

Abstract
Huntington disease (HD) is a neurodegenerative disorder caused by expansion of a CAG repeat within the Huntingtin (HTT) gene, though the clinical presentation of disease and age-of-onset are strongly influenced by ill-defined environmental factors. We recently reported a gene-environment interaction wherein expression of mutant HTT is associated with neuroprotection against manganese (Mn) toxicity. Here, we are testing the hypothesis that this interaction may be manifested by altered protein expression patterns in striatum, a primary target of both neurodegeneration in HD and neurotoxicity of Mn. To this end we compared striatal proteomes of wild-type and HD (YAC128Q) mice exposed to vehicle or Mn. Principal component analysis of proteomic data revealed that Mn exposure disrupted a segregation of WT versus mutant proteomes by the major principal component in vehicle-exposed mice. Identification of altered proteins revealed novel markers of Mn toxicity, particularly proteins involved in glycolysis, excitotoxicity and cytoskeletal dynamics. In addition, YAC128Q dependent changes suggest that axonal pathology may be an early feature in HD pathogenesis. Finally, for several proteins, genotype-specific responses to Mn were observed. These differences include increased sensitivity to exposure in YAC128Q mice (UBQLN1) and amelioration of some mutant HTT-induced alterations (SAE1, ENO1). We conclude that the interaction of Mn and mutant HTT may suppress proteomic phenotypes of YAC128Q mice, which could reveal potential targets in novel treatment strategies for HD.
Keywords: Huntington disease, Manganese toxicity, gene-environment interaction, striatum, mouse
Introduction
Huntington disease (HD) is an inherited, progressive, neurodegenerative disorder that impairs motor, cognitive, and psychiatric functions. It is manifested by choreiform movements that yield to rigidity and bradykinesia in late stages, as well as by memory deficits, disorientation, anxiety, apathy and depression.1 The principal neuropathological feature of HD is a progressive loss of medium spiny neurons in striatum.2 In later disease stages, continuous striatal degeneration is accompanied by neurodegeneration in other regions, like cortex,3 hypothalamus4 and cerebellum.5
HD is caused by expansion of glutamine-coding CAG triplet repeats within exon 1 of the Huntingtin (HTT) gene.6 The mutation is dominant and the disease symptoms may be observed when the number of CAG repeats exceeds 35, but full penetrance is associated with alleles containing more than 40 repeats.7 It is well established that HD age-of-onset is generally, in an inversely proportional manner, determined by the CAG triplet repeat length. Nevertheless, significant variability is often observed between patients with the same length, what suggests the existence of additional factors that may modify the disease phenotype.8 Analysis of the Venezuelan HD kindreds, the largest HD population in the world, revealed that about 40% of the remaining variability in age-of-onset is attributed to the genetic factors other than mutant HTT, and about 60% to unidentified environmental factors.9 Recently, in order to identify potential environmental factors that may modulate HD pathophysiology, we screened a set of neurotoxic metals for their interaction with mutant HTT in a striatal cell model of HD. We discovered interesting and unexpected gene-environment interaction wherein expression of mutant HTT partially protects the cells against manganese (Mn) neurotoxicity, and decreases intracellular accumulation of this metal. Importantly, we observed a striatal-specific deficit of Mn accumulation in the YAC128Q mouse model of HD following in vivo Mn exposure (three 50 mg/kg MnCl2 × 4 H2O subcutaneous injections over one week).10,11
Manganese, besides being an essential trace element, is neurotoxic when absorbed in excess.12 Prolonged exposure to occupational or environmental Mn leads to manganism, a neurological disorder characterized by symptoms similar to those observed in Parkinson's Disease (PD), including tremor, rigidity, bradykinesia, posture instability and psychosis.13 Experimental data indicate that this motor and behavioral dysfunction is related to disruption of dopaminergic14 and GABAergic15 neurotransmission by Mn. Neuropathological changes in Mn-exposed humans and in animal models of Mn toxicity is found mainly in basal ganglia structures, especially in globus pallidus and striatum, where preferential accumulation of excessive Mn is observed.16 The prominent role of striatum in pathogenesis of both HD and Mn neurotoxicity underlines the importance of striatal-specific interaction between mutant HTT and Mn.
In HD, similar to other neurodegenerative diseases, inappropriate protein folding is a key hallmark of pathogenesis. The expansion of the polyglutamine (polyQ) tract causes misfolding of HTT protein and results in gain of toxic function.17 HTT has numerous interacting partners, and disturbed interactions of these proteins with misfolded HTT may lead to changes in their expression, conformation and function.18 Moreover, accumulation of insoluble mutant HTT with disease progression may inhibit cellular protein quality control mechanisms like autophagy and proteasomes resulting in accumulation of other abnormally folded proteins.19,20 Finally, HTT is involved in gene expression, and transcriptional dysregulation mediated by aberrant interactions of mutant HTT with gene promoters21 and with different transcription factors has been shown to play important role in HD pathogenesis.22,23 All of these events may together lead to changes in the pattern of protein expression, post-translational modifications or conformation that may be investigated using a proteomic approach.
Here, using two-dimensional difference gel electrophoresis (2D DIGE), we compared striatal proteomic profiles between mice expressing mutant HTT and their wild-type (WT) littermates, and analyzed alterations in the patterns of these proteomes caused by a subchronic (1 week) exposure to Mn in order to test the hypothesis that influence of Mn exposure on protein expression may be different between WT and HD animals. We used YAC128Q mice, the transgenic rodent model that replicates many features of HD, like motor impairment, specific striatal and cortical neuropathology, HTT aggregation, and progression of the disease symptoms.24, 25 We decided to perform our analysis using 3 month old animals, because at this age the first subtle HD symptoms are observed,24 as well at this age the striatal Mn uptake deficit is also present.10,11 We anticipate that proteomic alterations found at this age may provide crucial information for understanding the initiation of disease pathogenesis.
Materials and Methods
Animals and experimental design
All experiments were approved by the Vanderbilt University Medical Center Institutional Animal Care and Use Committee, and were designed to minimize animal pain. The FVB-Tg (YAC128)53Hay/J mouse line (YAC128Q)24 was purchased from Jackson Laboratory (#004938, Bar Harbor, ME). These transgenic animals were crossed with WT FVB mice to maintain the line in its original genetic background in order to minimize variations induced by genetic factors other than transgene expression. The mice were genotyped by PCR according to a previously published protocol (#004938; Jackson Laboratory),24 and distributed into experimental groups across multiple litters. For the proteomic study only males were used, but for other experiments the genders in experimental groups were balanced. The exposure paradigm followed a previously published protocol.10,11 Briefly, 12-week-old animals were subcutaneously injected at the hind leg with MnCl2 × 4 H2O (50 mg/kg of body weight) or with vehicle (water, Veh) on the experimental days 0, 3, and 6. The body weight was measured prior to each injection and Mn dose was adjusted accordingly, however, overall no significant differences in the weights were observed over the time of experiment. Twenty-four hours after last injection (day 7, 13-week-old mice) or 3 weeks after last injection (day 28, 16-week-old mice), animals were sacrificed by cervical dislocation, striata and cerebral cortices were immediately dissected, snap frozen in liquid nitrogen and stored in −80°C.
Sample preparation
The samples were prepared by homogenization of tissues in Potter-Elvehjem glass tissue grinders with PTFE pestles (Kontes) in the buffers appropriate to the type of assay. RIPA buffer (50 mM Tris, 150 mM NaCl, 0.1% sodium dodecyl sulfate (SDS), 1% IGEPAL CA-630, 12 mM deoxycholic acid, pH 8.0) was used to prepare the samples for proteomic analysis and western blotting. Buffer used for the samples for global SUMOylation pattern analysis contained N-ethylmaleimide (NEM) that inhibits deSUMOylation of proteins (50 mM Tris, 150 mM NaCl, 1 mM EDTA, 0.1% SDS, 1% Triton-X100, 20 mM NEM; pH 7.4). Samples for enolase activity assay were prepared in PBS containing 0.8% Triton-X100. The buffers were supplemented with 1% protein inhibitor cocktail (Sigma) and 1% phosphatase inhibitor cocktails II and III (Sigma) directly before use. Frozen tissue was transferred to the grinder containing ice-cold buffer, and was immediately homogenized on ice. Homogenate was incubated on ice for 5 minutes, and centrifuged at 14 000 × g, for 10 min., at 4°C. Supernatant was transferred to fresh tube, snap-frozen with liquid nitrogen, and stored in −80°C. Prior to snap-freezing, an aliquot of 5 μl was taken and used for protein content measurement with DC protein assay kit (Bio-Rad)
2D DIGE
RIPA buffer homogenates of striata from 13-week-old males (six animals per each of four experimental groups) were used. The detailed techniques employed in this study were as described previously.26 Briefly, the proteins were minimally labeled with N-hydroxysuccinimide esters dyes (CyDyes). Cy3 and Cy5 were used for labeling of the individual samples, and Cy2 for labeling of internal standard (200 pmol of dye / 100 μg of protein). In order to minimize the effect of fluorophore-dependent variations on the results, three samples in each group were labeled with Cy3 and the other three with Cy5. Mixed internal standard was prepared by combining 50 μg of protein from each sample used in the experiment. Proteins were precipitated27 and the labeling reaction was carried out in labeling buffer (7 M urea, 2 M thiourea, 4% CHAPS, 30 mM Tris, 5 mM magnesium acetate) on ice in the dark for 30 min, and was stopped by 10 min incubation with 10 mM lysine followed by addition of rehydratation buffer (7 M urea, 2 M thiourea, 4% CHAPS, 4 mg/ml DTT). Pairs of Cy3- and Cy5-labeled samples (100 μg each) were combined with an aliquot of the Cy2-labeled internal standard (100 μg), and volume was adjusted to 450 μl with rehydratation buffer and 0.5% IPG buffer pH 4–7 (GE Healthcare). Using this scheme the 24 individual samples were resolved using 12 DIGE gels that were all coordinated by the same Cy2-labeled mixed-sample internal standard.
First dimension separation was performed using Ettan IPGphor isoelectric focusing system (GE Healthcare). The protein samples were applied to the 24-cm IPG strips (ph 4–7) and were subjected to isoelectric focusing according to manufacturer's recommendations. Prior to second dimension separation, the IPG strips were incubated at room temperature (RT) for 20 min in equilibration buffer (30% glycerol, 2% SDS, 6 M urea, 50 mM Tris; pH 8.8) containing 1% DTT and for another 20 min in equilibration buffer supplemented with 2.5% iodoacetamide. Second dimension separation was performed using SDS-polyacrylamide gel electrophoresis (SDS-PAGE). The strips were placed on the top of 12% polyacrylamide gels and electrophoresis was run using Ettan DALT 12 system (GE Healthcare) at <1 W/gel overnight and then at 15 W/gel until the bromophenol blue tracking dye had run off the gel.
Image analysis
The proteins in the gel were visualized at individual CyDye-specific excitation and emission wavelengths, the images were acquired at 100 μm resolution as 16-bit data files using Typhoon 9400 Variable-Mode Gel Imager (GE Healthcare), and the gels were stained with SYPRO Ruby stain (Invitrogen) according to manufacturer's instructions to reveal total protein content. Image analysis was performed using DeCyder 6.5 (GE Healthcare). Cy3 and Cy5 signal intensities were quantified relative to the intensity of Cy2 signal from the pooled standard for each resolved protein feature. The Cy2 values were then used to normalize the Cy3/Cy2 and Cy5/Cy2 ratios across all samples in all gels. 26
Protein identification
The spot features selected for protein identification were robotically picked using Ettan Spot Handling Workstation and in-gel digested with Trypsin Gold (Promega). Resulting peptides were subjected to C18 reverse-phase liquid chromatography coupled in-line with tandem mass spectrometry (LC-MS/MS) using an LTQ linear ion trap tandem mass spectrometer equipped with a MicroAS autosampler and Surveyor HPLC pump, nanospray source, and Xcalibur 2.0 instrument control (ThermoScientific, San Jose, CA). To determine the candidate proteins that formed individual spots of interest in the 2D gels, the tandem MS/MS data were searched against the UniProtKB database (www.uniprot.org) with the taxonomy tag “Mus musculus (Mouse) [10090]” acquired on October 20, 2006, which was concatenated with the database sequences in reverse to enable false-discovery rate calculations, and also contained common laboratory contaminants. Searches were performed using both the Sequest and X! Tandem algorithms, allowing for cysteine carbamidomethylation and partial methionine oxidation.
Results were validated and assembled into protein identifications using Scaffold (version Scaffold_3.1.2, Proteome Software Inc., Portland, OR). Peptide identifications were accepted if they could be established at greater than 95.0% probability as specified by the Peptide Prophet algorithm.28 Protein identifications were accepted if they could be established at greater than 99.0% probability and contained at least 2 identified peptides. Protein probabilities were assigned by the Protein Prophet algorithm.29 Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony.
Western blotting
RIPA homogenates of striata from 13- and 16-week-old mice, and of cortices from 13-week-old mice were used for western blot experiments. Prior to gel loading, the samples were diluted with appropriate buffer to equal protein concentrations, mixed (2:1) with 3 × sample buffer (60 mM Tris, 6% SDS, 30% glycerol, 15% β-mercaptoethanol, 0.015% bromophenol blue, pH 6.8) and incubated at 100°C for 5 min. Large (15 × 22 cm), 8%, 10%, 12% or 15% polyacrylamide gels were used for SDS-PAGE, depending on predicted molecular mass of analyzed protein. The electrophoresis was run overnight at 1250 V × h using CBS Scientific Vertical Electrophoresis System in running buffer (0.1% SDS, 20 mM Tris-HCl, 192 mM glycine). After SDS-PAGE the proteins were electrophoretically transferred from the gel to 0.2 μm pore size Protran nitrocellulose membrane (Whatman) in transfer buffer (20 mM Tris-HCl, 192 mM Glycine, 10% methanol), at 500 mA, at 4°C for at least 3 h, using CBS Scientific Electrophoretic Blotting System. Next, the membrane was stained with Ponceau S (Sigma) to visualize the protein bands before proceeding to the next steps. Ponceau was washed off with TBST buffer (150 mM NaCl, 56 mM Tris-HCl, 44 mM Trizma base, 0.05% Tween-20), and the membrane was blocked with 5% milk in TBST for 3 h at RT. The blocked membranes were incubated with primary antibodies diluted in 5% milk in TBST overnight in RT or for 72 h in 4°C. The following antibodies (source, dilution) were used: rabbit (Rb) × CA2 (Abcam, 1:100,000), Rb × PMM1 (Proteintech, 1:800), Rb × PMM2 (Proteintech, 1:1,000), mouse (Mo) × NFL (Millipore, 1:20,000), Mo × NFM (Millipore, 1:15,000), Rb × NFH (Millipore, 1:10,000), Rb × UBQLN1 (Abcam, 1:2,000), Rb × ENO1 (Abcam, 1:25,000), Rb × ARP3 (Millipore, 1:1,000), Rb × SAE1 (Abcam, 1:500), Mo × actin (Millipore, 1:100,000), Rb × SUMO1 (Cell Signalling, 1:500) and Rb × SUMO2/3 (Cell Signalling, 1:250). Prior to incubations with secondary antibodies the membranes were washed 6 × 5 min. with TBST. Anti-Mo (Jackson Immunoresearch Laboratories or KPL) and anti-Rb secondary antibodies (Jackson Immunoresearch Laboratories or Thermo Fisher Scientific) were used at 1:15,000 dilutions in 5% milk in TBST. After 1.5 h incubation in RT, the membranes were washed 6 × 5 min. with TBST and the blots were visualized with West Dura Extended Duration Chemiluminescent Substrate (Thermo Fisher Scientific). The analysis of the blots was performed using ImageJ (NIH), with the background correction calculated using a signal ratio error model.30 Actin was used as a reference protein for normalization (except SUMOylation assay, where normalization to Ponceau was performed).
Enolase activity assay
PBS/Triton homogenates of striata from 13-week-old mice (6 animals per experimental group) were used for enolase activity analysis. Bioluminescent assay was employed with slight modification to a published protocol,31 where phosphoenolpyruvate (PEP) produced by enolase is used by exogenously added pyruvate kinase for ATP synthesis, that is coupled with luciferin / luciferase system to generate light. Concentrations of the reagents were chosen so that PEP production was the only limiting factor in the three-step assay, thus luminescence was directly proportional to enolase activity. 10 μl of sample was mixed with 10 μl of luciferase reagent (Roche) and 50 μl of reactive mixture (19.68 u/ml pyruvate kinase (Sigma), 2.4 mg/ml bovine serum albumine, 24 mM MgCl2, 2.4 mM AMP, 1.2 mM ADP, 0.48 μM diadenosine pentaphosphate in 100 mM imidazole/acetate buffer, pH 6.8) in black 96-well plate with clear bottom, and luminescence resulting from endogenous PEP and/or ATP was monitored until reaching plateau, using MTX 880 Multimode Detector (Beckman Coulter). 50 μl of 60 mM 2-phosphoglycerate (2-PG) (Sigma) in imidazole buffer was then added and luminescence was measured for 30 min in 75 s intervals with integration time of 400 ms. Enolase activity, expressed as nmol of produced PEP/min/μg of protein, was calculated using standard curve generated with PEP (Sigma) added at known concentrations to the reaction mixture in the absence of sample and 2-PG.
Statistics
The DeCyder v6.5 suite of analytical software tools was used to analyze the DIGE data with univariate Student's t-test and ANOVA, and multivariate Principal Component Analysis (PCA). Detailed parameters used in these analyses are described in the results section. Univariate ANOVA of western blotting and enolase activity experiment was performed using SPSS Statistics 19 (IBM). Student's t-test pairwise comparisons between experimental groups were performed with Excel 2008 (Microsoft).
Results
2D gel electrophoresis
In order to identify proteomic markers of an in vivo interaction between Mn and mutant HTT we compared striatal proteomes of WT and YAC128Q mice exposed subcutaneously to MnCl2 × 4 H2O (50 mg / kg body weight) or vehicle (Veh) (Figure 1). Individual samples, pre-labeled with Cy3 or Cy5, were separated using 2D DIGE. The representative picture of a SYPRO Ruby-stained gel is shown in Figure 2. Signal intensities of each spot were normalized to the corresponding spot from Cy2-labeled mixed-sample internal standard resolved simultaneously in the same gel and were compared across WT-Veh, WT-Mn, YAC-Veh and YAC-Mn experimental groups. To enable analysis of the large dataset as a whole, we performed principal component analysis (PCA), which converts sets of potentially correlated variables into uncorrelated principal components. Because the biological variation appeared to be low in these samples, we limited PCA to the 41 features that differed significantly between experimental groups upon genotype, exposure, and the interaction term of these two factors (univariate two-way ANOVA, p<0.05, at 80% confidence interval). PC1 and PC2, that comprise the greatest sources of variation, accounted for 38.3% and 19.8% of the total variance in the data, respectively. The PC1 × PC2 score plot showed complete separation of the samples from vehicle-treated WT and YAC128Q mice, whereas Mn exposure grouped WT and YAC128Q samples together with strong tendency to form one cluster (only one sample of 6 in YAC-Mn group was found not to cluster with the WT-Mn samples) (Figure 3). The subsequent principal components (PC3–PC5) plotted against PC1 – PC5 failed to distinguish any of the other experimental groups (data not shown).
Figure 1.

Manganese exposure paradigm. Twelve-week-old mice were subcutaneously injected with MnCl2 × 4 H2O (50 mg / kg body weight) or vehicle at exposure day 0, 3 and 6 and sacrificed at day 7 (age of 13 weeks) or day 28 (age of 16 weeks) for further analysis.
Figure 2.

Representative image of one of the twelve DIGE gels used in this analysis (syproRuby post-stained), indicating features numbered 1–21 that were excised for protein identification using LC-MS/MS. First horizontal dimensional separation was by isoelectric focusing (24 cm, pH 4–7), second (vertical) dimensional separation indicates apparent molecular masses between approximately 10 and 200 kDa.
Figure 3.

Statistical analysis of striatal proteomic profiles of 13-week-old wild-type and YAC128Q mice exposed to vehicle or Mn. Forty-one protein spot features which intensities was significantly different between any of the four experimental groups (univariate two-way ANOVA, p<0.05 at 80% confidence interval) were subjected to principal component analysis (PCA). PC1 accounted for 38.3% and PC2 for 19.8% of the data variability. In the control conditions, wild-type (WT-Veh) and YAC128Q (YAC-Veh) samples were clearly separated on the PC1 × PC2 score plot, but Mn exposure made their proteomic profiles inseparable from each other. The grey colored ellipsis surrounding individual experimental groups do not represent any statistical certainty; they were drawn to facilitate visual analysis of the figure.
Protein identification
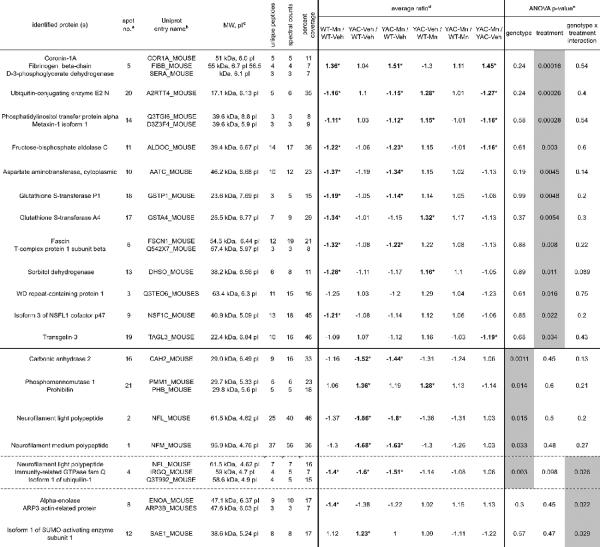
Twenty-one protein features that displayed the strongest (univariate ANOVA, p<0.05 at 95% confidence interval) alterations upon mutant HTT expression, Mn exposure, or exhibited genotype-exposure interaction effects were chosen for protein identification (Figure 2), 19 of which were identified including 6 with more than one protein. (Table I).
Table 1.
Summary of the proteins identified by MS analysis
|
The table lists proteins identified in the spots for which the main effect of exposure (top part of the table), main effect of genotype (middle part of the table) or interaction between genotype and treatment (bottom part of the table) were revealed by univariate two-way ANOVA (p<0.05 at 95% confidence interval).
The numbers correspond to the numbered features shown in Figure 2.
Uniprot KB mouse database (October 20, 2006; www.uniprot.org) was searched.
Theoretical molecular weight (MW) and isoelectric point (pl) were calculated from the database entries, which often contain unmodified precursor sequences not present in the mature form migrating on the gel.
Asterisks mark ratios of significantly different average spot intensities (two-tailed t-test, p<0.05).
Gray highlight indicates main effect or interaction of the factors (p<0.05 at 95% confidence interval, univariate two-way ANOVA)
Importantly, t-test comparisons between individual experimental groups confirmed the alterations of protein expression patterns found by PCA. No differences between YAC-Mn and WT-Mn animals were found for any of identified features, including even the ones for which ANOVA revealed a significant main effect of genotype. Moreover, Mn-induced changes in protein expression compared to basal values in vehicle-exposed animals were more prominent and occurred at greater frequency in WT than in YAC128Q mouse striata. Twelve features differed significantly in their intensity between WT-Veh and WT-Mn, while 5 differed between YAC-Veh and YAC-Mn (t-test, p<0.05).
The main effect of exposure was found for 12 features, comprising 16 identified proteins (Table I, upper part). The predominant effect of Mn in our experimental paradigm was a decrease in an optical density of protein features, since as many as 11 were down-regulated in the striata from Mn-exposed animals comparing to the vehicle-exposed. The only feature showing significant up-regulation by Mn exposure (no. 5) consisted of 3 different proteins (coronin-1A, fibrinogen beta-chain, and D-3-phosphoglycerate dehydrogenase).
Five features were found whose intensities were affected by the genotype (Table I, middle part). For one of these (no. 4), the effect of Mn exposure was found in WT mice, and ANOVA additionally identified a genotype-exposure interaction. This feature contained both immunity-related GTPase family Q and isoform 1 of ubiquilin-1 (UBQLN1), both near the limit of detection (Table I; the presence of NFL in this feature is likely due to the diffusion of this protein from feature no. 2). Similarly to Mn exposure, expression of mutant HTT resulted predominantly in decrease in protein expression (4 down-regulated spots versus 1 up-regulated).
Genotype-exposure interactions were found for 3 features, and as noted above, in one of these (no. 4) the main effect of genotype was also revealed (Table I). In this and in feature no. 8 (alpha-enolase; ENO1 and actin-related protein; ARP3), the interaction consisted of significant decrease in protein expression by Mn exposure in WT mice, and lack of Mn effect in YAC128Q animals. In feature no. 12 (isoform 1 of SUMO-activating enzyme subunit 1; SAE1) the pattern of changes was different – the expression of mutant HTT induced protein level increase, when Mn exposure resulted in strong trend toward down-regulation of protein expression in YAC128Q mice and a loss of difference between YAC128Q and WT animals.
Western blot analysis
We performed a western blot study to confirm a subset of our proteomic findings, and to distinguish which proteins are responsible for the changes observed in features containing more than one protein. We focused on the proteins identified in the features for which genotype-exposure interactions or main effects of genotype were found. For strong and reliable validation of the proteomic data, we used a new set of striatal samples from an independent cohort of mice subjected to the same exposure paradigm. Additionally to striata, cerebral cortices from age-matched animals were analyzed to assay protein expression in subsequent region of neurodegeneration in YAC128Q model of HD.32 Finally, to assess the persistence of changes after Mn cessation, we performed immunoblot analysis of selected proteins in striata from 16-week-old animals, 3 weeks after the last injection (experimental day 28; Figure 1), the time point when excess Mn is completely eliminated from the striata of Mn-exposed YAC128Q, but not yet entirely from Mn-exposed WT mice (about 1.5-fold above basal Mn levels; as opposed to the ~6-fold elevation in striatal Mn levels one day following the last exposure).11
Western blotting confirmed the changes in striatal CA2 at week 13 (decrease in protein expression in YAC128Q animals, and lack of Mn exposure effect), however the magnitude of the changes (decrease by ~20%, p=0.015) was not as great as that found by proteomic analysis (decrease by ~50%, p=0.0011) (Figure 4A). Although no significant alterations in PMM1 or NFM protein levels were detected by immunoblotting in striata at week 13 (Figure 4B, C, E), changes in these proteins trended in the same direction as revealed by 2D DIGE (increased levels of PMM1 in YAC128Q animals, and loss of NFM induced by Mn exposure or mutant HTT expression). Western blot for NFL partially confirmed changes revealed by DIGE (no. 2) – with the main effect of genotype (loss of NFL protein by ~25%, p=0.031), and the statistically significant difference between WT-Veh and YAC-Veh (but not between WT-Veh and YAC-Mn) (Figure 4D). UBQLN1 (no. 4) was similarly found to be altered in expression by western analysis for the occurrence of a genotype effect and a genotype-exposure interaction (Figure 4F), but the expression pattern alterations between the experimental groups were different. This may be due to the fact that another protein, immunity-related GTPase family Q was also found in this feature (as was NFL, likely due to the proximity of this protein in no. 2 nearby). Further experimentation is necessary to determine the relative influence these proteins have on the observed expression changes. SAE1, when measured by western blotting was found to follow closely the pattern of SAE1-positive spot no. 12 in 2D gel (Figure 4I). However, no significant differences were found by western analysis for PMM1 (or the related PMM2 protein), NFM, ENO1 or ARP3 in striata at week 13 (Figure 4B, C, E, G, H).
Figure 4.

Western blot analysis of selected proteins in striatum of 13-week-old mice. A, significant effect of genotype on carbonic anhydrase 2 levels was revealed (F(1,50)=6.36, p=0.015); a - statistically significant difference between WT-Mn and YAC-Veh was found (p=0.025); b - statistically significant difference between WT and YAC was found (p=0.015). B, no significant differences in phosphomannomutase 1 levels were found. C, no significant differences in phosphomannomutase 2 levels were found. D, significant effect of genotype on neurofilament light polypeptide levels was revealed (F(1,33)=5.14, p=0.031); a - statistically significant difference between WT-Veh and YAC-Veh was found (p=0.016); b – statistically significant difference between WT and YAC was found (p=0.022). E, no significant differences in neurofilament medium polypeptide levels were found. F, significant effect of genotype (F(1,50)=13.36, p=0.001) and genotype-exposure interaction (F(1,50)=15.32, p<0.001) on ubiquilin 1 levels were revealed; a, b, c - statistically significant differences between YAC-Mn and WT-Veh (p=0.004), WT-Mn (p<0.001), YAC-Veh (p=0.002) respectively were found; d - statistically significant difference between WT and YAC was found (p=0.004). G, no significant differences in alpha-anolase levels were found. H, no significant differences in actin-related protein 3 levels were found. I, significant effect of genotype-exposure interaction on SUMO-activating enzyme 1 levels was revealed (F(1,35)=4.81, p=0.036); a - statistically significant difference between YAC-Veh and YAC-Mn was found (p=0.016). Data are the means of actin-normalized optical densities ±SEM graphed as percentage of control (WT-Veh) group. The numbers of independent samples used in the experiments are indicated above the graphs. Bars are representing standard error, and the statistically significant differences between the experimental groups are indicated above the error bars (post hoc test; p<0.05).
Analysis of CA2 levels in striatum of 16-week-old animals revealed that the effect of genotype was progressive, since at this time point CA2 protein loss in YAC128Q mice was more pronounced (decrease by ~25%, p=0.006) (Figure 5A). Similarly to 13-week-old mice, no changes in total levels of PMM1, PMM2, or NFM were found by western blot in striata at week 16 (data not shown). In the case of NFL, the effect of genotype in mice at 16 weeks of age (loss of NFL protein by ~20%, p=0.019) (Figure 5B) was similar to the one at 13 weeks of age. Genotype-exposure interaction effects identified for UBQLN1 by western blotting in 3-week-old animals were transient or closely tied to the increased Mn levels in YAC128Q mouse striatum, since it was not present at week 16 (Figure 5C). Similarly, no changes were detected in 16-week-old mouse striatum in the levels of SAE1 (Figure 5D), another protein for which Mn × mutant HTT interactions were found at week 13. Importantly, we also performed western blot analysis for CA2, PMM1, PMM2, NFM, NFL, UBQLN1, ENO1 and SAE1 in cortex of 13-week-old mice, but we found no differences in any of these proteins (data not shown). This suggests that the alterations we identified in striatum may be specific for this region.
Figure 5.

Western blot analysis of selected proteins in striatum of 16-week-old mice. A, significant effect of genotype on carbonic anhydrase 2 levels was revealed (F(1,22)=9.75, p=0.006); a - statistically significant difference between WT-Veh and YAC-Mn was found (p=0.025); b - statistically significant difference between WT-Mn and YAC-Mn was found (p=0.022), c - statistically significant difference between WT and YAC was found (p=0.004). B, significant effect of genotype on neurofilament light polypeptide levels was revealed (F(1,24)=0.019); a - statistically significant difference between WT-Mn and YAC-Mn was found (p=0.023); b- statistically significant difference between WT and YAC was found (p=0.018). C, no significant differences in ubiquilin 1 levels were found. D, no significant differences in SUMO-activating enzyme 1 levels were found. Data are the means of actin-normalized optical densities ±SEM graphed as percentage of control (WT-Veh) group. The numbers of independent samples used in the experiments are indicated above the graphs. Bars are representing standard error, and the statistically significant differences between the experimental groups are indicated above the error bars (post hoc test; p<0.05).
Functional consequences of protein alterations related to Mn × HTT interaction
SAE1 is an enzyme necessary for post-translational modification of proteins by the small ubiquitin-like modifier protein (SUMO),33 we therefore tested if these changes in SAE1 levels may have an effect on protein SUMOylation pattern in striatum of 13-week-old mice. To this end we analyzed striatal lysates by immunoblotting with antibodies against SUMO1 and SUMO2/3 (these two isoforms are immunochemically indistinguishable because of their high homology), but we failed to observe any differences between the experimental groups (Figure 6)
Figure 6.

Global SUMOylation pattern in striatum of 13-week-old mice. No significant differences between experimental groups in the pattern of protein modification by neither SUMO 1 nor SUMO 2/3 were found. Data are the means of optical densities normalized to Ponceau S staining ±SEM graphed as percentage of control (WT-Veh) group. Bars are representing standard error.
To test functional consequences of potential ENO1 alterations revealed by proteomic study in striatum of 13-week-old mice, we measured enzymatic activity of enolase. We found that activity of this enzyme strictly followed the pattern of changes identified in ENO1-positive feature no. 8: a genotype-exposure interaction was detected, as well as significant loss of enzymatic activity in WT-Mn mice compared to WT-Veh. Additionally, we identified decreased activity of enolase in YAC-Veh animals relative to control group (Figure 7). Therefore, our proteomic analysis identified at least one gene-environment interaction affect between Mn-exposure and HD that resulted in a change in functional activity.
Figure 7.

Enzymatic activity of enolase in striatum of 13-week-old mice. Significant effect of genotype-exposure interaction on activity of enolase was revealed (F(1,23)=5.61, p=0.028); a - statistically significant difference between WT-Veh and WT-Mn was found (p=0.031); b - statistically significant difference between WT-Veh and YAC-Veh was found (p=0.01). Data are the means ±SEM of enolase enzymatic activity in six independent samples expressed as nanomoles of phosphoenolpyruvate produced in minute normalized to total protein content. Bars are representing standard error, and the statistically significant differences between the experimental groups are indicated above the error bars (post hoc test; p<0.05).
Discussion
Gene-environment interactions play a substantial role in the pathogenesis of many neurodegenerative diseases.34,35,36 Metals are particularly interesting in this context, because they promote protein misfolding and aggregation, processes that are fundamental in neurodegeneration.37,38,39 HD is caused by mutation in a single gene, HTT, but environmental factors may strongly modulate the age-of-onset and progression of the disease.9 Herein, to test the hypothesis that the neurotoxic metal, Mn, alters protein expression patterns in HD, we investigated striatal proteomes of WT and YAC128Q mice after subchronic exposure to Mn versus vehicle. We also employed western blot analysis to follow up selected 2D DIGE findings. Immunoblotting confirmed some subset of these findings, however usually the differences between experimental groups identified by proteomic approach were stronger than those confirmed by western analysis. This could suggest that 2D DIGE, where the signal is measured after direct labeling of proteins with CyDyes may be more sensitive than western blot. It should be noted, however, that western blot analyses were performed following one dimensional (1D) gel electrophoresis whereas proteomic study employed 2D gel, with protein separation on the basis of both molecular weights (as 1D gel does) and pI. As such many of the proteins may have distributed in 2D gel to multiple spots, if they exist in alternate pI isoforms. Thus a shift in the pI subpopulations, usually due to post-translational modifications, may be manifested as protein spot density differences between experimental groups even when total protein levels are not altered. In such a case, 1D gels followed by western blotting would be unable to identify these kinds of differences.
Subchronic Mn exposure suppresses proteomic phenotype of YAC128Q mice
PCA revealed the separation of the samples on PC1 × PC2 score plot into three clusters – (i) WT-Veh, (ii) YAC-Veh and (iii) WT-Mn together with YAC-Mn. This finding indicates that Mn exposure makes WT and YAC128Q mouse striatal proteomes more similar, while there is a distinct difference between the proteomes of these two genotypes in vehicle exposed animals. To our knowledge this is the first example where interactions of HD with an environmental factor have been investigated using a systems biology approach. Regardless of the general suppression effect of Mn on the YAC128Q proteome, exposure-induced alterations in the proteome of mutant mice were less pronounced than in WT animals. Among the differentially-expressed features analyzed by our proteomics approach, t-tests highlighted 12 that differed between WT-Veh and WT-Mn groups, but only 5 between YAC-Veh and YAC-Mn. This difference may be due to the impaired post-exposure Mn accumulation in striata of YAC128Q that was recently reported by our group.10,11 It should also be noted that the changes induced by Mn and by expression of mutant HTT tended often to the same direction, as was revealed by the PC1 × PC2 score plot. This may suggest that the changes in striatal proteome manifest the strongest possible response to Mn, since the altered proteins are reaching the same point, despite starting from different basal levels in WT comparing to YAC128Q animals. This hypothesis may be supported by the fact that Mn exposure paradigm as used in current study caused very high, about 6-fold, increases in striatal Mn levels.11 It should be noted that the dose used in our experiments is well above typical environmental exposure to Mn in people. However, while usually human exposures have lower doses, they are often chronic. Such exposures are difficult to accurately reproduce in animal models.40 Thus subcutaneous or intraperitoneal injections with doses similar to used in our study are often employed in rodent studies.41,42,43
Subchronic Mn exposure alters proteins related to energy metabolism, cytoskeletal dynamics and glutamatergic neurotransmission
ANOVA identified 12 resolved features altered by Mn exposure that contained a total of 16 identified proteins by mass spectrometry. To our knowledge this is the first proteomic analysis of the effect of Mn exposure on the mammalian neural striatum, and most of the proteins reported here have not previously been found associated with Mn over-exposure. While proteins from a variety of cell physiological pathways were identified, proteins related to either energy metabolism or cellular transport/cytoskeleton stand out in the list. For example, three enzymes involved in glucose (Glc) metabolism were found in DIGE features affected by Mn exposure: fructosebisphosphate aldolase C,44 sorbitol dehydrogenase45 and phosphoglycerate dehydrogenase (PHGDH).46 Likewise, four proteins involved in the regulation of cytoskeletal dynamics were found in the DIGE features altered by Mn exposure (i.e. fascin,47 WD repeat-containing protein,48 coronin-1A,49 and isoform 3 of NSFL1 cofactor p4750). Finally, two proteins related to glutamate (Glu) neurotransmission were identified: cytoplasmic aspartate aminotransferase, known to regulate Glu metabolism in excitatory axon terminals51 and PHGDH, which is a regulatory enzyme for synthesis pathway of two potent glutamatergic NMDA receptor co-agonists – glycine and D-serine.52,53,54 The direction of alterations seen for these two proteins suggest Glu neurotransmission up-regulation in striatum, this could enhance excitotoxicity in concert with other pro-glutamatergic events observed following Mn exposure.55,56 Further work is needed to evaluate the contribution of dysfunction in these pathways to Mn neuotoxicity.
Proteins altered in YAC128Q mice suggest axonal impairment
Several reports have been published in which proteomes of CSF,57 plasma,58 and postmortem brain of HD patients,59,60,61 as well as brain of the R6/2 mouse model of HD59,61,62,63,64,65 have been analyzed. Our study is the first proteomic analysis of YAC128Q mice, a mouse HD model that accurately recapitulates the course of human disease.24
A main effect of genotype was detected for 5 features. The strongest effect was found for CA2 (ANOVA, p=0.0011). Importantly, we confirmed these proteomic findings by western blot analysis, and found that loss of this protein in striatum is progressive, since in 16-week-old animals the effect was stronger comparing to 13-week-old mice. Western blot analysis of cortical CA2 revealed no differences between WT and YAC128Q animals, suggesting that the genotype effect is specific to striatum, the primary region of degeneration in HD. CA2 is the most abundant brain isoform of carbonic anhydrase, an enzyme responsible for maintenance of equilibrium between bicarbonate and carbon dioxide.66 CA2 is expressed in glial cells, mainly in oligodendrocytes,67, and its loss is known to precede demyelination.68 In HD patients, myelin breakdown in striata is observed early before symptom onset.69 CA2 decrease found in our study may be thus a marker of early demyelination. Besides myelin formation, CA2 has been demonstrated to play important role in several cellular processes: maintenance of pH, expiration of CO2, and regulation of H2 and ion equilibrium.66,70 Loss of CA2 may deregulate these crucial physiological processes and contribute to HD pathogenesis. Importantly, decrease in CA2 transcript,71 and protein64 has been found in brain of R6/2 mice. Interestingly, alterations in CA2 protein levels, post-translational modifications and enzymatic activity were found in Alzheimer's Disease (AD) and Parkinson's Disease patients and mouse models,72,73,74 what indicates that this protein may be a general marker of neurodegeneration.
PMM1 was found in the feature that was up-regulated in YAC128Q mice (Table I), but western blot analysis failed to find any differences in total protein levels of either PMM1 or the structurally related PMM2 enzyme between the experimental groups. It is not unexpected that alterations detected by DIGE on resolved protein isoforms may not be reflected in a western analysis that is directed against many/all forms of a given protein. PMM1 is an enzyme responsible for hydrolysis of Glc-1,6-bisphosphate,75 a cofactor of variety of sugar metabolism enzymes,76,77,78 when PMM2 catalyzes production of mannose-1-phosphate, the intermediate in the pathway of synthesis of GDP-mannose, a substrate for protein glycosyltransferases.79 Since western blot analysis didn't confirm the proteomic findings, further studies are required to identify the role of PMM(s) in HD pathogenesis.
Finally, we identified axonal cytoskeletal proteins NFM and NFL as down-regulated in YAC128Q mice. Loss of NFL (but not of NFM) was confirmed by western analysis in striatum of both 13- and 16-week-old mice. Neurofilaments (NFs) are abundant in neurons, where they provide mechanical support and regulate diameter of axons.80 Several reports demonstrate negative effect of mutant HTT on NFs. It was shown that expanded polyQ domain binds to NF and disassemble its network.81 Other reports show loss of NFL in R6/2 HD mouse brain in rat primary cell cultures transfected with polyQ-expanded N-terminal fragment of HTT.82 NF network disruption may affect axonal integrity and contribute to the processes that play prominent role in the disease pathogenesis, like impairment of axonal transport and neurotransmission.
Summarizing, we have shown loss of oligodendrocyte/myelin protein, CA2 and axonal proteins, NFs in striata of 13-week-old YAC128Q mice, this suggests that axonal pathology may be an early step in HD pathogenesis.
Alterations specific to Mn × mutant HTT interaction in mouse striatum may ameliorate some of HD-related cellular abnormalities
Three features containing six proteins were affected by genotype-exposure interaction in our experimental paradigm. Further analysis suggested that three of these proteins - UBQLN1, ENO1 and SAE1, each representing different features, were the most probable candidates for markers of mutant HTT × Mn exposure interaction.
UBQLN1 belongs to the family of proteins involved in protein degradation. Ubiquilins recognize ubiqutylated proteins and deliver them to the proteasome for degradation.83. Moreover, ubiquilin is involved in endoplasmic reticulum-associated protein degradation,84 and in macroautophagy.85 Here, we identified an effect of Mn × HTT interaction on UBQLN1. Western blot analysis confirmed occurrence of the interaction, but revealed different pattern of changes comparing to the DIGE results. This difference was likely due to the fact that this feature also contained immunity-related GTPase family Q protein, and both of these proteins were found near the limit of detection using mass spectrometry. These changes in total UBQLN1 are interesting in the context of cellular role of UBQLN1 in neurodegenerative diseases. It has been shown that polymorphisms in the UBQLN1 gene are associated with AD,86 and that UBQLN1 protein interacts with protein aggregates in different neurodegenerative diseases87,88 and finally with HTT aggregates in cellular model of HD and in R6/2 mice.89 Moreover, it was shown that UBQLN1 overexpression reduces aggregate formation and cell death in the cells transfected with polyQ HTT, and diminishes motility defect in C. elegans expressing mutant HTT.90 Increase in UBQLN1 levels following Mn exposure specifically in YAC128Q mouse striatum may be therefore interesting neuroprotective mechanism that can facilitate degradation of mutant HTT.
ENO1 and ARP3 did not show differences in total levels between experimental groups by western analysis. Further analysis, however, revealed that enzymatic activity of enolase followed the pattern of changes found by DIGE (i.e. decrease in WT-Mn and YAC-Veh but not in YAC-Mn mice comparing to WT-Veh animals). The enolase activity assay measures the sum of activities of both enolase isoforms – non-neuronal ENO1 and neuron-specific ENO2. ENO1, however is more abundant in brain,91 but less stable in its active, dimeric form and more sensitive to different inactivating conditions,92 therefore ENO2 activity may be negligible comparing to ENO1 and/or not affected in our experimental conditions. The lack of alterations found by western analysis indicate that the identified feature likely contained only the active form of the enzyme, and that both Mn exposure and mutant HTT expression affected equilibrium between active and inactive forms of ENO1 rather than total protein content. ENO1, a glycolytic enzyme catalyzing conversion of 2-phosphoglycerate to phosphoenolpyruvate, in CNS is expressed predominantly in astrocytes.93 Down-regulation of ENO1 caused by either Mn exposure or expression of mutant HTT may indicate decreased glycolytic activity in striatal astrocytes in both conditions. A Mn effect on ENO1 is in agreement with our hypothesis that glycolysis may be prominent factor in Mn toxicity and it underlies the role of astrocytes in this mechanism.
SAE1 is a component of the E1 heterodimer that is responsible for activation of SUMO protein - the first step in SUMOylation pathway,33 a post-translational modification that regulates different cellular processes including transcription, cellular transport, apoptosis, stress response, neuronal development and synaptic transmission.94 Our proteomic analysis showed that mutant HTT expression resulted in increased levels of SAE1. While western analysis did not confirm this finding (though trended in the same direct), a gene-environment interaction was observed by both proteomic and western analyses. This interaction was manifested as a loss of SAE1 protein after Mn exposure in YAC128Q mice, in the absence of an effect in WT animals. It is known that SUMOylation may be controlled by regulation of SAE1 expression,95 and that global SUMOylation is elevated in different stress conditions.96 Nevertheless, we failed to find any differences in striatal protein SUMOylation pattern between the experimental groups. This is in contrast to findings by others that the SUMO1 pathway is activated in affected brain regions in the patients suffering from different polyQ diseases (including HD).97 It was also shown that overexpression of SAE1 partner, SAE2 enhances neurodegenerative phenotype in fly model of other polyQ disease, spinobulbar muscular atrophy.98 Moreover, experiments by Steffan et al revealed that mutant HTT may be SUMOylated, which decreases its clearance and enhances degenerative phenotype.99 All these data suggest that SUMO pathway may contribute to polyQ-dependent neurodegeneration, therefore down-regulation of SAE1 by Mn in striatum of YAC128Q may be potentially neuroprotective.
Conclusions
We employed 2D DIGE followed by mass spectrometry to identify markers of subchronic Mn exposure, presymptomatic HD and interaction of these two conditions in mouse striatum. We found that Mn induces protein alterations that may promote inhibition of glycolysis and energy metabolism, support excitotoxicity and cause dysregulation of cytoskeletal integrity. The findings in YAC128Q mice suggest that axonal degeneration may be early pathological event in HD. Interestingly, despite a weaker effect of Mn exposure on proteome of YAC128Q mice comparing to WT animals, Mn suppress the striatal proteomic signature of early HD. Protein changes specific for YAC128Q mouse striatum following Mn exposure may potentially ameliorate different neurodegeneration pathways known to contribute to HD pathogenesis. Thorough examination of the mechanisms responsible for this genotype-exposure interaction may reveal new strategies of early intervention in HD.
Acknowledgments
This work was supported by NIH grant ES016931 and NICHD grant P30HD15052. The authors would like to thank Sarah Stuart and Christopher Jetter for excellent technical assistance. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH or NICHD.
References
- 1.Walker FO. Huntington's disease. Lancet. 2007;369(9557):218–28. doi: 10.1016/S0140-6736(07)60111-1. [DOI] [PubMed] [Google Scholar]
- 2.Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP., Jr. Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol. 1985;44(6):559–77. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- 3.Hedreen JC, Peyser CE, Folstein SE, Ross CA. Neuronal loss in layers V and VI of cerebral cortex in Huntington's disease. Neurosci Lett. 1991;133(2):257–61. doi: 10.1016/0304-3940(91)90583-f. [DOI] [PubMed] [Google Scholar]
- 4.Kremer HP, Roos RA, Dingjan G, Marani E, Bots GT. Atrophy of the hypothalamic lateral tuberal nucleus in Huntington's disease. J Neuropathol Exp Neurol. 1990;49(4):371–82. doi: 10.1097/00005072-199007000-00002. [DOI] [PubMed] [Google Scholar]
- 5.Rodda RA. Cerebellar atrophy in Huntington's disease. J Neurol Sci. 1981;50(1):147–57. doi: 10.1016/0022-510x(81)90049-6. [DOI] [PubMed] [Google Scholar]
- 6.A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell. 1993;72(6):971–83. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 7.Langbehn DR, Brinkman RR, Falush D, Paulsen JS, Hayden MR. A new model for prediction of the age of onset and penetrance for Huntington's disease based on CAG length. Clin Genet. 2004;65(4):267–77. doi: 10.1111/j.1399-0004.2004.00241.x. [DOI] [PubMed] [Google Scholar]
- 8.Andrew SE, Goldberg YP, Kremer B, Telenius H, Theilmann J, Adam S, Starr E, Squitieri F, Lin B, Kalchman MA, et al. The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington's disease. Nat Genet. 1993;4(4):398–403. doi: 10.1038/ng0893-398. [DOI] [PubMed] [Google Scholar]
- 9.Wexler NS, Lorimer J, Porter J, Gomez F, Moskowitz C, Shackell E, Marder K, Penchaszadeh G, Roberts SA, Gayan J, Brocklebank D, Cherny SS, Cardon LR, Gray J, Dlouhy SR, Wiktorski S, Hodes ME, Conneally PM, Penney JB, Gusella J, Cha JH, Irizarry M, Rosas D, Hersch S, Hollingsworth Z, MacDonald M, Young AB, Andresen JM, Housman DE, De Young MM, Bonilla E, Stillings T, Negrette A, Snodgrass SR, Martinez-Jaurrieta MD, Ramos-Arroyo MA, Bickham J, Ramos JS, Marshall F, Shoulson I, Rey GJ, Feigin A, Arnheim N, Acevedo-Cruz A, Acosta L, Alvir J, Fischbeck K, Thompson LM, Young A, Dure L, O'Brien CJ, Paulsen J, Brickman A, Krch D, Peery S, Hogarth P, Higgins DS, Jr., Landwehrmeyer B. Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington's disease age of onset. Proc Natl Acad Sci U S A. 2004;101(10):3498–503. doi: 10.1073/pnas.0308679101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Williams BB, Li D, Wegrzynowicz M, Vadodaria BK, Anderson JG, Kwakye GF, Aschner M, Erikson KM, Bowman AB. Disease-toxicant screen reveals a neuroprotective interaction between Huntington's disease and manganese exposure. J Neurochem. 2010;112(1):227–37. doi: 10.1111/j.1471-4159.2009.06445.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Madison JL, Wegrzynowicz M, Aschner M, Bowman AB. Early changes in medium spiny neuron morphology and striatal neurochemistry in Huntington disease mice are influenced by Mn exposure. 2011 submitted. [Google Scholar]
- 12.Aschner M, Guilarte TR, Schneider JS, Zheng W. Manganese: recent advances in understanding its transport and neurotoxicity. Toxicol Appl Pharmacol. 2007;221(2):131–47. doi: 10.1016/j.taap.2007.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perl DP, Olanow CW. The neuropathology of manganese-induced Parkinsonism. J Neuropathol Exp Neurol. 2007;66(8):675–82. doi: 10.1097/nen.0b013e31812503cf. [DOI] [PubMed] [Google Scholar]
- 14.Bird ED, Anton AH, Bullock B. The effect of manganese inhalation on basal ganglia dopamine concentrations in rhesus monkey. Neurotoxicology. 1984;5(1):59–65. [PubMed] [Google Scholar]
- 15.Gianutsos G, Murray MT. Alterations in brain dopamine and GABA following inorganic or organic manganese administration. Neurotoxicology. 1982;3(3):75–81. [PubMed] [Google Scholar]
- 16.Yamada M, Ohno S, Okayasu I, Okeda R, Hatakeyama S, Watanabe H, Ushio K, Tsukagoshi H. Chronic manganese poisoning: a neuropathological study with determination of manganese distribution in the brain. Acta Neuropathol. 1986;70(3–4):273–8. doi: 10.1007/BF00686083. [DOI] [PubMed] [Google Scholar]
- 17.Tobin AJ, Signer ER. Huntington's disease: the challenge for cell biologists. Trends Cell Biol. 2000;10(12):531–6. doi: 10.1016/s0962-8924(00)01853-5. [DOI] [PubMed] [Google Scholar]
- 18.Li SH, Li XJ. Huntingtin-protein interactions and the pathogenesis of Huntington's disease. Trends Genet. 2004;20(3):146–54. doi: 10.1016/j.tig.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 19.Bennett EJ, Bence NF, Jayakumar R, Kopito RR. Global impairment of the ubiquitinproteasome system by nuclear or cytoplasmic protein aggregates precedes inclusion body formation. Mol Cell. 2005;17(3):351–65. doi: 10.1016/j.molcel.2004.12.021. [DOI] [PubMed] [Google Scholar]
- 20.Martinez-Vicente M, Talloczy Z, Wong E, Tang G, Koga H, Kaushik S, de Vries R, Arias E, Harris S, Sulzer D, Cuervo AM. Cargo recognition failure is responsible for inefficient autophagy in Huntington's disease. Nat Neurosci. 2010;13(5):567–76. doi: 10.1038/nn.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Benn CL, Sun T, Sadri-Vakili G, McFarland KN, DiRocco DP, Yohrling GJ, Clark TW, Bouzou B, Cha JH. Huntingtin modulates transcription, occupies gene promoters in vivo, and binds directly to DNA in a polyglutamine-dependent manner. J Neurosci. 2008;28(42):10720–33. doi: 10.1523/JNEUROSCI.2126-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boutell JM, Thomas P, Neal JW, Weston VJ, Duce J, Harper PS, Jones AL. Aberrant interactions of transcriptional repressor proteins with the Huntington's disease gene product, huntingtin. Hum Mol Genet. 1999;8(9):1647–55. doi: 10.1093/hmg/8.9.1647. [DOI] [PubMed] [Google Scholar]
- 23.Zhai W, Jeong H, Cui L, Krainc D, Tjian R. In vitro analysis of huntingtin-mediated transcriptional repression reveals multiple transcription factor targets. Cell. 2005;123(7):1241–53. doi: 10.1016/j.cell.2005.10.030. [DOI] [PubMed] [Google Scholar]
- 24.Slow EJ, van Raamsdonk J, Rogers D, Coleman SH, Graham RK, Deng Y, Oh R, Bissada N, Hossain SM, Yang YZ, Li XJ, Simpson EM, Gutekunst CA, Leavitt BR, Hayden MR. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum Mol Genet. 2003;12(13):1555–67. doi: 10.1093/hmg/ddg169. [DOI] [PubMed] [Google Scholar]
- 25.Van Raamsdonk JM, Pearson J, Slow EJ, Hossain SM, Leavitt BR, Hayden MR. Cognitive dysfunction precedes neuropathology and motor abnormalities in the YAC128 mouse model of Huntington's disease. J Neurosci. 2005;25(16):4169–80. doi: 10.1523/JNEUROSCI.0590-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Franco AT, Friedman DB, Nagy TA, Romero-Gallo J, Krishna U, Kendall A, Israel DA, Tegtmeyer N, Washington MK, Peek RM., Jr. Delineation of a carcinogenic Helicobacter pylori proteome. Mol Cell Proteomics. 2009;8(8):1947–58. doi: 10.1074/mcp.M900139-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wessel D, Flugge UI. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal Biochem. 1984;138(1):141–143. doi: 10.1016/0003-2697(84)90782-6. [DOI] [PubMed] [Google Scholar]
- 28.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem. 2002;74(20):5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 29.Nesvizhskii AI, Keller A, Kolker E, Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem. 2003;75(17):4646–4658. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- 30.Kreutz C, Bartolome Rodriguez MM, Maiwald T, Seidl M, Blum HE, Mohr L, Timmer J. An error model for protein quantification. Bioinformatics. 2007;23(20):2747–53. doi: 10.1093/bioinformatics/btm397. [DOI] [PubMed] [Google Scholar]
- 31.Wevers RA, Jacobs AA, Hommes OR. A bioluminescent assay for enolase (EC 4.2.1.11) activity in human serum and cerebrospinal fluid. Clin Chim Acta. 1983;135(2):159–68. doi: 10.1016/0009-8981(83)90131-6. [DOI] [PubMed] [Google Scholar]
- 32.Van Raamsdonk JM, Murphy Z, Slow EJ, Leavitt BR, Hayden MR. Selective degeneration and nuclear localization of mutant huntingtin in the YAC128 mouse model of Huntington disease. Hum Mol Genet. 2005;14(24):3823–35. doi: 10.1093/hmg/ddi407. [DOI] [PubMed] [Google Scholar]
- 33.Lois LM, Lima CD. Structures of the SUMO E1 provide mechanistic insights into SUMO activation and E2 recruitment to E1. EMBO J. 2005;24(3):439–51. doi: 10.1038/sj.emboj.7600552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Manolakou K, Beaton J, McConnell I, Farquar C, Manson J, Hastie ND, Bruce M, Jackson IJ. Genetic and environmental factors modify bovine spongiform encephalopathy incubation period in mice. Proc Natl Acad Sci U S A. 2001;98(13):7402–7. doi: 10.1073/pnas.121172098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dick FD, De Palma G, Ahmadi A, Osborne A, Scott NW, Prescott GJ, Bennett J, Semple S, Dick S, Mozzoni P, Haites N, Wettinger SB, Mutti A, Otelea M, Seaton A, Soderkvist P, Felice A. Gene-environment interactions in parkinsonism and Parkinson's disease: the Geoparkinson study. Occup Environ Med. 2007;64(10):673–80. doi: 10.1136/oem.2006.032078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ghebranious N, Mukesh B, Giampietro PF, Glurich I, Mickel SF, Waring SC, McCarty CA. A pilot study of gene/gene and gene/environment interactions in Alzheimer disease. Clin Med Res. 2011;9(1):17–25. doi: 10.3121/cmr.2010.894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mizoroki T, Meshitsuka S, Maeda S, Murayama M, Sahara N, Takashima A. Aluminum induces tau aggregation in vitro but not in vivo. J Alzheimers Dis. 2007;11(4):419–27. doi: 10.3233/jad-2007-11401. [DOI] [PubMed] [Google Scholar]
- 38.Binolfi A, Lamberto GR, Duran R, Quintanar L, Bertoncini CW, Souza JM, Cervenansky C, Zweckstetter M, Griesinger C, Fernandez CO. Site-specific interactions of Cu(II) with alpha and beta-synuclein: bridging the molecular gap between metal binding and aggregation. J Am Chem Soc. 2008;130(35):11801–12. doi: 10.1021/ja803494v. [DOI] [PubMed] [Google Scholar]
- 39.Mo ZY, Zhu YZ, Zhu HL, Fan JB, Chen J, Liang Y. Low micromolar zinc accelerates the fibrillization of human tau via bridging of Cys-291 and Cys-322. J Biol Chem. 2009;284(50):34648–57. doi: 10.1074/jbc.M109.058883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lucchini RG, Martin CJ, Doney BC. From manganism to manganese-induced parkinsonism: a conceptual model based on the evolution of exposure. Neuromolecular Med. 2009;11(4):311–21. doi: 10.1007/s12017-009-8108-8. [DOI] [PubMed] [Google Scholar]
- 41.Gianutsos G, Seltzer MD, Saymeh R, Wu ML, Michel RG. Brain manganese accumulation following systemic administration of different forms. Arch Toxicol. 1985;57(4):272–5. doi: 10.1007/BF00324791. [DOI] [PubMed] [Google Scholar]
- 42.Milatovic D, Zaja-Milatovic S, Gupta RC, Yu Y, Aschner M. Oxidative damage and neurodegeneration in manganese-induced neurotoxicity. Toxicol Appl Pharmacol. 2009;240(2):219–25. doi: 10.1016/j.taap.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dodd CA, Ward DL, Klein BG. Basal Ganglia accumulation and motor assessment following manganese chloride exposure in the C57BL/6 mouse. Int J Toxicol. 2005;24(6):389–97. doi: 10.1080/10915810500366500. [DOI] [PubMed] [Google Scholar]
- 44.Kusakabe T, Motoki K, Hori K. Human aldolase C: characterization of the recombinant enzyme expressed in Escherichia coli. J Biochem. 1994;115(6):1172–7. doi: 10.1093/oxfordjournals.jbchem.a124475. [DOI] [PubMed] [Google Scholar]
- 45.Jeffery J, Jornvall H. Enzyme relationships in a sorbitol pathway that bypasses glycolysis and pentose phosphates in glucose metabolism. Proc Natl Acad Sci U S A. 1983;80(4):901–5. doi: 10.1073/pnas.80.4.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Greenberg DM, Ichihara A. Further studies on the pathway of serine formation from carbohydrate. J Biol Chem. 1957;224(1):331–40. [PubMed] [Google Scholar]
- 47.Yamashiro S, Yamakita Y, Ono S, Matsumura F. Fascin, an actin-bundling protein, induces membrane protrusions and increases cell motility of epithelial cells. Mol Biol Cell. 1998;9(5):993–1006. doi: 10.1091/mbc.9.5.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rodal AA, Tetreault JW, Lappalainen P, Drubin DG, Amberg DC. Aip1p interacts with cofilin to disassemble actin filaments. J Cell Biol. 1999;145(6):1251–64. doi: 10.1083/jcb.145.6.1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ahmed Z, Shaw G, Sharma VP, Yang C, McGowan E, Dickson DW. Actin-binding proteins coronin-1a and IBA-1 are effective microglial markers for immunohistochemistry. J Histochem Cytochem. 2007;55(7):687–700. doi: 10.1369/jhc.6A7156.2007. [DOI] [PubMed] [Google Scholar]
- 50.Uchiyama K, Kondo H. p97/p47-Mediated biogenesis of Golgi and ER. J Biochem. 2005;137(2):115–9. doi: 10.1093/jb/mvi028. [DOI] [PubMed] [Google Scholar]
- 51.Salganicoff L, Derobertis E. Subcellular Distribution of the Enzymes of the Glutamic Acid, Glutamine and Gamma-Aminobutyric Acid Cycles in Rat Brain. J Neurochem. 1965;12:287–309. doi: 10.1111/j.1471-4159.1965.tb06766.x. [DOI] [PubMed] [Google Scholar]
- 52.Klomp LW, de Koning TJ, Malingre HE, van Beurden EA, Brink M, Opdam FL, Duran M, Jaeken J, Pineda M, Van Maldergem L, Poll-The BT, van den Berg IE, Berger R. Molecular characterization of 3-phosphoglycerate dehydrogenase deficiency--a neurometabolic disorder associated with reduced L-serine biosynthesis. Am J Hum Genet. 2000;67(6):1389–99. doi: 10.1086/316886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang JH, Wada A, Yoshida K, Miyoshi Y, Sayano T, Esaki K, Kinoshita MO, Tomonaga S, Azuma N, Watanabe M, Hamase K, Zaitsu K, Machida T, Messing A, Itohara S, Hirabayashi Y, Furuya S. Brain-specific Phgdh deletion reveals a pivotal role for L-serine biosynthesis in controlling the level of D-serine, an N-methyl-D-aspartate receptor co-agonist, in adult brain. J Biol Chem. 2010;285(53):41380–90. doi: 10.1074/jbc.M110.187443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Henneberger C, Papouin T, Oliet SH, Rusakov DA. Long-term potentiation depends on release of D-serine from astrocytes. Nature. 2010;463(7278):232–6. doi: 10.1038/nature08673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Crooks DR, Welch N, Smith DR. Low-level manganese exposure alters glutamate metabolism in GABAergic AF5 cells. Neurotoxicology. 2007;28(3):548–54. doi: 10.1016/j.neuro.2007.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee ES, Sidoryk M, Jiang H, Yin Z, Aschner M. Estrogen and tamoxifen reverse manganese-induced glutamate transporter impairment in astrocytes. J Neurochem. 2009;110(2):530–44. doi: 10.1111/j.1471-4159.2009.06105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fang Q, Strand A, Law W, Faca VM, Fitzgibbon MP, Hamel N, Houle B, Liu X, May DH, Poschmann G, Roy L, Stuhler K, Ying W, Zhang J, Zheng Z, Bergeron JJ, Hanash S, He F, Leavitt BR, Meyer HE, Qian X, McIntosh MW. Brain-specific proteins decline in the cerebrospinal fluid of humans with Huntington disease. Mol Cell Proteomics. 2009;8(3):451–66. doi: 10.1074/mcp.M800231-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dalrymple A, Wild EJ, Joubert R, Sathasivam K, Bjorkqvist M, Petersen A, Jackson GS, Isaacs JD, Kristiansen M, Bates GP, Leavitt BR, Keir G, Ward M, Tabrizi SJ. Proteomic profiling of plasma in Huntington's disease reveals neuroinflammatory activation and biomarker candidates. J Proteome Res. 2007;6(7):2833–40. doi: 10.1021/pr0700753. [DOI] [PubMed] [Google Scholar]
- 59.Zabel C, Klose J. Influence of Huntington's disease on the human and mouse proteome. Int Rev Neurobiol. 2004;61:241–83. doi: 10.1016/S0074-7742(04)61010-5. [DOI] [PubMed] [Google Scholar]
- 60.Sorolla MA, Reverter-Branchat G, Tamarit J, Ferrer I, Ros J, Cabiscol E. Proteomic and oxidative stress analysis in human brain samples of Huntington disease. Free Radic Biol Med. 2008;45(5):667–78. doi: 10.1016/j.freeradbiomed.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 61.Zabel C, Chamrad DC, Priller J, Woodman B, Meyer HE, Bates GP, Klose J. Alterations in the mouse and human proteome caused by Huntington's disease. Mol Cell Proteomics. 2002;1(5):366–75. doi: 10.1074/mcp.m200016-mcp200. [DOI] [PubMed] [Google Scholar]
- 62.Zabel C, Sagi D, Kaindl AM, Steireif N, Klare Y, Mao L, Peters H, Wacker MA, Kleene R, Klose J. Comparative proteomics in neurodegenerative and non-neurodegenerative diseases suggest nodal point proteins in regulatory networking. J Proteome Res. 2006;5(8):1948–58. doi: 10.1021/pr0601077. [DOI] [PubMed] [Google Scholar]
- 63.Zabel C, Mao L, Woodman B, Rohe M, Wacker MA, Klare Y, Koppelstatter A, Nebrich G, Klein O, Grams S, Strand A, Luthi-Carter R, Hartl D, Klose J, Bates GP. A large number of protein expression changes occur early in life and precede phenotype onset in a mouse model for huntington disease. Mol Cell Proteomics. 2009;8(4):720–34. doi: 10.1074/mcp.M800277-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu X, Miller BR, Rebec GV, Clemmer DE. Protein expression in the striatum and cortex regions of the brain for a mouse model of Huntington's disease. J Proteome Res. 2007;6(8):3134–42. doi: 10.1021/pr070092s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Perluigi M, Poon HF, Maragos W, Pierce WM, Klein JB, Calabrese V, Cini C, De Marco C, Butterfield DA. Proteomic analysis of protein expression and oxidative modification in r6/2 transgenic mice: a model of Huntington disease. Mol Cell Proteomics. 2005;4(12):1849–61. doi: 10.1074/mcp.M500090-MCP200. [DOI] [PubMed] [Google Scholar]
- 66.Sly WS, Hu PY. Human carbonic anhydrases and carbonic anhydrase deficiencies. Annu Rev Biochem. 1995;64:375–401. doi: 10.1146/annurev.bi.64.070195.002111. [DOI] [PubMed] [Google Scholar]
- 67.Cammer W, Bieler L, Fredman T, Norton WT. Quantitation of myelin carbonic anhydrase-development and subfractionation of rat brain myelin and comparison with myelin from other species. Brain Res. 1977;138(1):17–28. doi: 10.1016/0006-8993(77)90781-8. [DOI] [PubMed] [Google Scholar]
- 68.Komoly S, Jeyasingham MD, Pratt OE, Lantos PL. Decrease in oligodendrocyte carbonic anhydrase activity preceding myelin degeneration in cuprizone induced demyelination. J Neurol Sci. 1987;79(1–2):141–8. doi: 10.1016/0022-510x(87)90268-1. [DOI] [PubMed] [Google Scholar]
- 69.Bartzokis G, Lu PH, Tishler TA, Fong SM, Oluwadara B, Finn JP, Huang D, Bordelon Y, Mintz J, Perlman S. Myelin breakdown and iron changes in Huntington's disease: pathogenesis and treatment implications. Neurochem Res. 2007;32(10):1655–64. doi: 10.1007/s11064-007-9352-7. [DOI] [PubMed] [Google Scholar]
- 70.Sun MK, Alkon DL. Carbonic anhydrase gating of attention: memory therapy and enhancement. Trends Pharmacol Sci. 2002;23(2):83–9. doi: 10.1016/s0165-6147(02)01899-0. [DOI] [PubMed] [Google Scholar]
- 71.Luthi-Carter R, Strand A, Peters NL, Solano SM, Hollingsworth ZR, Menon AS, Frey AS, Spektor BS, Penney EB, Schilling G, Ross CA, Borchelt DR, Tapscott SJ, Young AB, Cha JH, Olson JM. Decreased expression of striatal signaling genes in a mouse model of Huntington's disease. Hum Mol Genet. 2000;9(9):1259–71. doi: 10.1093/hmg/9.9.1259. [DOI] [PubMed] [Google Scholar]
- 72.Sultana R, Perluigi M, Newman SF, Pierce WM, Cini C, Coccia R, Butterfield DA. Redox proteomic analysis of carbonylated brain proteins in mild cognitive impairment and early Alzheimer's disease. Antioxid Redox Signal. 2010;12(3):327–36. doi: 10.1089/ars.2009.2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sultana R, Poon HF, Cai J, Pierce WM, Merchant M, Klein JB, Markesbery WR, Butterfield DA. Identification of nitrated proteins in Alzheimer's disease brain using a redox proteomics approach. Neurobiol Dis. 2006;22(1):76–87. doi: 10.1016/j.nbd.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 74.Poon HF, Frasier M, Shreve N, Calabrese V, Wolozin B, Butterfield DA. Mitochondrial associated metabolic proteins are selectively oxidized in A30P alpha-synuclein transgenic mice--a model of familial Parkinson's disease. Neurobiol Dis. 2005;18(3):492–8. doi: 10.1016/j.nbd.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 75.Veiga-da-Cunha M, Vleugels W, Maliekal P, Matthijs G, Van Schaftingen E. Mammalian phosphomannomutase PMM1 is the brain IMP-sensitive glucose-1,6-bisphosphatase. J Biol Chem. 2008;283(49):33988–93. doi: 10.1074/jbc.M805224200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Krzanowski J, Matschinsky FM. Regulation of phosphofructokinase by phosphocreatine and phosphorylated glycolytic intermediates. Biochem Biophys Res Commun. 1969;34(6):816–23. doi: 10.1016/0006-291x(69)90253-8. [DOI] [PubMed] [Google Scholar]
- 77.Beitner R, Haberman S, Livni L. Complementarity in the regulation of phosphoglucomutase, phosphofructokinase and hexokinase; the role of glucose 1,6-bisphosphate. Biochim Biophys Acta. 1975;397(2):355–69. doi: 10.1016/0005-2744(75)90125-4. [DOI] [PubMed] [Google Scholar]
- 78.Koster JF, Slee RG, Staal GE, van Berkel TJ. The influence of glucose I,6-diphosphate on the enzymatic activity of pyruvate kinase. Biochim Biophys Acta. 1972;258(3):763–8. doi: 10.1016/0005-2744(72)90177-5. [DOI] [PubMed] [Google Scholar]
- 79.Hansen SH, Frank SR, Casanova JE. Cloning and characterization of human phosphomannomutase, a mammalian homologue of yeast SEC53. Glycobiology. 1997;7(6):829–34. doi: 10.1093/glycob/7.6.829. [DOI] [PubMed] [Google Scholar]
- 80.Nguyen MD, Lariviere RC, Julien JP. Reduction of axonal caliber does not alleviate motor neuron disease caused by mutant superoxide dismutase 1. Proc Natl Acad Sci U S A. 2000;97(22):12306–11. doi: 10.1073/pnas.97.22.12306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nagai Y, Onodera O, Chun J, Strittmatter WJ, Burke JR. Expanded polyglutamine domain proteins bind neurofilament and alter the neurofilament network. Exp Neurol. 1999;155(2):195–203. doi: 10.1006/exnr.1998.6991. [DOI] [PubMed] [Google Scholar]
- 82.Zala D, Benchoua A, Brouillet E, Perrin V, Gaillard MC, Zurn AD, Aebischer P, Deglon N. Progressive and selective striatal degeneration in primary neuronal cultures using lentiviral vector coding for a mutant huntingtin fragment. Neurobiol Dis. 2005;20(3):785–98. doi: 10.1016/j.nbd.2005.05.017. [DOI] [PubMed] [Google Scholar]
- 83.Kleijnen MF, Shih AH, Zhou P, Kumar S, Soccio RE, Kedersha NL, Gill G, Howley PM. The hPLIC proteins may provide a link between the ubiquitination machinery and the proteasome. Mol Cell. 2000;6(2):409–19. doi: 10.1016/s1097-2765(00)00040-x. [DOI] [PubMed] [Google Scholar]
- 84.Lim PJ, Danner R, Liang J, Doong H, Harman C, Srinivasan D, Rothenberg C, Wang H, Ye Y, Fang S, Monteiro MJ. Ubiquilin and p97/VCP bind erasin, forming a complex involved in ERAD. J Cell Biol. 2009;187(2):201–17. doi: 10.1083/jcb.200903024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rothenberg C, Srinivasan D, Mah L, Kaushik S, Peterhoff CM, Ugolino J, Fang S, Cuervo AM, Nixon RA, Monteiro MJ. Ubiquilin functions in autophagy and is degraded by chaperone-mediated autophagy. Hum Mol Genet. 2010;19(16):3219–32. doi: 10.1093/hmg/ddq231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bertram L, Hiltunen M, Parkinson M, Ingelsson M, Lange C, Ramasamy K, Mullin K, Menon R, Sampson AJ, Hsiao MY, Elliott KJ, Velicelebi G, Moscarillo T, Hyman BT, Wagner SL, Becker KD, Blacker D, Tanzi RE. Family-based association between Alzheimer's disease and variants in UBQLN1. N Engl J Med. 2005;352(9):884–94. doi: 10.1056/NEJMoa042765. [DOI] [PubMed] [Google Scholar]
- 87.Mah AL, Perry G, Smith MA, Monteiro MJ. Identification of ubiquilin, a novel presenilin interactor that increases presenilin protein accumulation. J Cell Biol. 2000;151(4):847–62. doi: 10.1083/jcb.151.4.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kim SH, Shi Y, Hanson KA, Williams LM, Sakasai R, Bowler MJ, Tibbetts RS. Potentiation of amyotrophic lateral sclerosis (ALS)-associated TDP-43 aggregation by the proteasome-targeting factor, ubiquilin 1. J Biol Chem. 2009;284(12):8083–92. doi: 10.1074/jbc.M808064200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Doi H, Mitsui K, Kurosawa M, Machida Y, Kuroiwa Y, Nukina N. Identification of ubiquitin-interacting proteins in purified polyglutamine aggregates. FEBS Lett. 2004;571(1–3):171–6. doi: 10.1016/j.febslet.2004.06.077. [DOI] [PubMed] [Google Scholar]
- 90.Wang H, Lim PJ, Yin C, Rieckher M, Vogel BE, Monteiro MJ. Suppression of polyglutamine-induced toxicity in cell and animal models of Huntington's disease by ubiquilin. Hum Mol Genet. 2006;15(6):1025–41. doi: 10.1093/hmg/ddl017. [DOI] [PubMed] [Google Scholar]
- 91.Joseph J, Cruz-Sanchez FF, Carreras J. Enolase activity and isoenzyme distribution in human brain regions and tumors. J Neurochem. 1996;66(6):2484–90. doi: 10.1046/j.1471-4159.1996.66062484.x. [DOI] [PubMed] [Google Scholar]
- 92.Marangos PJ, Parma AM, Goodwin FK. Functional properties of neuronal and glial isoenzymes of brain enolase. J Neurochem. 1978;31(3):727–32. doi: 10.1111/j.1471-4159.1978.tb07847.x. [DOI] [PubMed] [Google Scholar]
- 93.Royds JA, Parsons MA, Taylor CB, Timperley WR. Enolase isoenzyme distribution in the human brain and its tumours. J Pathol. 1982;137(1):37–49. doi: 10.1002/path.1711370105. [DOI] [PubMed] [Google Scholar]
- 94.Scheschonka A, Tang Z, Betz H. Sumoylation in neurons: nuclear and synaptic roles? Trends Neurosci. 2007;30(3):85–91. doi: 10.1016/j.tins.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 95.Boggio R, Passafaro A, Chiocca S. Targeting SUMO E1 to ubiquitin ligases: a viral strategy to counteract sumoylation. J Biol Chem. 2007;282(21):15376–82. doi: 10.1074/jbc.M700889200. [DOI] [PubMed] [Google Scholar]
- 96.Zhou W, Ryan JJ, Zhou H. Global analyses of sumoylated proteins in Saccharomyces cerevisiae. Induction of protein sumoylation by cellular stresses. J Biol Chem. 2004;279(31):32262–8. doi: 10.1074/jbc.M404173200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ueda H, Goto J, Hashida H, Lin X, Oyanagi K, Kawano H, Zoghbi HY, Kanazawa I, Okazawa H. Enhanced SUMOylation in polyglutamine diseases. Biochem Biophys Res Commun. 2002;293(1):307–13. doi: 10.1016/S0006-291X(02)00211-5. [DOI] [PubMed] [Google Scholar]
- 98.Chan HY, Warrick JM, Andriola I, Merry D, Bonini NM. Genetic modulation of polyglutamine toxicity by protein conjugation pathways in Drosophila. Hum Mol Genet. 2002;11(23):2895–904. doi: 10.1093/hmg/11.23.2895. [DOI] [PubMed] [Google Scholar]
- 99.Steffan JS, Agrawal N, Pallos J, Rockabrand E, Trotman LC, Slepko N, Illes K, Lukacsovich T, Zhu YZ, Cattaneo E, Pandolfi PP, Thompson LM, Marsh JL. SUMO modification of Huntingtin and Huntington's disease pathology. Science. 2004;304(5667):100–4. doi: 10.1126/science.1092194. [DOI] [PubMed] [Google Scholar]