

The site-selective, catalytic modification of complex molecules is a long-standing goal for chemists. Given the high efficiency of many fermentation processes, the use of natural products as scaffolds for diversification is part of many epic drug discovery programs.[i] Yet, it is generally accepted that these research efforts are challenging, often requiring multistep syntheses of the requisite analogs,[ ii ] re-engineered biosynthetic pathways,[ iii] or enzymatic modification of the natural product itself.[iv ] The use of chemical catalysts to alter complex scaffolds may represent a growing field with potential to impact these interdisciplinary research efforts.[v,vi ] The challenges at the front end of this approach include the identification of not only relevant reactions to explore, but also the inherent architectural and stereochemical molecular features that the catalyst-substrate complexes ultimately embody.
Among the more comprehensively studied scaffold-modifying processes in nature are oxidation reactions.[vii ] Chemists can parallel nature’s course, which tends to increase the overall oxidation state of a substrate during the course of a biosynthesis.[viii, ix ] But, how might chemists turn the biosynthetic clock back? Can chemists take nature’s oxidized scaffolds, and catalytically and selectively achieve deoxygenated analogues directly, without de novo synthesis of a deoxygenated, complex molecule? While enzymes that deoxygenate polyols are known,[x] they are perhaps less prevalent than their oxidase counterparts. Recognizing the power of chemical deoxygenation, chemists have developed a lexicon of deoxygenation protocols.[xi] However, it might reflect the state of the art that very few involve catalysis, and even fewer address selectivity in polyfunctional molecules. One approach to these issues is described below.
Our laboratory has been exploring methods for the site-selective manipulation of polyols.[xii ] Among the reactions we have explored, site-selective deoxygenation is one of the more challenging.[xiii] We reported that peptides containing modified histidine (1) could indeed effect catalytic, site-selective thiocarbonylation of several simple polyols as a prelude to deoxygenation (2→3→4; Figure 1a).[xiv ] However, a number of limitations exist. For example, when the approach is applied to erythromycin A (Ery A), side reactions can reduce efficiency. While direct thiocarbonylation of Ery A under our previously developed conditions can deliver thiocarbonate 5 in 76% yield (Figure 1b), the same conditions applied to 2′-acetyl-Ery A results in dealkylation of the amino group (6, Figure 1c).[xv ] Because natural products decorated with amino sugars are common,[ xvi ] we suspected that such side reactions would be a consistent liability with carbonyl-based electrophiles when applied to this class of structures.
Figure 1.

(a) Peptide-catalyzed site-selective thiocarbonylation as a precursor to radical deoxygenation. (b) Thiocarbonylation of erythromycin A with N-methyl imidazole catalyst. (c) Thiocarbamate formation on 2′-acetyl erythromycin A.
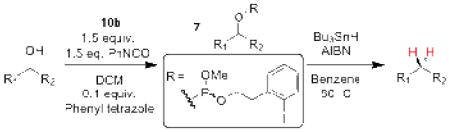
In order to address limitations such as these, we undertook the exploration of a new catalytic cycle for site-selective deoxygenation of polyols. With the goal of achieving the mildest of conditions, such that diverse, complex natural product functionality could be tolerated, we were drawn to the efficient deoxygenation of hydroxyl groups pioneered by Koreeda and coworkers.[xvii ] In this chemistry, hydroxyl groups are converted to the corresponding phosphites, via phosphitylation with P(III)-based chlorophosphites that contain a pendant iodoarene (7, Figure 2a). Then, radical deoxygenation occurs under standard conditions. While powerful, this P(III)-based approach is not readily adaptable to catalysis, given the high reactivity of chlorophosphites. Thus, we wondered if it might be adapted to a catalytic phosphoramidite transfer process we had recently reported.[xviii] In this work (Figure 2b), we showed that the venerable phosphoramidite method described by Caruthers[xix] could indeed be performed with peptide-embedded tetrazoles as catalysts (e.g., 8; ~5 mol% of the catalyst is typical for high conversion). Moreover, we showed that the approach was amenable to the transfer of stereochemical information, with desymmetrization of inositol derivative 9 as an example. The question thus became whether or not these approaches could be merged to enable catalytic, site-selective phosphoramidite transfer. The key advance required the development of previously unknown phosphoramidite 10b (Figure 2c), and the demonstration that it is amenable to selective transfer and subsequent deoxygenation.
Figure 2.

(a) Dichlorophosphite strategy to form phosphite-based deoxygenation precursors. (b) Peptide-catalyzed, enantio- and regioselective phosphitylation employing phosphoramidites. (c) Peptide-catalyzed site-selective formation of phosphite-based deoxygenation precursors employing phosphoramidites.
Initially, we attempted to adapt the same catalytic strategy described in Figure 2b employing the diethylamino variant of the phosphoramidite (10a) and 10 Å molecular sieves.[ xx ] These conditions, however, proved to be incompatible with low molecular weight substrate 11, and sluggish for the phosphitylation of 2′-Acetyl Ery A. Furthermore, phosphoramidite 10a, in the absence of molecular sieves, was highly prone to hydrolysis, and decomposed in the presence of an alternative amine scavenger, phenyl isocyanate.[xxi ]
Therefore, we pursued the synthesis of the much more stable phosphoramidite 10b, based on diisopropylamino substitution, for use in combination with phenyl isocyanate as an amine scavenger (Figure 2c). On a multigram scale, phosphoramidite 10b can be synthesized from 2-(2-iodophenyl)ethanol and methyl tetraisopropyl phosphordiamidite in greater than 90% yield.[xxii] Not only did 10b prove to be compatible with tetrazole catalysis (Table 1), it was also stable to both hydrolysis and oxidation over the course of several months without any special precautions.
Table 1.
Catalytic phosphitylation and deoxygenation of alcohol substrates.
 | |||
|---|---|---|---|
| Entry | Alcohol | Phosphite | Deoxygenated Product |
| 1 |
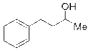 11 |
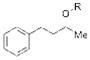 13 >95% NMR Conversion |
Not Determined |
| 2 |
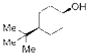 12 |
14 >95% NMR Conversion |
Not Determined |
| 3 |
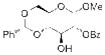 15 |
Not Isolated |
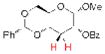 18, 77% yielda |
| 4 |
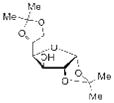 16 |
Not Isolated |
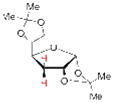 19 68% yielda |
| 5 |
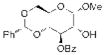 17 |
Not Isolated |
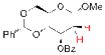 20 67% yielda |
| 6 |
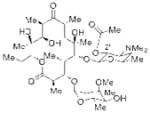 2′-Acetyl Ery A |
21 (4″-Phosphite) 83% isolated yield |
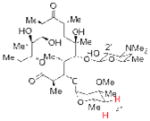 22 4″-deoxy Ery A 84% yieldb |
Isolated yields of the deoxygenated species are given for the two-step process.
Isolated yield determined from phosphite 21 after methanol cleavage of 2′-acetyl protecting group.
We initially examined the catalytic phosphitylation of simple alcohols 11 and 12 in the presence of only 10 mol % phenyl tetrazole as catalyst, and 1.5 equivalents each of 10b and phenyl isocyanate (Table 1, entries 1 and 2). Both substrates 11 and 12 were completely consumed within 4 hours, and 1H NMR spectra of the resulting crude material indicated a very clean transformation with no remaining starting alcohol and evidence for the formation of the corresponding phosphites (13 and 14, respectively), typically to greater than 95 percent conversion (1H NMR analysis).[22] Although it was clear that the catalytic phosphitylation reaction proceeded with great efficiency, isolation of the corresponding phosphites by silica gel chromatography proved to be much more problematic due to the persistent co-elution with the side product urea derived from reaction of the liberated diisopropylamine with the isocyanate amine scavenger. Purification was further hampered by the overall instability of the phosphite products.
Having demonstrated the feasibility of the catalytic phosphitylation reaction with simple substrates, we next sought to assess the overall deoxygenation strategy by investigating whether the crude phosphite products of more relevant, highly oxygenated substrates (Table 1, 15, 16, 17; entries 3–5) could be carried through the deoxygenation step without isolation and purification of the intermediate phosphites. Such a strategy would potentially streamline the process and minimize the loss of material. Moreover, in the context of chiral natural products, the structure of the deoxygenated products would be easier to assign than the epimeric P-chiral phosphites. We therefore subjected compounds 15–17 to the same catalytic phosphitylation conditions described for 11 and 12. Then, after basic workup, each was immediately treated with the standard radical deoxygenation conditions. We were delighted to find that the two-step strategy gave the corresponding deoxygenated products 18, 19 and 20, respectively, in good overall yields. Moreover, as shown in Table 1 (entry 6), we applied the new catalytic phosphitylation strategy to 2′-Acetyl-Ery A, observing clean converstion to the 4″-phosphite product 21, which was isolated in 83% yield. This phosphite product was readily deoxygenated under standard conditions, and after methanolic deacylation, gave 4″-deoxy-Ery A (22) in 84% yield.[xxiii]
We next wished to assess prospects for catalyst-dependent, site-selective phosphitylation/ deoxygenation of a complex, native polyol with the newly developed protocol. Our hypothesis was that a peptide-based appendage to the catalytically active tetrazole moiety would provide a handle for tuning site-selectivity, in analogy to our early observations with other group transfer processes.[ xxiv ] The goal was to examine whether catalyst control might overcome the well-known high reactivity of the 2′-hydroxyl of Ery A.[xxv ] It is well precedented that the 2′-hydroxyl reacts rapidly with many carbonyl-based electrophiles, presumably through a neighboring base-assisted mechanism. As such, methods to modify the other hydroxyls of erythromycin A generally require protection of this position. To test this prospect for catalyst-dependent reversal of site-selectivity, without recourse to the use of a protective acetyl group, we subjected Ery A to the phosphitylation conditions described above. Upon workup, we analyzed the product distribution by 31P-NMR. As indicated in Figure 3a, the simple catalyst phenyl tetrazole gave a distribution of phosphite products, with four intense chemical shifts. By analogy to the previously isolated 2′-acetyl-4″-phosphite of Ery A (cf. Table 1, entry 6, 21), we believe that the two 31P chemical shifts at 143 and 144 ppm correspond to the two 31P-epimers of the 4″-regioisomer. The other most significant product of the reaction, with 31P-chemical shifts at 140.0 and 140.5 ppm, would appear to be the alternative 2′-phosphite. Subjection of this mixture to deoxygenation conditions gives a corresponding mixture of deoxygenation products. Of the two major products that are formed, one is indeed identical to 23, that was obtained from the deoxygenation of thiocarbonyl 5 (Figure 1b). The other may be assigned as the 4″-regioisomer (22), albeit at low relative abundance. Notably, this catalytic protocol delivers the mixture in an approximate 3:1 ratio (1H NMR analysis), reflecting an inherent preference for the 2′-deoxy isomer (23), with the simple phenyl tetrazole catalyst.[22]
Figure 3.

(a) Non-selective, phenyl tetrazole-catalyzed phosphitylation gives multiple phosphite products by 31P-NMR (crude sample). Deoxygenation yields 23 as the major product. (b) N-methyl imidazole catalyzed thiocarbonylation followed by radical deoxygenation gives 23. (c) Peptide 24 directly delivers the 4″-phosphite product (31P-NMR, crude sample) for efficient deoxygenation to give 4″-deoxyerythromycin A (22). A larger scale representation of the NMR data is presented in the Supporting Information as Supplemental Figure 3.
On the other hand, the peptide-embedded tetrazole-based catalyst 24 provides the putative 4″-regioisomer with near complete selectivity (Figure 3c). Furthermore direct radical deoxygenation of the corresponding crude reaction mixture gives 4″-deoxy-Ery A (22), in 50–60% isolated yield, from Ery A.
It is notable that the peptide-catalyzed phosphitylation/deoxygenation protocol results in both clean reactions and altered selectivity in comparison to the results achieved with the simple phenyl tetrazole catalyst. In addition, it is also relevant to our efforts that the most efficient paths to either the 2′-deoxy Ery A 23 or the 4″-deoxy Ery A 22 turn out to employ different protocols – thiocarbonylation with a simple catalyst in the former case, and phosphitylation with a peptide-embedded tetrazole in the latter case. These findings emphasize the need to evaluate multiple approaches in parallel for the goal of site-selective deoxygenation of complex polyols, in pursuit of multiple and various deoxygenated analogs.
We conclude this report with several thoughts regarding these findings. First, we are intrigued by our ongoing observation of catalyst-dependent reversal of inherent selectivity in derivatization reactions of this type.[5] From the pragmatic perspective, it may be that this approach will represent an addition to the tools available to chemists for the direct manipulation of complex structures. Notably, our work in this area has revealed that there is a premium on the use of catalysts for complex molecule derivatizations since improvements in selectivity can greatly facilitate isolation of pure materials and, eventually, their large-scale synthesis. Maybe most important, however, is the identification of orthogonal catalysts and protocols that provide access to unique natural product analogs. Perhaps we can aspire to comprehensive and general approaches to problems like catalytic, site-selective deoxygenation of complex structures. But, our studies in this area reveal that, at present, diverse approaches may be most enabling. In this vein, we also anticipate that libraries of catalysts, for example those bearing the tetrazolylalanine, will be needed to generalize these findings to other classes of compounds. Our extension of these studies to other families of complex molecule deoxygenations are ongoing and shall be reported in the future.
Supplementary Material
Acknowledgments
We are grateful to the National Institutes of General Medical Sciences of the National Institute of Health Foundation (GM-068649) for support.
References
- i.Newman DJ, Cragg GM. J Nat Prod. 2007;70:461–477. doi: 10.1021/np068054v. [DOI] [PubMed] [Google Scholar]
- ii.Wilson RM, Danishefsky SJ. J Org Chem. 2006;71:8329–8351. doi: 10.1021/jo0610053. [DOI] [PubMed] [Google Scholar]
- iii.a) McGlinchey RP, Nett M, Eustáquio AS, Asolkar RN, Fenical W, Moore BS. J Am Chem Soc. 2008;130:7822–7823. doi: 10.1021/ja8029398. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Eustáquio AS, O’Hagan D, Moore BS. J Nat Prod. 2010;73:378–382. doi: 10.1021/np900719u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- iv.a) Mortison JD, Sherman DH. J Org Chem. 2010;75:7041–7051. doi: 10.1021/jo101124n. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Langenhan JM, Griffith BR, Thorson JS. J Nat Prod. 2005;68:1696–1711. doi: 10.1021/np0502084. [DOI] [PubMed] [Google Scholar]; c) Williams GJ, Zhang C, Thorson JS. Nat Chem Biol. 2007;10:657–662. doi: 10.1038/nchembio.2007.28. [DOI] [PubMed] [Google Scholar]; d) Shi R, Lamb SS, Zakeri B, Proteau A, Cui Q, Sulea T, Matte A, Wright GD. Chem Biol. 2009;16:401–410. doi: 10.1016/j.chembiol.2009.02.007. [DOI] [PubMed] [Google Scholar]; d) Lamb SS, Patel T, Koteva KP, Wright GD. Chem Biol. 2006;13:171–181. doi: 10.1016/j.chembiol.2005.12.003. [DOI] [PubMed] [Google Scholar]
- v.a) Lewis CA, Longcore KE, Miller SJ, Wender PA. J Nat Prod. 2009;11:4318–4321. doi: 10.1021/np9004932. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lewis CA, Merkel J, Miller SJ. Bioorg Med Chem Lett. 2008;18:6007–6011. doi: 10.1016/j.bmcl.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Lewis CA, Miller SJ. Angew Chem Int Ed. 2006;45:5616–5619. doi: 10.1002/anie.200601490. [DOI] [PubMed] [Google Scholar]
- vi.Chamni S, He QL, Dang Y, Bhat S, Liu JO, Romo D. ACS Chem Biol. 2011;6:1175–1181. doi: 10.1021/cb2002686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- vii.Mendoza A, Ishihara Y, Baran PS. Nat Chem. 2012;4:21–25. doi: 10.1038/nchem.1196. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Davis EM, Croteau R. Top Curr Chem. 2000;209:53–95. [Google Scholar]; c) Jennewein S, Rithner CD, Williams RM, Croteau RB. Proc Natl Acad Sci U S A. 2001;24:13595–13600. doi: 10.1073/pnas.251539398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- viii.Chen K, Baran PS. Nature. 2009;459:824–828. doi: 10.1038/nature08043. [DOI] [PubMed] [Google Scholar]
- ix.a) Chen MS, White MC. Science. 2007;318:783–787. doi: 10.1126/science.1148597. [DOI] [PubMed] [Google Scholar]; b) Bigi MA, Reed SA, White MC. Nat Chem. 2011;3:216–222. doi: 10.1038/nchem.967. [DOI] [PubMed] [Google Scholar]; c) Stang EM, White MC. Nat Chem. 2009;1:547–551. doi: 10.1038/nchem.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- x.Stubbe J, Riggs-Gelasco P. Trends Biochem Sci. 1998;23:438–443. doi: 10.1016/s0968-0004(98)01296-1. [DOI] [PubMed] [Google Scholar]
- xi.Barton DHR, McCombie SW. J Chem Soc, Perkin Trans. 1975;1:1574–1585.Crich D, Quintero L. Chem Rev. 1989;89:1413–1432.Studer A, Amrein S. Synthesis. 2002;7:835–849.For an example, in a complex molecular setting, see: Palacios DS, Anderson TM, Burke MD. J Am Chem Soc. 2007;129:13804–13805. doi: 10.1021/ja075739o.
- xii.For example, see: Sculimbrene BR, Morgan AJ, Miller SJ. J Am Chem Soc. 2002;124:11653–11656. doi: 10.1021/ja027402m.Griswold KS, Miller SJ. Tetrahedron. 2003;59:8869–8875.Fiori KW, Puchlopek ALA, Miller SJ. Nat Chem. 2009;1:630–634. doi: 10.1038/nchem.410.d) Reference 5.
- xiii.Morgan AJ, Wang YK, Roberts MF, Miller SJ. J Am Chem Soc. 2004;126:15370–15371. doi: 10.1021/ja047360x.For a pioneering application of thiocarboylation of carbohydrates for deoxygenation, see: Haque ME, Kikuchi T, Kanemitsu K, Tsuda Y. Chem Pharm Bull. 1987;35:1016–1029.
- xiv.Sánchez-Roselló M, Puchlopek ALA, Morgan AJ, Miller SJ. J Org Chem. 2008;73:1774–1782. doi: 10.1021/jo702334z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xv.For previous precendents of chlorocarbonate chemistry with erythromycin see: Hengeveld JE, Gupta AK, Kemp AH, Thomas AV. Tetrahedron Lett. 1999;40:2497–2500.Flynn EH, Murphy HW, McMahon RE. J Am Chem Soc. 1955;77:3104–3106.Flynn EH, Sigal MV, Jr, Wiley PF, Gerzon K. J Am Chem Soc. 1954;76:3121–3131.
- xvi.Weymouth-Wilson AC. Nat Prod Rep. 1997;14:99–110. doi: 10.1039/np9971400099. [DOI] [PubMed] [Google Scholar]
- xvii.Zhang L, Koreeda M. J Am Chem Soc. 2004;126:13190–13191. doi: 10.1021/ja0462777. [DOI] [PubMed] [Google Scholar]
- xviii.Jordan PA, Kayser-Bricker KJ, Miller SJ. Proc Natl Acad Sci U S A. 2010;107:20620–20624. doi: 10.1073/pnas.1001111107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xix.Caruthers MH. Acc Chem Res. 1991;24:278–284. [Google Scholar]
- xx.(a) Due to the displacement of the basic dialkyamine moiety in the acid catalyzed reaction, an amine scavenger is necessary for catalyst turnover, see: Dahl BH, Nielsen J, Dahl O. Nucleic Acids Res. 1987;15:1729–1743. doi: 10.1093/nar/15.4.1729.Dahl BH, Nielsen J, Dahl O. Nucleosides Nucleotides. 1987;6:457–460.(b) The use of 10Å molecular sieves as a scavenger necessitated diethylamine as a leaving group. See: reference 18.
- xxi.Brady PB, Morris EM, Fenton OS, Sculimbrene BR. Tetrahedron Lett. 2009;50:975–978. [Google Scholar]
- xxii.See supporting information.
- xxiii.4″-Deoxyerythromycin A was referenced previously in the following patent. See: Fujiwara T, Honda E, Hirano T, Sakakibara H. 58-49396. (TOYO JOZO CO LTD) JP. 1983For various analogues employing this method see: Lartey PA, Nellans HN, Faghih R, Petersen A, Edwards CM, Freiberg L, Quigley S, Marsh K, Klein LL, Plattner JJ. J Med Chem. 1995;38:1793–1798. doi: 10.1021/jm00010a024.
- xxiv.See reference 5.
- xxv.Our observations of the preferential reactivity of the 2′-hydroxyl group of Ery A under various conditions are in accord with long-standing observations from Abbott laboratories. See: Jones PH, Perun TJ, Rowley EK, Baker EJ. J Med Chem. 1972;15:631–634. doi: 10.1021/jm00276a017.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.