
- 87.Slattery ML, Sweeney C, Murtaugh M, Ma KN, Wolff RK, et al. Associations between ERalpha, ERbeta, and AR genotypes and colon and rectal cancer. Cancer Epidemiol Biomarkers Prev. 2005;14:2936–2942. doi: 10.1158/1055-9965.EPI-05-0514. [DOI] [PubMed] [Google Scholar]
- 88.Johnson SA, Dubeau L, Kawalek M, Dervan A, Schonthal AH, et al. Increased expression of TATA-binding protein, the central transcription factor, can contribute to oncogenesis. Mol Cell Biol. 2003;23:3043–3051. doi: 10.1128/MCB.23.9.3043-3051.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hiranuma C, Kawakami K, Oyama K, Ota N, Omura K, et al. Hypermethylation of the MYOD1 gene is a novel prognostic factor in patients with colorectal cancer. Int J Mol Med. 2004;13:413–417. [PubMed] [Google Scholar]
- 90.Medina MW. The relationship between HMGCR genetic variation, alternative splicing, and statin efficacy. Discov Med. 2010;9:495–499. [PubMed] [Google Scholar]
- 91.Lipkin SM, Chao EC, Moreno V, Rozek LS, Rennert H, et al. Genetic variation in 3-hydroxy-3-methylglutaryl CoA reductase modifies the chemopreventive activity of statins for colorectal cancer. Cancer Prev Res (Phila) 2010;3:597–603. doi: 10.1158/1940-6207.CAPR-10-0007. [DOI] [PubMed] [Google Scholar]
- 92.Diamandis EP, Yu H. New biological functions of prostate-specific antigen? J Clin Endocrinol Metab. 1995;80:1515–1517. doi: 10.1210/jcem.80.5.7538144. [DOI] [PubMed] [Google Scholar]
- 93.Levesque M, Hu H, D'Costa M, Diamandis EP. Prostate-specific antigen expression by various tumors. J Clin Lab Anal. 1995;9:123–128. doi: 10.1002/jcla.1860090209. [DOI] [PubMed] [Google Scholar]
- 94.Borgono CA, Diamandis EP. The emerging roles of human tissue kallikreins in cancer. Nat Rev Cancer. 2004;4:876–890. doi: 10.1038/nrc1474. [DOI] [PubMed] [Google Scholar]
- 95.Diamandis EP, Yousef GM. Human tissue kallikreins: a family of new cancer biomarkers. Clin Chem. 2002;48:1198–1205. [PubMed] [Google Scholar]
- 96.Talieri M, Mathioudaki K, Prezas P, Alexopoulou DK, Diamandis EP, et al. Clinical significance of kallikrein-related peptidase 7 (KLK7) in colorectal cancer. Thromb Haemost. 2009;101:741–747. [PubMed] [Google Scholar]
- 97.Kim MS, Chung NG, Kang MR, Yoo NJ, Lee SH. Genetic and expressional alterations of CHD genes in gastric and colorectal cancers. Histopathology. 2011;58:660–668. doi: 10.1111/j.1365-2559.2011.03819.x. [DOI] [PubMed] [Google Scholar]
- 98.Thomas G, Jacobs KB, Yeager M, Kraft P, Wacholder S, et al. Multiple loci identified in a genome-wide association study of prostate cancer. Nat Genet. 2008;40:310–315. doi: 10.1038/ng.91. [DOI] [PubMed] [Google Scholar]
- 99.Ohkubo I, Tada T, Ochiai Y, Ueyama H, Eimoto T, et al. Human seminal plasma beta-microseminoprotein: its purification, characterization, and immunohistochemical localization. Int J Biochem Cell Biol. 1995;27:603–611. doi: 10.1016/1357-2725(95)00021-G. [DOI] [PubMed] [Google Scholar]
- 100.Katoh M. Human FOX gene family (Review). Int J Oncol. 2004;25:1495–1500. [PubMed] [Google Scholar]
- 101.Niittymaki I, Tuupanen S, Li Y, Jarvinen H, Mecklin JP, et al. Systematic search for enhancer elements and somatic allelic imbalance at seven low-penetrance colorectal cancer predisposition loci. BMC Med Genet. 2011;12:23. doi: 10.1186/1471-2350-12-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kim IJ, Kang HC, Jang SG, Kim K, Ahn SA, et al. Oligonucleotide microarray analysis of distinct gene expression patterns in colorectal cancer tissues harboring BRAF and K-ras mutations. Carcinogenesis. 2006;27:392–404. doi: 10.1093/carcin/bgi237. [DOI] [PubMed] [Google Scholar]
- 103.Lagger G, Doetzlhofer A, Schuettengruber B, Haidweger E, Simboeck E, et al. The tumor suppressor p53 and histone deacetylase 1 are antagonistic regulators of the cyclin-dependent kinase inhibitor p21/WAF1/CIP1 gene. Mol Cell Biol. 2003;23:2669–2679. doi: 10.1128/MCB.23.8.2669-2679.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Thangaraju M, Carswell KN, Prasad PD, Ganapathy V. Colon cancer cells maintain low levels of pyruvate to avoid cell death caused by inhibition of HDAC1/HDAC3. Biochem J. 2009;417:379–389. doi: 10.1042/BJ20081132. [DOI] [PubMed] [Google Scholar]
- 105.Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 2009;41:178–186. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Landi D, Gemignani F, Naccarati A, Pardini B, Vodicka P, et al. Polymorphisms within micro-RNA-binding sites and risk of sporadic colorectal cancer. Carcinogenesis. 2008;29:579–584. doi: 10.1093/carcin/bgm304. [DOI] [PubMed] [Google Scholar]
- 107.Team RDC. R: A Language and Environment for Statistical Computing. 2008.
- 108.Huang T, Wan S, Xu Z, Zheng Y, Feng KY, et al. Analysis and prediction of translation rate based on sequence and functional features of the mRNA. PLoS One. 2011;6:e16036. doi: 10.1371/journal.pone.0016036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Huang T, Chen L, Liu X-J, Cai Y-D. Predicting triplet of transcription factor - mediating enzyme - target gene by functional profiles. Neurocomputing. 2011;74:3677–3681. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The cancer-related gene list and the two colorectal cancer-related gene lists.
(XLS)
The MaxRel features table and mRMR features table.
(XLS)
Feature numbers and the first order accuracy which the IFS curve plot was based on.
(XLS)
The 15 shortest paths with the lowest cost presented with protein and gene, respectively.
(DOC)
The overlap between the 41 candidate genes and the three other datasets and the corresponding Fisher's exact test.
(DOC)
Five gene sets. First gene set is our 41 selected genes. The second gene set is the top 41 mRMR genes. The third gene set is the top 41 t-test genes that have the smallest t-test p values. The second and third gene sets were from gene expression profiles alone. Our 41 gene were selected based on both gene expression profiles and protein interaction network. The fourth gene set is the 742 cancer genes. The fifth gene set is the combined known colorectal cancer related genes.
(XLS)