

Abstract
Synchronous activation of neural networks is an important physiological mechanism, and dysregulation of synchrony forms the basis of epilepsy. We analyzed the propagation of synchronous activity through chronically epileptic neural networks. Electrocorticographic recordings from epileptic patients demonstrate remarkable variance in the pathways of propagation between sequential interictal spikes (IISs). Calcium imaging in chronically epileptic slice cultures demonstrates that pathway variance depends on the presence of GABAergic inhibition and that spike propagation becomes stereotyped following GABA receptor blockade. Computer modeling suggests that GABAergic quenching of local network activations leaves behind regions of refractory neurons, whose late recruitment forms the anatomical basis of variability during subsequent network activation. Targeted path scanning of slice cultures confirmed local activations, while ex vivo recordings of human epileptic tissue confirmed the dependence of interspike variance on GABA-mediated inhibition. These data support the hypothesis that the paths by which synchronous activity spreads through an epileptic network change with each activation, based on the recent history of localized activity that has been successfully inhibited.
Introduction
Synchronization among neurons is critical for information processing (Uhlhaas et al., 2009). It is becoming clear that the spatial components of oscillations are as critical as the temporal components for both physiological (Lubenov and Siapas, 2009) and pathological (Ibarz et al., 2010) network operations. Yet, surprisingly little is known concerning the mechanisms that govern the spatial spread of synchronization through neural networks, particularly at the temporal resolution relevant for higher network oscillation frequencies. Recent insights suggest that the ongoing barrage of cortical synaptic activity may define assemblies of neurons that transiently become ready to fire as a consequence of the momentary balance of excitatory and inhibitory input (Haider and McCormick, 2009). Such assemblies may represent important sensory and cognitive constructs (Mazor and Laurent, 2005; Pastalkova et al., 2008). Elements of these assemblies are recognizable from one instantiation to the next (Ikegaya et al., 2004; Kerr et al., 2007), although the state of the network, manifest as the background level of synaptic activity, imposes a substantial degree of variance (Kerr et al., 2007).
Perhaps the first signatures of cortical assemblies to be recognized were interictal spikes (IISs) (Marsan, 1961; Bourien et al., 2005). IISs are an electrographic hallmark of epilepsy, a disorder characterized by prolonged epochs of spontaneous, widespread, sustained, pathologically synchronous activity termed seizures. IISs occur much more frequently than seizures and are comprised of brief synchronous discharges of neuronal populations that are large enough to be manifest as voltage transients in the EEG activity recorded from the scalps of epileptic patients (Worrell et al., 2002). However, beyond the features of the electrographic waveforms and their cooccurrence with seizures in epilepsy, very little is known about IISs. Their variance limits their utility as biomarkers for the location of the seizure onset zone in the planning of the surgical resection of intractable epileptic foci (Hufnagel et al., 2000; Otsubo et al., 2001). Why spikes vary has not been examined, but in light of what has been discovered previously about cortical activity (Haider and McCormick, 2009), we studied the propagation of IISs and the variance of propagation.
The mechanisms by which IISs initiate and spread through neural networks (Traub et al., 1987a,b; Traub and Miles, 1991; Ulbert et al., 2004; Wittner and Miles, 2007) are not yet defined. Although a deficit of inhibition relative to excitation is thought to underlie this spread (McNamara, 1999), this tenet is under scrutiny following observations of increased inhibitory activity during IISs (Aradi and Maccaferri, 2004; Esclapez et al., 1997; Trevelyan et al., 2006), reduced firing rates in subpopulations of neurons during epileptiform activity (Bower and Buckmaster, 2008; Keller et al., 2010), and the proposal that interneurons may synchronize both physiological and pathological oscillations in neural networks (Klaassen et al., 2006; Beenhakker and Huguenard, 2009).
Here we use approaches that provide complimentary spatial and temporal resolutions including human electrocorticographic recordings, multiphoton microscopy of calcium-sensitive fluorophores in in vitro chronic epilepsy models, and ex vivo recordings from resected human epileptic foci to examine the mechanisms underlying spike-to-spike variability in the propagation of IISs through cortical networks that range in scale from micrometers to many centimeters.
Materials and Methods
Electrocorticographic data.
Electrocorticographic records from five patients (four males, one female; mean age at surgery, 34.2 years; minimum age of 25; maximum age of 52) with long-standing pharmacoresistant seizures (Alarcon et al., 1997) were analyzed. Patients selected were known to have seizures with focal onset and typical complex partial events often with secondary generalization. Recordings were performed using a standard clinical recording system (Xltek; Natus Medical) with a 500 Hz sampling rate. Analysis of the data from these patients was performed retrospectively under protocols monitored by the local institutional review boards according to NIH guidelines. Two-dimensional subdural electrode arrays (64 channels, 1 cm electrode spacing; Ad-tech Medical) were placed to confirm the hypothesized seizure focus and locate epileptogenic tissue in relation to essential cortex, thus directing surgical treatment. The grids of electrodes used in this study covered regions of temporal, frontal, and parietal lobes (see Fig. 1A). The decision to implant, the electrode targets, and the duration of implantation were made entirely on clinical grounds without reference to this research study. Etiologies represented include mesial temporal sclerosis (MTS) as diagnosed by history, seizure semiology, imaging, and pathology (n = 1) and cortical dysplastic lesions confirmed with pathology (n = 2) (see Fig. 2). In two of the patients, an underlying etiology was never ascertained. IISs were visually identified by an experienced electroencephalographer using standard amplitude and duration criteria (Chatrian et al., 1974). We selected spikes that appeared to initiate from a single location based on earliest activity being detected at the same electrode in the electrode grid. We compared 25–50 IISs per patient. To map IIS trajectory, each electrode was considered a single point in an eight-by-eight matrix (see Fig. 1B). For each electrode, we calculated the rate of voltage change ΔV over a time step of Δt = 32 ms as an estimate of the time derivative of the voltage signal (Fig. 1C). Using these ΔV values, we calculated spatial activity maps (Fig. 1D1). At each time step, we considered electrodes as active only if ΔV values were positive (representing increasing voltage values). While inclusion of negative values gives qualitatively similar results, we analyzed signals at electrodes where the voltage was increasing to maximize the correspondence with the in vitro calcium imaging in which the spatial averages were based on increases in intracellular calcium. To generate activity trajectories, the spatial average of all active electrodes was calculated at each time step, weighted by the value of ΔV for each active electrode. The location of the trajectory on the x–y plane was determined by the spatial average, or center of mass, of all active points at each time point (Chao et al., 2007), and the magnitude of the spatial average was represented by the trajectory diameter (see Fig. 1D1,D2, white rings 1–5). Activity trajectories were created by stacking each spatial average location and diameter for each time point along the z-axis (Fig. 1D2). During periods with no IISs, the spatial average resides in the center as it includes only small contributions from background EEG activity, evidenced as variance in the baseline of each IIS trajectory. The spatial averaging procedure was also minimally affected by background EEG activity during IIS propagation because of the large differences in amplitude between the ΔV during the IIS versus the ΔV from the background EEG activity. Trajectories from sequential spikes for a single patient (Fig. 1E1) were aligned to the maximum peak value of the voltage trace of the electrode determined to be nearest the onset location. Arrows (1–5) in Figure 1C denote the temporal location of the frames shown in Figure 1, D1 and D2. Spatial variance in pathway activation (pathway variance) was quantified by measuring the mean distance between all trajectories for each patient (Fig. 1E2). Statistical analyses were conducted in Matlab and SigmaPlot.
Figure 1.

IISs propagate via multiple pathways in epileptic patients. A, Three-dimensional MRI reconstruction showing the position of a 64 channel subdural electrode grid placed to confirm seizure focus and locate epileptogenic cortex in an example patient. B, One second electrocorticographic recording of a single IIS acquired simultaneously at all electrodes. C, Expanded view of the IISs depicted in B. Voltage data for all 64 electrodes are overlaid for the time interval from 80 ms before to 80 ms after the peak on channel E4 (blue traces). We calculated the rate of change of the EEG signal as the time derivative using a time step of 32 ms (red traces) to analyze spatial propagation of each IIS. The rate of change is thus proportional to the slope of the voltage signal and is maximal at the steepest segment of the blue voltage traces before the peak of the spike. Time points 1 through 5 (arrows) represent the points depicted in D1. To map the propagation of an IIS through cortex, the weighted spatial average was calculated from the increase in voltage at each time point (see Materials and Methods) (Chao et al., 2007). White rings (1 through 5) are centered on the spatial mean of all active points, and ring diameter corresponds to intensity. D2, Trajectories were created by stacking the weighted averages for each time point (times 1 through 5 are represented in the trajectory). E1, Trajectories from eight IISs in the example patient demonstrate substantial spatial variance. E2, Path variability in network activation was quantified by calculating the average distance between trajectories within each patient and demonstrated an initial decrease in variability from prespike baseline during spike initiation (due to single initiation locus), followed by an increase in variability as the spike followed multiple routes of propagation through the network (Wilcoxon signed rank test, p < 0.001; 150 IISs from 5 patients).
Figure 2.
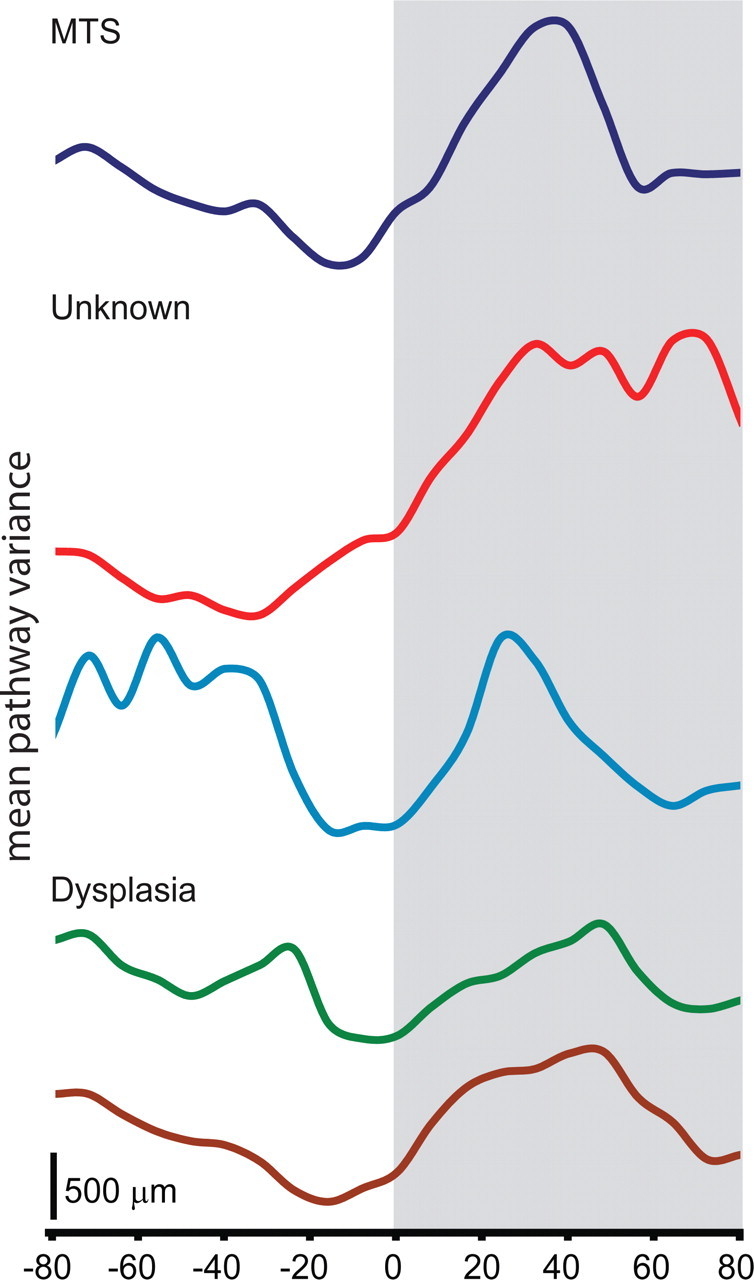
Etiology may determine the degree of IIS-related variability in spike propagation. The mean pathway variance (average distance between each trajectory) for n = 25 spike trajectories for each patient is shown. The 0 ms time point corresponds with IIS peak. Patients with identified dysplasias showed less spike-related increases in variance than patients with MTS or unknown etiologies.
In vitro imaging and electrophysiology.
Rat hippocampal slices (350 μm thick) were cultured according to Gähwiler (1981), with the following modifications: After 48 h in culture, 20 μl antimitotics (30 μg/ml cytosine-β-d-arabino-funanoside, 30 μg/ml uridine, and 30 μg/ml 5-fluro-2′-deoxyuridine) were added to each culture tube to inhibit glial overgrowth. After 24 h exposure, the media was replaced with Neurobasal A/B27 supplemented with 0.5 mm GlutaMAX (Sigma) and 30 μg/ml gentamicin. Subsequently, growth medium was changed every 7 d. Following 2–3 weeks incubation, cultures exhibit spontaneous ictal- and interictal-like activity when recorded in culture media (McBain et al., 1989; Bausch et al., 2006; Dyhrfjeld-Johnsen et al., 2010). All experiments were conducted in modified neurobasal media (see Culture media, below). Cultures were bath incubated in 5 μm Oregon Green BAPTA-1 AM (OGB1) in HEPES-buffered artificial CSF (ACSF) for 60 min at room temperature, followed by 30 min in probenecid (Invitrogen), and were then transferred to the recording chamber where spontaneous activity was recorded and imaged in modified neurobasal media (see Culture media, below). Epifluorescence microscopy was used to image calcium transients associated with epileptiform events (X-cite 120 arc lamp, chroma FITC/Texas Red filters, Olympus BX 51WI microscope). An XLUMPlanFl 20× objective (0.95 NA) in combination with 0.5× magnification changer provided a “virtual” 10× magnification and sufficiently wide field of view to visualize the entire CA3 subfield. Images (see Fig. 3A) were collected at 125 frames per second using a VS4–1845 intensifier (Cooke Camera) and an eight-bit Fastec 250 Complementary metal-oxide semiconductor camera (640 × 480 pixels) and processed using custom software (available on request). Synchronized electrical field recordings for GABA receptor (GABA-R) blockade experiments were collected using a 30 μm tungsten electrode (see Figs. 3A,B, 4A1–A3,B1–B3) and custom software based on the study of (White et al., 2010). GABAA receptor (GABAAR) and GABABR were blocked by addition of 100 μm picrotoxin and 1 μm CGP 55845(C18H22CI2NO3P·HCI) to the bath. Partial blockades of AMPA receptors and voltage-dependent sodium channels (see Fig. 6A,B) were accomplished with 100 nm DNQX and 50 nm tetrodotoxin, respectively. These concentrations were expected to block currents by ∼50% (Mennerick and Zorumski, 1996; Espinosa and Kavalali, 2009; Zhang et al., 2009) and were empirically chosen to reduce but not block bursting. We limited our analysis to events with a duration <2 s and a minimum of 2 s since the end of the previous event. To focus on fast changes associated with neuronal signaling, reduce the inclusion of slow glial changes, and account for photobleaching, images were temporally filtered by pixelwise subtraction of the frame obtained 32 ms prior from the current frame (that is, Fn - Fn − 4, where Δt between frames is 8 ms). We applied the same weighted spatial averaging technique described above for the EEG signals for the 640 × 480 pixel images to create trajectories (Figs. 3C,D, 4A3,B3). Trajectories for multiple spike events were aligned to the first frame in which the electrical field amplitude exceeded three SDs (time, 0 ms) (see Fig. 4A3,B3). To examine changes in electrical variance, we aligned each IIS by peak (see Fig. 4A2,B2) and compared the SD of the electrical waveforms at each time point (see Fig. 4C). To quantify spatial variance in pathway activation (pathway variance), we measured mean distance between all trajectories for each slice (Fig. 4D). Statistical analyses were conducted in Matlab and SigmaPlot.
Figure 3.

Tracking IIS propagation in vitro. A, To investigate the network determinants of IIS variability in epileptic tissue, we loaded organotypic slice cultures with the calcium indicator OGB1 and imaged hippocampal region CA3. B, Each IIS event (top) is accompanied by an increase in intracellular calcium (times 1–3 correspond with time points shown in C). C, D, Calcium transients corresponding to IIS events were used to create propagation trajectories (D) of IIS events by applying the same weighted spatial averaging technique used in Figure 1 to the 640 × 480 pixel calcium images.
Figure 4.
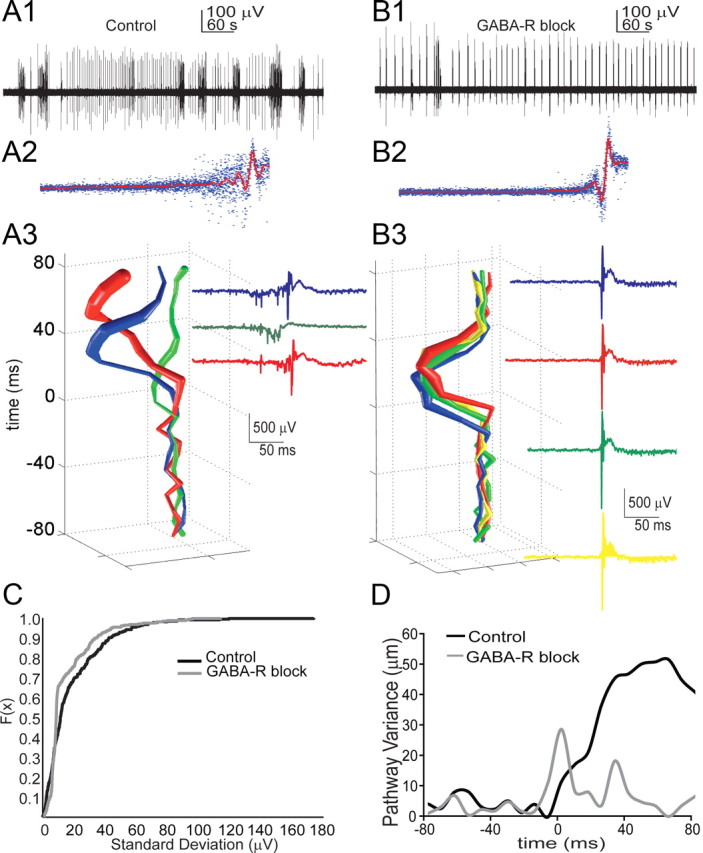
Interneurons sculpt the spread of IIS events. A1, Electrical field record of spontaneous interictal- and ictal-like activity in a slice culture. A2, Peaks for each individual spike were aligned (blue decimated traces, 87 events), and the mean waveform (red trace) was calculated for control conditions. The spread of the blue traces demonstrates the degree of variability present in the spike waveforms within a single culture. A3, Applying the spatial weighted averaging technique to IIS-related increases in calcium signal reveals that multiple spontaneous IIS events from a single culture propagate via distinct pathways. Electrical traces of each event are shown and color coded to correspond with propagation trajectories. B1, B2, Following GABA-R blockade (100 μm picrotoxin, 1 μm CGP 55845), interictal-like activity persisted and waveform variability decreased (B2; 107 events). B3, D, Following GABA-R blockade, the propagation patterns for the culture shown in A3 become stereotyped and the average distance between trajectories after GABA block is sharply reduced (D). C, A two-sample KS test on the SD of the electrical waveforms at each time point revealed distinct distributions for control vs GABA block <0.001; n > = 300 bursts, 4 cultures). Blocking GABA-R significantly reduced pathway variance (Mann–Whitney rank sum test, p = 0.006; n = 18 control; n = 17 GABA blocked bursts, 4 cultures).
Figure 6.
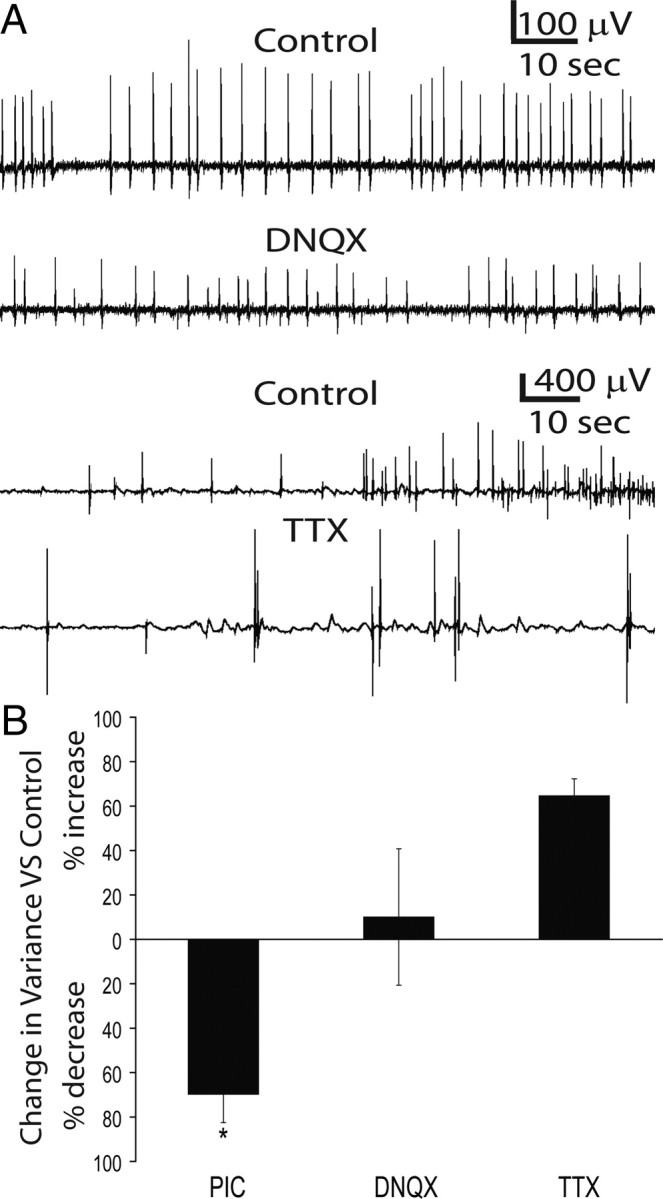
Reductions in IIS variability are specific for GABA-R blockade. A, Electrical field potential recorded from a spontaneously epileptiform culture demonstrates that IISs persist following application of the AMPA blocker DNQX (100 nm) and inactivation of Na channels with 50 nm TTX. B, GABA-R blockade robustly (70 ± 11% SEM) and significantly (t test, p = 0.003) decreases the electrical waveform variance of IISs compared to control conditions. In contrast, DNQX and TTX both produced nonsignificant increases in waveform variance (p = 0.27 and 0.17, respectively) over control conditions.
To compare waveform variance in TTX and DNQX (see Fig. 6), all events were aligned to their peak as in Figure 4, A2 and B2, and the mean and SD for the population of waveforms was calculated for each condition. To ensure that amplitude changes were not biasing waveform variance estimates, we normalized each event dividing by the peak to trough amplitude. We then calculated the area under the curve for the SD occurring in the 50 ms leading up to the peak value (see Fig. 6B). Significance was assessed using t tests (SigmaPlot).
Culture media.
Neurobasal medium was modified to reduce the fluorescence in the emission range of OGB1 (∼500 to 540 nm) by removing B vitamins. Modified neurobasal components were as follows (in mm): 1.8 CaCl2, 5.36 KCl, 0.812 MgCl2, 5.3 NaCl, 26 NaHCO3, 0.9 NaH2PO4, 25 d-glucose, 10 HEPES, 0.23 sodium pyruvate, 0.020 l-alanine, 0.4 l-arginine, 0.500 glutamax, 0.400 glycine, 0.200 l-histidine HCl, 0.005 l-lysine, 0.200 l-methinone, 0.067 l-proline, 0.400 l-serine, 0.800 l-threonine, 0.800 l-valine, 0.008 d-Ca pantothenate, 0.028 choline chloride, 0.040 l-inositol, 0.030 niacinamide, 0.020 pyridoxal HCl, 0.010 thiamine HCl, 0.800 l-isoleucine, 0.400 l-pheneylalanine, 0.080 l-tryptophan, and 0.400 l-tyrosine (obtained from Sigma-Aldrich).
Targeted path scanning.
Two-photon targeted path scan (TPS) imaging was performed as described previously (Lillis et al., 2008) using a custom-built control interface to a Radiance MP2000 scanhead (Bio-Rad Laboratories). Briefly, cells were selected from a high-resolution image of the sample (acquired using a classic raster scan). The laser was then scanned along a path that bisected each selected cell for 25 pixels and accelerated/decelerated maximally between selected cells. This allowed us to sample fluorescence with single-cell resolution at rates high enough to record calcium transients associated with epileptiform activity (see Fig. 5A–C). Field recordings during targeted path scanning were conducted using a glass electrode positioned in the cell layer and Labview software (see Fig. 5D). Calcium dynamics were recorded by preparing organotypic slice cultures from mice that express the FRET-based calcium-sensitive protein Yellow Cameleon 3.6 (YC3.6) in all cells that expressed nestin during development (essentially all neurons, glia, and blood vessels in the hippocampus). YC3.6 was excited at 860 nm with a Mai Tai Ti-sapphire laser (Spectra-Physics Lasers), and relative calcium changes were recorded as a ratio of YFP (530–560 nm) to CFP (410–490 nm). Trajectories were created by taking the spatial average of all neurons activating at each time step (26 ms) (see Fig. 5D). Pathway variance was quantified by measuring the mean distance between all trajectories.
Figure 5.

Targeted path scanning in Yellow Cameleon 3.6 cultures confirms variable IIS trajectories. A, Raster plot of calcium traces (corresponding to the burst shown in B) illustrates variable activation times for individual cells. B, C, Panels depict the unique activation order of cells (colored circles) during two of the bursts represented in the trajectories. Cell color encodes the time of initial spike-related increase in calcium relative to the peak of the population spike in the field recording (0 ms). D, Neurons activate in unique sequences during subsequent IISs. Calcium imaging data from the events marked with asterisks in the electrical field record (top) were plotted as trajectories by calculating the spatial average of all activating cells at each time step (26 ms).
We analyzed partial spontaneous synchronizations preceding full IISs using targeted path scanning of Y3.6 fluorescence in CA3 neurons. We calculated the rate of change of somatic Ca2+ in 72 ms time intervals to remove slow signal changes such as bleaching from each cell's signal. Figure 9A illustrates a raster plot of calcium traces for individual cells. We calculated the mean rate of Ca2+ change in all cells (see Fig. 9B) for manually selected partial synchronizations (blue lines a–e) and IIS initiations (f). We created a six-by-five activity matrix of 30 recorded cells in which each pixel represented single cell. Although the location of cells in the matrix had no correspondence to slice location, each pixel represented the same cell from frame to frame. Cells were considered active if the recorded calcium level was >2 SDs above mean. Figure 9C illustrates cell activity during five partial synchronizations (a–e) and IIS initiation (f). To estimate which cells were most likely to be refractory at the time of IIS onset, we calculated a time average of each cell's activity, with activity weighted in inverse proportion to the time remaining to the next IIS (see Fig. 9D). To predict least refractory cells, i.e., neurons most ready to participate in the next IIS initiation, we inverted the time average of the activity. In this inverted matrix, high values (red) indicated areas of probable initiation for the next IIS, and low values (blue) indicated recently active areas that were less likely to participate. We then compared inverted mapping and IIS initiation matrices by calculating the summed differences between them. Since we were focused on cells that should not be active during next IIS initiation, we calculated the error as the sum of cells with initiation values larger than predicted values (i.e., cells that participated early in the IIS despite being recently active) and divided it by the total number of cells.
Figure 9.
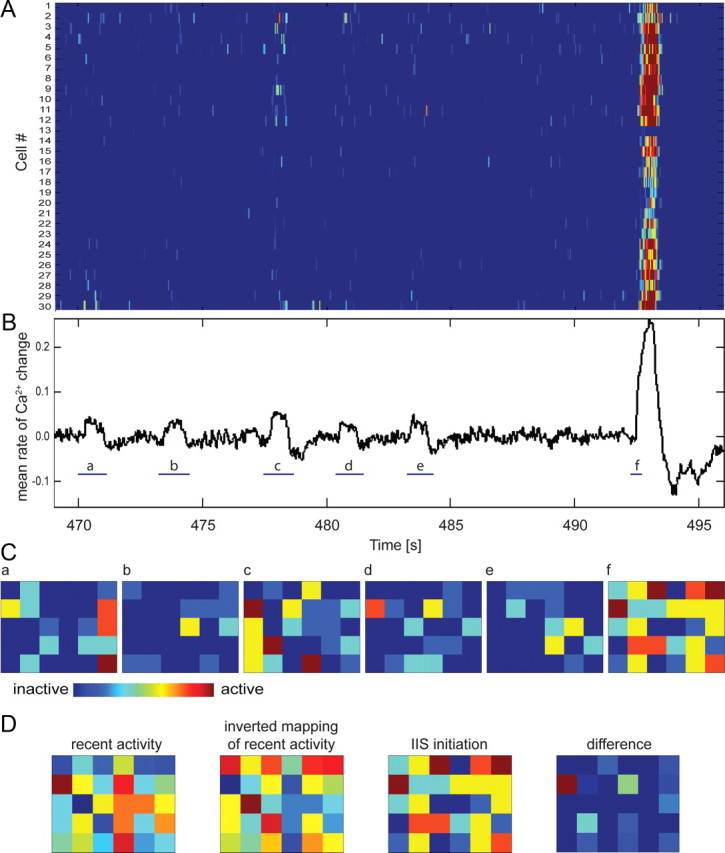
Partial spontaneous synchronizations precede full IISs. A, Raster plot of calcium increases in 30 cells imaged using TPS in Y3.6 slice cultures illustrates the variable activation of neurons preceding IIS. B, Mean rate of Ca2+ increase in all cells. Spontaneous partial synchronizations (a–e) and IIS initiation (f) were selected manually (blue lines). C, Cell activity matrices of six-by-five cells (30 recorded cells) during five partial synchronizations (a–e) and IIS initiation (f). Each pixel represents a single cell. The location of cells in the matrix has no correspondence to their location within the culture but is consistent between matrices. A cell was considered active if the recorded calcium level was above a threshold of 3 SDs from the mean baseline noise level. D, Calculation of prediction error (from left to right): recent activity estimate matrix, inverted mapping of recent activity, IIS initiation matrix, and difference matrix. The recent activity matrix was calculated as a weighted sum of each cell in the partial synchronization matrices a through e. Weight was inversely proportional to the time remaining between activity and the next IIS. The inverted mapping reversed the weighted averages of recent activity of each cell in the matrix. The difference matrix shows only positive values of difference between IIS initiation times and inverted mapping of recent activity.
Computational modeling.
Model connectivity was based on the hippocampal circuitry as described by Traub and Miles (1991), including the 1:10 ratio of interneurons to principal cells (Miles et al., 1988; Wittner and Miles, 2007), and consisted of a two-dimensional network of pyramidal cells (100 × 100) and interneurons (33 × 33) (see Fig. 7A). Both pyramidal cells and interneurons were recurrently connected and implemented as integrate-and-fire neurons. All cells of the same type had the same properties; however, network connectivity was not homogenous. The probability of a synaptic connection between any two neurons was inversely proportional to the distance between the cells and was calculated using the normal distribution. Every cell was assigned a maximum connectivity radius that limited the distance of connections. This radius was calculated as the sum of the minimum connectivity radius (of three cells) and a random value from 1 to 100 using the normal distribution. The result was a scale-free network with the majority of cells connected locally and a small number of cells reaching far through the network (Buzsáki, 2006; Morgan and Soltesz, 2008; Bonifazi et al., 2009). The spatial distribution of the most heavily connected neurons thus varied each time a new network was created, but this variability did not affect the spike-to-spike variance that all networks exhibited (see Figs. 7, 8). The average probability of any two pyramidal cells being synaptically connected was 2.7%, independent of distance (Debanne et al., 1995; Pavlidis and Madison, 1999). Synapses expressed a connection-length-dependent time delay and could result in multiple spikes propagating along the axon. Synapses between pyramidal cells expressed activity-dependent depression with a time constant for recovery from depression of 10 s (Staley et al., 2001). Spontaneous glutamate release rate at baseline was 0.001 releases per second per synapse, but was dependent on the level of available glutamate (Dobrunz and Stevens 1997). The rates of spontaneous glutamate release and recovery from short-term depression were as described previously (Jones et al., 2007; Swiercz et al., 2007). This was the main source of noise in the model causing synaptic transmission failures and randomized spontaneous transmitter release. GABAergic synapses had a high probability of action potential-dependent release, and no spontaneous GABA release (Swiercz et al., 2007). The dynamics of synaptic excitation and inhibition in the model were based on AMPA and GABAA mechanisms, respectively; NMDA and GABAB receptors were not included in the model. Time constants for postsynaptic potentials and for short-term synaptic activity depression and recovery for excitatory synapses are shown in Table 1 (Traub and Miles, 1991; Wittner and Miles, 2007).
Figure 7.
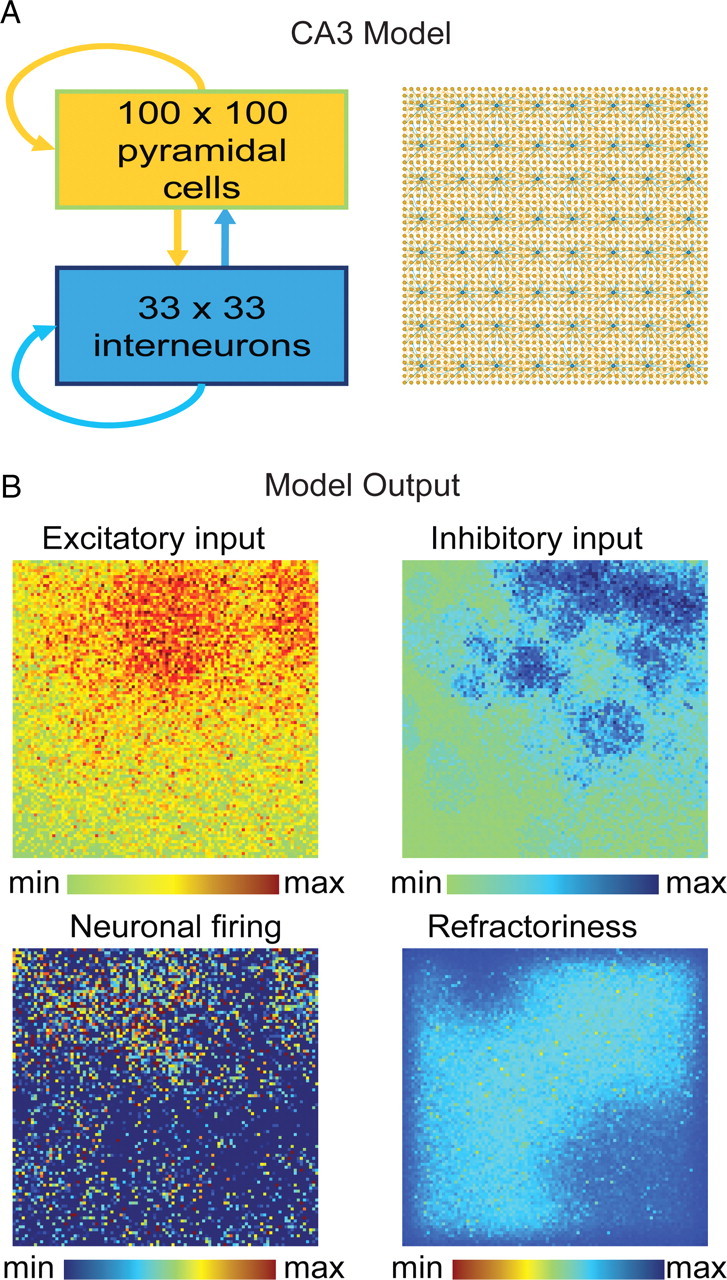
CA3 network model and outputs. A, Network connectivity diagram of our CA3 computational model. B, Model output consists of panels showing 100 × 100 pixel images representing the entire area of the network and allows for visualization of the development and spatial spread of excitatory and inhibitory inputs, neuronal firing, and refractoriness due to glutamate depletion.
Figure 8.
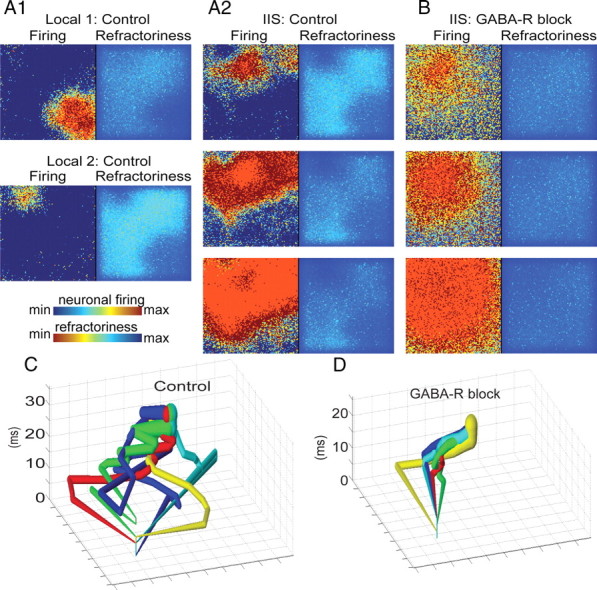
GABAergic networks determine the IIS pathways via the creation of refractory pockets. A1, A2, When inhibition was intact in our model of epileptiform activity (control), spontaneous IIS events were preceded by a series of local events (A1) in which spatially restricted, transiently synchronous activity (red regions, left) was quenched by local activation of GABAergic synapses, resulting in pockets of refractoriness due to activity-dependent, short-term depression of glutamatergic synapses (dark blue regions, right). Upon initiation of an IIS (A2, top), activity propagates (center and bottom) evenly through nonrefractory areas (light blue and green regions, right), but poorly through refractory regions. B, When GABAergic synapses were blocked locally, synchronized, nonpropagating events no longer occurred; rather, the first synchronization spread evenly through the entire neural network. C, D, We applied the spatial averaging technique described in Figure 1 to the neuronal firing output of our computational model to track the propagation of IISs initiating from a single spatial location before and after GABA-R blockade. In agreement with our in vitro data, GABA blockade sharply reduced the spatial variance of the propagation of synchronous activity through the network (average distance between each trajectory control vs GABA-R block; t test, p < 0.01).
Table 1.
Post-synaptic potential time constants depend on synapse type
| e→e | e→i | i→e and i→i | |
|---|---|---|---|
| τr | 3 ms | 1 ms | 1 ms |
| τd | 3 ms | 7 ms | 1 ms |
| λd | 30 ms | 30 ms | |
| λr | 10 s | 10 s |
e, Excitatory synapse; i, inhibitory synapse; τr, raise time constant; τd, decay time constant; λd, short-term synaptic activity depression time constant; λr, short-term synaptic activity recovery time constant.
We included real edges to facilitate visualization of network activation. Simulations were run at 1 ms time increments as described previously (Swiercz et al., 2006, 2007).
The model generates two-dimensional outputs of the following network parameters: excitatory and inhibitory synaptic input levels, neuronal firing and potentiation, and refractoriness of synapses (see Fig. 7B). We applied the same spatial averaging technique described for the electrocorticographic (ECoG) and calcium-imaging data to the neuronal firing output of the model and mapped the onset trajectories in control and GABA-R block. Pathway variance was compared by calculating the mean distance between trajectories initiating from a single location for the two conditions.
Human ex vivo recordings.
Temporal lobe tissue containing the subiculum was obtained after operations on two patients with pharmacoresistant mesial temporal lobe epilepsies associated with hippocampal sclerosis. Patients gave their written consent, and the Comité Consultatif National d'Ethique approved the protocol. Postsurgical tissue was transported in a cold, oxygenated solution containing the following (in mm): 248 d-sucrose, 26 NaHCO3, 1 KCl, 1 CaCl2, 10 MgCl2, and 10 d-glucose, equilibrated with 5% CO2 in 95% O2. Slices (400 μm thick) were cut with a vibratome (HM650V; Microm) and maintained at 37°C, equilibrated with 5% CO2 in 95% O2, in an interface chamber perfused with a solution containing the following (mm): 124 NaCl, 26 NaHCO3, 4 KCl, 2 MgCl2, 2 CaCl2, and 10 d-glucose. Synchronous population activity was initiated as in the study by Huberfeld et al. (2011) by increasing extracellular K+ to 8 mm and lowering extracellular Mg2+ and Ca2+ to 0.5 mm. Extracellular records were made with tungsten electrodes etched to a tip diameter of ∼5 μm. Signals were amplified 1000× and filtered to pass frequencies of 0.1 Hz to 10 kHz (A-M Systems, Model 1700). Recordings were analyzed with Clampfit software (Molecular Devices). Extracellular signals were low-pass filtered at 100 Hz. Epileptiform discharges were detected and aligned with a template search algorithm. Propagation delay was measured from the difference in onset of the field potential at two electrodes separated by 3 mm (see Fig. 10A). Changes in the variance of all variables before and after blocking GABAA receptors were examined in Matlab and Sigmaplot.
Figure 10.
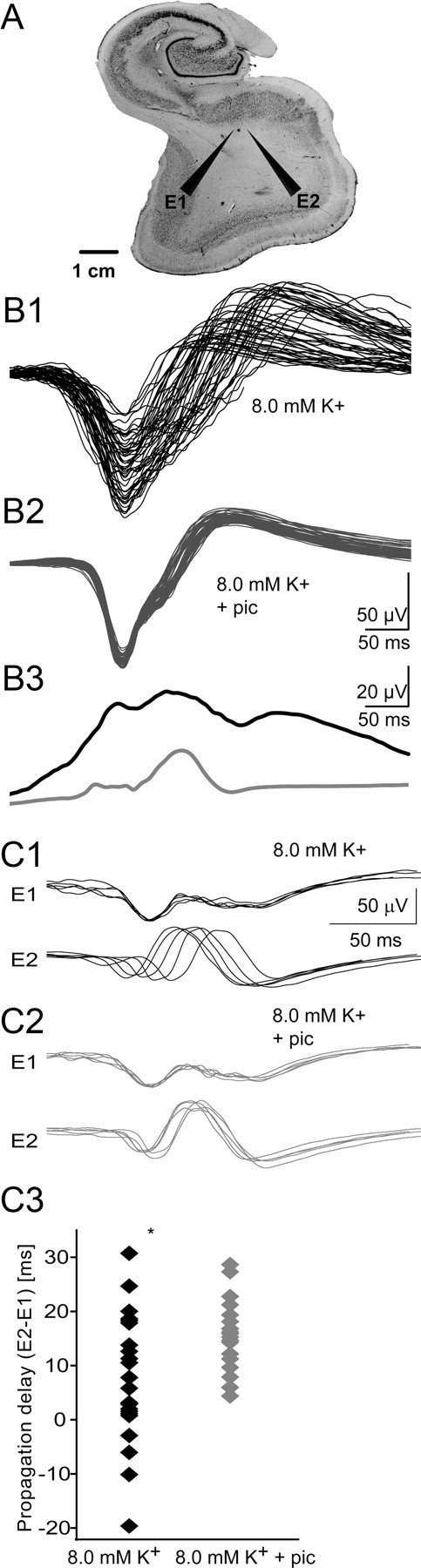
Interneurons contribute to IIS variance in chronically epileptic human tissue. A, We examined the contribution of interneurons to IIS variability in the subiculum of brain slices resected from patients for control of intractable temporal lobe epilepsy. B1, B2, Population discharges were readily recorded in ACSF containing (in mm) 8 [K+]o 0.5 [Ca2+]o, and 0.5 [Mg2+]o. IIS waveform variability from a single electrode was compared for 50 IISs before (B1) and after (B2) bath application of 50 μm picrotoxin. B3, Variability was significantly reduced following blockade of GABAA receptors (light gray trace; KS test on SD, p < 0.001). C1–C3, Spatial propagation of IISs across two electrodes (E1 and E2) separated by as much as 3 mm becomes stereotyped following GABAA block. Before GABA block (C1), IISs propagated in both directions (E1 to E2 or E2 to E1). Following blockade (C2), all spikes propagated from E1 to E2 (all positive values on scatter plot; C3), and the propagation time became significantly more consistent (Levene's test, p = 0.016), suggesting that interneurons determine the IIS pathway across a minimum of 3 mm.
Results
IISs take variable propagation pathways through epileptic cortex
We analyzed the spread of IISs using electrocorticographic recordings from subdural electrode arrays on human epileptic neocortex (Fig. 1A,B) (peak value at 500 ms on electrode E4) to determine the variance in spatial propagation of spike activity through cortex. We compared sequential IIS waveforms recorded from each of the 64 electrodes on an implanted grid. Aligning voltage traces for one spike recorded in all electrodes (Fig. 1C, blue traces) demonstrates the temporal variation in spike onset at each electrode. However, spatial variance in propagation is not readily discerned by visual inspection of the ECoG waveforms in Figure 1C. Tracking spike propagation using 3D trajectories provides a robust description of spike-to-spike variability. We therefore used propagation trajectories, calculated from the array of time-series data shown in Figure 1B as described in the materials and methods section. To minimize sources of spatial variance, the analysis was restricted to spikes that originated from a single electrode. Trajectories calculated for sequential IISs in a single patient demonstrated substantial spatial variance (Fig. 1E1). We quantified the variability in network activation by calculating the average distance between trajectories in each patient (Fig. 2). Figure 1E2 shows the average spatial variance as a function of time for 150 IISs from five patients. Pathway variability initially decreased from baseline as a consequence of the process of selecting spikes with onset in a particular electrode (see Materials and Methods), which was done to focus on the variance in propagation through the network, rather than variance in the anatomical onset. As IISs propagate along cortex, pathway variability increases significantly from the prespike baseline (Wilcoxon signed rank test, p < 0.001), demonstrating that IISs have variable propagation trajectories. This raises the possibility, investigated in vitro in the following sections, that even when the same cortical areas participate in multiple IISs, they are recruited in a unique sequence for each IIS. The patterns suggest that the degree of variance may provide a potentially useful insight into the nature of the epileptic focus.
Following IISs in vitro: GABAergic networks provide a major contribution to variance
To study the network determinants underlying IIS variability pharmacologically, we turned to a reduced experimental model of chronic posttraumatic epilepsy. As a consequence of luxuriant axonal sprouting (Del Turco and Deller 2007), hippocampal roller tube slice cultures (Gähwiler 1981) develop robust, spontaneous ictal- and interictal-like epileptiform activity over 2–3 weeks in vitro (see Materials and Methods) (McBain et al., 1989; Bausch et al., 2006; Berdichevsky et al., 2009; Dyhrfjeld-Johnsen et al., 2010). We simultaneously recorded field potentials and Ca2+ signals in chronically epileptic hippocampal organotypic slice cultures (Fig. 3A,B). Spontaneous ictal and IIS-like events were readily identified in field potential recordings in control conditions (Fig. 4A1) (see Materials and Methods). Each IIS event was associated with an increase in calcium signal. Interictal events spread throughout area CA3 to reach peak average calcium levels within 40–100 ms (n = 40). To examine the variance in individual IIS field potential waveforms (Fig. 4A2), we aligned individual IIS events (blue decimated traces) to their peaks and calculated the mean waveform (red trace). Variability in IIS waveforms is evident in the spread of the blue versus red waveforms. We then applied the spatial averaging technique described in Materials and Methods to the calcium signals from sequential spontaneous IIS events recorded in our cultures (Fig. 3C,D). Similar to the ECoG findings, spontaneous IIS events in vitro exhibited distinct propagation trajectories (Fig. 4A3). Electrical field records for each spike are shown, color coded to indicate the accompanying trajectory. Calcium imaging and field potential recordings of IISs in hippocampal slice cultures demonstrated multiple pathways of network activation during sequential IIS-like events (n = 24 bursts, 6 cultures).
Interneurons synchronize physiological activity in the hippocampus (Buzsáki and Chrobak, 1995; Cobb et al., 1995; Bonifazi et al., 2009), although their role in pathological synchronization is less clear (Beenhakker and Huguenard 2009). Earlier experimental and modeling studies predicted that GABA-mediated inhibition was an important determinant of the pattern of network activation during synchronous hippocampal discharges (Traub et al., 1987a,b). To test the influence of interneuron activity on the pattern of network activation during IISs, we blocked GABAAR and GABABR and tracked IIS onsets in a given culture under control and disinhibited conditions. IISs were readily identified in the electrical field recordings under both conditions (Fig. 4A1,B1). To compare the variance in the electrical waveforms for control (Fig. 4A2) versus GABA-R block conditions (Fig. 4B2), we calculated the SD of the electrical waveforms at each time point and compared the two distributions using Kolmogorov–Smirnov (KS) test for two populations. Figure 4C shows the two populations represented as cumulative frequency distribution. GABA-R block significantly reduced waveform variance (maximum SD, control, 180 μV vs GABA-R block 110 μV; KS test, p < 0.001; n = 300 bursts, recorded from 4 cultures) and led to a correspondingly sharp decrease in propagation trajectory variance (Fig. 4B3). We quantified trajectory variance to compare the average distance between trajectories in control conditions and under GABA-R blockade. Figure 4D demonstrates the GABA-R antagonist induced decrease in trajectory for the culture shown in Figure 4, A3 and B3. Overall, GABA-R blockade significantly reduced propagation variance (Mann–Whitney rank sum test, p = 0.006; control, n = 18; GABA block, n = 17, recorded from 4 cultures) and led to stereotyped activation pathways.
Thus, block of GABA-R reduces the path variance of interictal epileptiform activity, manifest both in extracellular records and in spatially resolved calcium imaging of the trajectory of network activation. These findings highlight the role of interneuron networks in shaping the spatial spread of synchronized activity through epileptic cortex, suggesting these networks are key determinants in shaping spatial propagation.
To distinguish spike-to-spike variance in the sequence of neuronal activation during IISs from spike-to-spike variance in the amplitude of calcium transients in individual neurons, we used targeted path scanning (Lillis et al., 2008) in organotypic hippocampal slice cultures from transgenic mice expressing Yellow Cameleon 3.6. The laser path was directed through a manually selected sample of cells in a single x–y plane, and IISs were analyzed to characterize the sequence of activation of each neuron relative to the peak of the population spike. Figure 5A illustrates variable activation times for individual cells on a raster plot of calcium traces (corresponding to the burst shown Fig. 5B). Figure 5, B and C, exemplifies the unique sequence of neuronal activation for two spike events, confirming variable activation of the network on sequential IISs on the single-cell level. Figure 5D demonstrates variable trajectories corresponding to the 5 IIS events marked in the electrical trace (n = 4 slices).
To determine whether the decrease in pathway variability was due to specific effects of GABA-R inactivation or was a nonspecific consequence of simplification of the network circuitry due to removal of one signaling mechanism, we recorded electrical activity in spontaneously epileptiform organotypic slices before and after partial blockade of AMPA receptors with 100 nm DNQX. IIS activity was still present in all three cultures (Fig. 6A). Events were aligned to their peak value, and waveform variance was calculated for each condition (see Materials and Methods). DNQX produced a nonsignificant (mean, 10 ± 8% SEM; p = 0.273) increase in waveform variability (Fig. 6B) over the control condition. We also partially blocked Na+ channels with 50 nm TTX. In three cultures, we observed a nonsignificant increase in waveform variance (mean, 65 ± 39% SEM; p = 0.17) over control conditions. In contrast, GABA-R blockade resulted in a significant decrease in variability (x = 70 ± 11% SEM; p = 0.003). Importantly, neither blocking AMPA receptors nor sodium channels decreased variance, supporting the idea that the decrease in pathway variance is a specific consequence of GABA-R blockade.
Computational modeling: how does GABA influence the variance of network activation?
To evaluate mechanisms by which interneurons might sculpt the trajectory of network activation, we used a computational model of the hippocampal CA3 network (Fig. 7A) (Swiercz et al., 2007). We first simulated a nonepileptic network with limited recurrent excitatory connections. Spontaneous activity was regulated by robust activation of interneurons, and although local areas of synchrony developed, they did not propagate or persist (Cohen et al., 2006; de la Prida et al., 2006), and no epileptiform activity was observed.
Increasing excitability, either by increases in [K+]o or increasing recurrent excitatory synaptic connectivity to mimic axon sprouting, resulted in spontaneous synchronized population events. Under these conditions, interictal-like population events were preceded by a series of spatially restricted events, similar to the smaller amplitude, nonpropagating local synchronies preceding IISs during in vitro electrical recordings (de la Prida et al., 2006). Plots of GABAergic synaptic activity demonstrated that these spatially restricted local events were quenched by local activation of GABAergic synapses. After the nascent IISs were quenched, the involved regions were left in a relatively refractory state due to activity-dependent depression of the glutamatergic synapses within a local region (Fig. 8A1). Consequently, when a successful activation of the entire network occurred, it spread preferentially through nonrefractory areas (Fig. 8A2). Thus, the areas that were most recently involved in locally synchronous activity were the last to become involved in widespread network activation. The timing and location of nonpropagating local events was extremely variable, and because these events determined the propagation pathway of network-wide events, the trajectories of IIS-like activity were also highly variable.
When GABAergic synapses were blocked in the model, transient, self-limited locally synchronized events were abolished; however, the first local synchronization continued to spread to activate the entire neural network (Fig. 8B). We observed a similar abolishment of smaller amplitude local synchronies following GABA-R blockade in vitro (Fig. 4B3, electrical insets). We compared burst trajectories from a single onset location under spontaneous bursting and GABA-R blockade conditions in the model (Fig. 8C,D) using the same technique described in Figure 1. As in vitro, GABA-R blockade sharply reduced the spatial variance (t test, average pathway variance, p < 0.01).
Targeted path scanning reveals local network activations preceding IISs
As described above (Fig. 5), all recorded cells were activated during IISs; however, the activation order varied between IISs. Spontaneous partial activations of the network preceded IISs in slices recorded before 11 DIV (Fig. 9A,B). Analysis of these partial activations demonstrated that each partial synchronization was comprised of a different group of cells (Fig. 9C). The pattern of activity during partial synchronizations preceding the IIS was compared to the initiation pattern of the IIS (Fig. 9D). Cells that participated in IIS initiation were less active during partial synchronizations (84 ± 5% prediction accuracy; n = 7 IISs in 5 slices), supporting the findings from our modeling experiments (Fig. 8).
Human ex vivo recordings support the role of GABA in IIS variance
To test whether the insights derived from our experimental and modeling data were relevant to propagation of synchronous IISs through human epileptic cortex, we examined the variability of IIS-like activity in slices of brain tissue resected from patients for control of intractable temporal lobe epilepsy (Cohen et al., 2002; Huberfeld et al., 2007, 2011) (Fig. 10A). Epileptiform discharges were induced by increasing extracellular K+ to 8 mm and lowering extracellular Mg2+ and Ca2+ to 0.5 mm. In four slices from two patients, the variability in field potential waveform was measured for epileptiform discharges recorded before (Fig. 10B1) and after GABAAR were blocked using 50 μm picrotoxin (Fig. 10B2). Blocking GABAAR significantly reduced the waveform variability of population spikes (Fig. 10B3) (two-sample Kolmogorov–Smirnov test on SD of waveform at each time point, p < 0.001) and led to a significant reduction in propagation variance (Levene's test, p = 0.016) and stereotyped propagation of spikes across electrodes spaced 3 mm apart (Fig. 10C1,C2). Before GABA-R block, spikes occurred first at either electrode, but following GABA-R block, all spikes propagated from electrode 1 to electrode 2 (Fig. 10C3). The reduction in variability of the electrical traces by GABAAR antagonists is similar to the findings in organotypic slice cultures (Fig. 4C), supporting the idea that interneuron activity governs propagation pathways of IISs in human epileptic tissue.
Discussion
These experiments demonstrate that IISs spread via multiple pathways in epileptic patients (Asano et al., 2003; Bast et al., 2004). Direct visualization of the spread of spontaneous network activity in an in vitro model of chronic epilepsy and ex vivo recordings from chronically epileptic human brain demonstrate that GABA-R mediated activity is a key determinant of the variability in IIS propagation. This experimental result as well as the presence of heterogeneous clusters of activity preceding the burst were predicted by previous modeling studies (Traub et al., 1987a,b). Our computational model (Fig. 8) suggests that pathway variance is due to the quenching of small, local bursts of network activity by GABAergic inhibition. This idea is supported by data from the targeted path scanning experiments (Fig. 9) demonstrating partial network activations precede IISs (Traub et al., 1987a,b). We propose that the quenched small, local activations leave behind spatially restricted pockets of refractoriness. The locations of these pockets then determine the propagation pathway of the subsequent IIS in a manner analogous to pins guiding the trajectory of a pinball. Our findings offer a mechanistic explanation of the spike-to-spike variance of IISs and account for the similarity of IISs following focal application of GABA-R blockers (Dichter and Spencer 1969; Schwartzkroin and Prince 1977; Traub et al., 1987a,b).
Interneurons have been proposed to create neuronal assemblies by synchronizing the activity of populations of neurons (Buzsáki and Chrobak 1995; Cobb et al., 1995). Recent findings suggest that interneurons exert the sort of localized control necessary to define subpopulations of neurons that would influence the spatial propagation of synchronous network activity (Glickfeld et al., 2009; Bazelot et al., 2010). These assemblies (Pastalkova et al., 2008), or their remnants in chronically epileptic tissue, could form the anatomical basis for the pockets of refractory neurons formed by nonpropagating local synchronous activity. The resulting pathway variance illuminates a fundamental feature of interictal spikes: waveforms are different not because of spike-to-spike variance in individual neuronal responses, but rather the variance in the sequence of neuronal responses; the wiring diagram of each individual interictal spike is different. The proposed mechanism of spike variance opens several avenues of future investigation. What are the spatial distributions of the local, nonpropagated activity patterns? Electrographic evidence of local activations can be recognized in epileptic networks (de la Prida et al., 2006; Bazelot et al., 2010), and local stimuli alter the pattern of activation of epileptic networks (Miles and Wong 1983; Staley et al., 2001; de la Prida et al., 2006), but visualizing the spatial extent of these activations, perhaps using chloride-sensitive fluorophores (Berglund et al., 2008), will provide important experimental validation of the models proposed in Figures 7 and 8. What are the patterns on interneuron connectivity in normal and epileptic hippocampus? Are there particular spatial patterns of refractory pockets that predispose to sustained network activation, i.e., a seizure? If only a small minority of the possible patterns of refractoriness led to seizures, this would explain the relative paucity of seizures versus interictal spikes: in analogy to a combination lock, only particular patterns of activation would lead to seizures. The number of these ictogenic patterns compared to the total number of pathways of activation (as well as the frequency with which each pathway is activated) would govern the abundance of spikes compared to seizures. This could be tested by recording large numbers of spikes and seizures at a sufficiently high spatial and temporal resolution.
The reduction in spatial variance following blockade of inhibition coincided with a reduction in the variance in the interspike interval (Fig. 4, compare A1, B1). A perfectly homogenous disinhibited network would be expected to initiate bursts from a wide array of identical neurons; under such conditions, the spatial variance might be expected to increase, while the temporal variance might decrease. The reduced spatial and temporal variance after disinhibition may reflect the preferential activation of a subset of pathways in the absence of inhibition. In our modeling experiments, the use of small-world networks including hub cells promoted this reduction in variance following disinhibition. Although evidence for small-world networks in the brain remains quite modest, we did notice a small minority of cells that were more frequently involved in network activations in the targeted path scanning experiments (Fig. 9). Whether these cells are interneurons or more heavily connected principal cells will require much larger sampling and cell-type-specific labeling.
Our data underscore the possible differences between epileptic discharges recorded from chronically epileptic tissue versus normal tissue in which epileptiform activity is induced by acute blockade of inhibitory conductances. The different variances in the network activation pathways (Fig. 4A3,B3) indicate that convulsants can induce patterns of activity that are not seen in chronically epileptic networks. These differences in network activation in disinhibited versus spontaneously active tissue could underlie clinically important phenomena such as the similarly incomplete spectra of efficacy of anticonvulsants discovered by screening acutely disinhibited animals (French et al., 2004; Brodie, 2010). They may also explain why antiepileptic strategies that are effective in acutely disinhibited in vitro preparations (Bains et al., 1999) are not effective in chronically epileptic animals (Hellier et al., 2009).
Clinically, the dependence of spatial IIS variance on GABAergic mechanisms implies that this variance could provide insight into the integrity of GABAergic inhibition in the epileptic network, which may be of utility for prognosis and selection of therapeutic approach. If the spatial variance of IIS propagation can be detected in the EEG recorded from the scalp, this would comprise a new, noninvasive insight into the nature of the epileptic focus.
While spikes observed in ECoG records occur over much larger spatial scales (centimeters) than our in vitro data, two arguments can be made in support of the study of networks of widely different scales. First is the remarkable agreement of earlier modeling studies (Traub et al., 1987a,b) with the current results, despite the fact that the earlier study was accomplished using a network that was two orders of magnitude smaller than the computational and experimental preparations studied here. Second, the recent descriptions of “microseizures” lend support to the idea that smaller ensembles of neurons can reproduce some of the phenomena that have typically been observed at larger scales (Schevon et al., 2009; Stead et al., 2010).
Our finding that blockade of GABA-R reduces spike variance supports the role of GABAergic interneurons as major contributors to spike variance in larger cortical networks. Although our data were derived from pathological tissue, our findings converge with recent insights into the mechanisms guiding the flow of synchronization through cortical networks (Haider and McCormick 2009). GABAergic networks are recognized as providing temporal control during sharp waves and ripple oscillations (Buzsáki and Chrobak 1995; Ylinen et al., 1995) and the spread of epileptic activity (Prince and Wilder 1967; Schwartz and Bonhoeffer 2001; Trevelyan et al., 2006). Here we demonstrate that GABAergic networks play an important role in determining the spatial spread of synchrony by writing the recent history of the network activation into the pattern of locally refractory network elements.
Footnotes
This work was supported by NIH Grants 5R01NS034700 and 1R01NS074772.
References
- Alarcon G, Garcia Seoane JJ, Binnie CD, Martin Miguel MC, Juler J, Polkey CE, Elwes RD, Ortiz Blasco JM. Origin and propagation of interictal discharges in the acute electrocorticogram. Implications for pathophysiology and surgical treatment of temporal lobe epilepsy. Brain. 1997;120:2259–2282. doi: 10.1093/brain/120.12.2259. [DOI] [PubMed] [Google Scholar]
- Aradi I, Maccaferri G. Cell type-specific synaptic dynamics of synchronized bursting in the juvenile CA3 rat hippocampus. J Neurosci. 2004;24:9681–9692. doi: 10.1523/JNEUROSCI.2800-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano E, Muzik O, Shah A, Juhász C, Chugani DC, Sood S, Janisse J, Ergun EL, Ahn-Ewing J, Shen C, Gotman J, Chugani HT. Quantitative interictal subdural EEG analyses in children with neocortical epilepsy. Epilepsia. 2003;44:425–434. doi: 10.1046/j.1528-1157.2003.38902.x. [DOI] [PubMed] [Google Scholar]
- Bains JS, Longacher JM, Staley KJ. Reciprocal interactions between CA3 network activity and strength of recurrent collateral synapses. Nat Neurosci. 1999;2:720–726. doi: 10.1038/11184. [DOI] [PubMed] [Google Scholar]
- Bast T, Oezkan O, Rona S, Stippich C, Seitz A, Rupp A, Fauser S, Zentner J, Rating D, Scherg M. EEG and MEG source analysis of single and averaged interictal spikes reveals intrinsic epileptogenicity in focal cortical dysplasia. Epilepsia. 2004;45:621–631. doi: 10.1111/j.0013-9580.2004.56503.x. [DOI] [PubMed] [Google Scholar]
- Bausch SB, He S, Petrova Y, Wang XM, McNamara JO. Plasticity of both excitatory and inhibitory synapses is associated with seizures induced by removal of chronic blockade of activity in cultured hippocampus. J Neurophysiol. 2006;96:2151–2167. doi: 10.1152/jn.00355.2006. [DOI] [PubMed] [Google Scholar]
- Bazelot M, Dinocourt C, Cohen I, Miles R. Unitary inhibitory field potentials in the CA3 region of rat hippocampus. J Physiol. 2010;588:2077–2090. doi: 10.1113/jphysiol.2009.185918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beenhakker MP, Huguenard JR. Neurons that fire together also conspire together: is normal sleep circuitry hijacked to generate epilepsy? Neuron. 2009;62:612–632. doi: 10.1016/j.neuron.2009.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berdichevsky Y, Sabolek H, Levine JB, Staley KJ, Yarmush ML. Microfluidics and multielectrode array-compatible organotypic slice culture method. J Neurosci Methods. 2009;178:59–64. doi: 10.1016/j.jneumeth.2008.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berglund K, Schleich W, Wang H, Feng G, Hall WC, Kuner T, Augustine GJ. Imaging synaptic inhibition throughout the brain via genetically targeted Clomeleon. Brain Cell Biol. 2008;36:101–118. doi: 10.1007/s11068-008-9031-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifazi P, Goldin M, Picardo MA, Jorquera I, Cattani A, Bianconi G, Represa A, Ben-Ari Y, Cossart R. GABAergic hub neurons orchestrate synchrony in developing hippocampal networks. Science. 2009;326:1419–1424. doi: 10.1126/science.1175509. [DOI] [PubMed] [Google Scholar]
- Bourien J, Bartolomei F, Bellanger JJ, Gavaret M, Chauvel P, Wendling F. A method to identify reproducible subsets of co-activated structures during interictal spikes. Application to intracerebral EEG in temporal lobe epilepsy. Clin Neurophysiol. 2005;116:443–455. doi: 10.1016/j.clinph.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Bower MR, Buckmaster PS. Changes in granule cell firing rates precede locally recorded spontaneous seizures by minutes in an animal model of temporal lobe epilepsy. J Neurophysiol. 2008;99:2431–2442. doi: 10.1152/jn.01369.2007. [DOI] [PubMed] [Google Scholar]
- Brodie MJ. Antiepileptic drug therapy the story so far. Seizure. 2010;19:650–655. doi: 10.1016/j.seizure.2010.10.027. [DOI] [PubMed] [Google Scholar]
- Buzsáki G. Rhythms of the brain. New York: Oxford UP; 2006. [Google Scholar]
- Buzsáki G, Chrobak JJ. Temporal structure in spatially organized neuronal ensembles: a role for interneuronal networks. Curr Opin Neurobiol. 1995;5:504–510. doi: 10.1016/0959-4388(95)80012-3. [DOI] [PubMed] [Google Scholar]
- Chao ZC, Bakkum DJ, Potter SM. Region-specific network plasticity in simulated and living cortical networks: comparison of the center of activity trajectory (CAT) with other statistics. J Neural Eng. 2007;4:294–308. doi: 10.1088/1741-2560/4/3/015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatrian G, Bergamini L, Dondey M, Klass DW, Lennox-Buchthal M, Petersén I. A glossary of terms most commonly used by clinical electroencephalographers. Electroencephalogr Clin Neurophysiol. 1974;37:538–548. doi: 10.1016/0013-4694(74)90099-6. [DOI] [PubMed] [Google Scholar]
- Cobb SR, Buhl EH, Halasy K, Paulsen O, Somogyi P. Synchronization of neuronal activity in hippocampus by individual GABAergic interneurons. Nature. 1995;378:75–78. doi: 10.1038/378075a0. [DOI] [PubMed] [Google Scholar]
- Cohen I, Navarro V, Clemenceau S, Baulac M, Miles R. On the origin of interictal activity in human temporal lobe epilepsy in vitro. Science. 2002;298:1418–1421. doi: 10.1126/science.1076510. [DOI] [PubMed] [Google Scholar]
- Cohen I, Huberfeld G, Miles R. Emergence of disinhibition-induced synchrony in the CA3 region of the guinea pig hippocampus in vitro. J Physiol. 2006;570:583–594. doi: 10.1113/jphysiol.2005.097899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Prida LM, Huberfeld G, Cohen I, Miles R. Threshold behavior in the initiation of hippocampal population bursts. Neuron. 2006;49:131–142. doi: 10.1016/j.neuron.2005.10.034. [DOI] [PubMed] [Google Scholar]
- Debanne D, Guérineau NC, Gähwiler BH, Thompson SM. Physiology and pharmacology of unitary synaptic connections between pairs of cells in areas CA3 and CA1 of rat hippocampal slice cultures. J Neurophysiol. 1995;73:1282–1294. doi: 10.1152/jn.1995.73.3.1282. [DOI] [PubMed] [Google Scholar]
- Del Turco D, Deller T. Organotypic entorhino-hippocampal slice cultures—a tool to study the molecular and cellular regulation of axonal regeneration and collateral sprouting in vitro. Methods Mol Biol. 2007;399:55–66. doi: 10.1007/978-1-59745-504-6_5. [DOI] [PubMed] [Google Scholar]
- Dichter M, Spencer WA. Penicillin-induced interictal discharges from the cat hippocampus. I. Characteristics and topographical features. J Neurophysiol. 1969;32:649–662. doi: 10.1152/jn.1969.32.5.649. [DOI] [PubMed] [Google Scholar]
- Dobrunz LE, Stevens CF. Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron. 1997;18:995–1008. doi: 10.1016/s0896-6273(00)80338-4. [DOI] [PubMed] [Google Scholar]
- Dyhrfjeld-Johnsen J, Berdichevsky Y, Swiercz W, Sabolek H, Staley KJ. Interictal spikes precede ictal discharges in an organotypic hippocampal slice culture model of epileptogenesis. J Clinical Neurophysiol. 2010;27:418–424. doi: 10.1097/WNP.0b013e3181fe0709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esclapez M, Hirsch JC, Khazipov R, Ben-Ari Y, Bernard C. Operative GABAergic inhibition in hippocampal CA1 pyramidal neurons in experimental epilepsy. Proc Natl Acad Sci U S A. 1997;94:12151–12156. doi: 10.1073/pnas.94.22.12151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa F, Kavalali ET. NMDA receptor activation by spontaneous glutamatergic neurotransmission. J Neurophysiol. 2009;101:2290–2296. doi: 10.1152/jn.90754.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French JA, Kanner AM, Bautista J, Abou-Khalil B, Browne T, Harden CL, Theodore WH, Bazil C, Stern J, Schachter SC, Bergen D, Hirtz D, Montouris GD, Nespeca M, Gidal B, Marks WJ, Jr, Turk WR, Fischer JH, Bourgeois B, Wilner A. Efficacy and tolerability of the new antiepileptic drugs II: treatment of refractory epilepsy: report of the Therapeutics and Technology Assessment Subcommittee and Quality Standards Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology. 2004;62:1261–1273. doi: 10.1212/01.wnl.0000123695.22623.32. [DOI] [PubMed] [Google Scholar]
- Gähwiler BH. Organotypic monolayer cultures of nervous tissue. J Neurosci Methods. 1981;4:329–342. doi: 10.1016/0165-0270(81)90003-0. [DOI] [PubMed] [Google Scholar]
- Glickfeld LL, Roberts JD, Somogyi P, Scanziani M. Interneurons hyperpolarize pyramidal cells along their entire somatodendritic axis. Nat Neurosci. 2009;12:21–23. doi: 10.1038/nn.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haider B, McCormick DA. Rapid neocortical dynamics: cellular and network mechanisms. Neuron. 2009;62:171–189. doi: 10.1016/j.neuron.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellier JL, White A, Williams PA, Dudek FE, Staley KJ. NMDA receptor-mediated long-term alterations in epileptiform activity in experimental chronic epilepsy. Neuropharmacology. 2009;56:414–421. doi: 10.1016/j.neuropharm.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huberfeld G, Wittner L, Clemenceau S, Baulac M, Kaila K, Miles R, Rivera C. Perturbed chloride homeostasis and GABAergic signaling in human temporal lobe epilepsy. J Neurosci. 2007;27:9866–9873. doi: 10.1523/JNEUROSCI.2761-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huberfeld G, Menendez de la Prida L, Pallud J, Cohen I, Le Van Quyen M, Adam C, Clemenceau S, Baulac M, Miles R. Glutamatergic pre-ictal discharges emerge at the transition to seizure in human epilepsy. Nat Neurosci. 2011;14:627–634. doi: 10.1038/nn.2790. [DOI] [PubMed] [Google Scholar]
- Hufnagel A, Dümpelmann M, Zentner J, Schijns O, Elger CE. Clinical relevance of quantified intracranial interictal spike activity in presurgical evaluation of epilepsy. Epilepsia. 2000;41:467–478. doi: 10.1111/j.1528-1157.2000.tb00191.x. [DOI] [PubMed] [Google Scholar]
- Ibarz JM, Foffani G, Cid E, Inostroza M, Menendez de la Prida L. Emergent dynamics of fast ripples in the epileptic hippocampus. J Neurosci. 2010;30:16249–16261. doi: 10.1523/JNEUROSCI.3357-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikegaya Y, Aaron G, Cossart R, Aronov D, Lampl I, Ferster D, Yuste R. Synfire chains and cortical songs: temporal modules of cortical activity. Science. 2004;304:559–564. doi: 10.1126/science.1093173. [DOI] [PubMed] [Google Scholar]
- Jones J, Stubblefield EA, Benke TA, Staley KJ. Desynchronization of glutamate release prolongs synchronous CA3 network activity. J Neurophysiol. 2007;97:3812–3818. doi: 10.1152/jn.01310.2006. [DOI] [PubMed] [Google Scholar]
- Keller CJ, Truccolo W, Gale JT, Eskandar E, Thesen T, Carlson C, Devinsky O, Kuzniecky R, Doyle WK, Madsen JR, Schomer DL, Mehta AD, Brown EN, Hochberg LR, Ulbert I, Halgren E, Cash SS. Heterogeneous neuronal firing patterns during interictal epileptiform discharges in the human cortex. Brain. 2010;133:1668–1681. doi: 10.1093/brain/awq112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr JN, de Kock CP, Greenberg DS, Bruno RM, Sakmann B, Helmchen F. Spatial organization of neuronal population responses in layer 2/3 of rat barrel cortex. J Neurosci. 2007;27:13316–13328. doi: 10.1523/JNEUROSCI.2210-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaassen A, Glykys J, Maguire J, Labarca C, Mody I, Boulter J. Seizures and enhanced cortical GABAergic inhibition in two mouse models of human autosomal dominant nocturnal frontal lobe epilepsy. Proc Natl Acad Sci U S A. 2006;103:19152–19157. doi: 10.1073/pnas.0608215103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lillis KP, Eng A, White JA, Mertz J. Two-photon imaging of spatially extended neuronal network dynamics with high temporal resolution. J Neurosci Methods. 2008;172:178–184. doi: 10.1016/j.jneumeth.2008.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubenov EV, Siapas AG. Hippocampal theta oscillations are travelling waves. Nature. 2009;459:534–539. doi: 10.1038/nature08010. [DOI] [PubMed] [Google Scholar]
- Marsan CA. Electrographic aspects of “epileptic” neuronal aggregates. Epilepsia. 1961;2:22–23. doi: 10.1111/j.1528-1167.1961.tb06244.x. [DOI] [PubMed] [Google Scholar]
- Mazor O, Laurent G. Transient dynamics versus fixed points in odor representations by locust antennal lobe projection neurons. Neuron. 2005;48:661–673. doi: 10.1016/j.neuron.2005.09.032. [DOI] [PubMed] [Google Scholar]
- McBain CJ, Boden P, Hill RG. Rat hippocampal slices “in vitro” display spontaneous epileptiform activity following long-term organotypic culture. J Neurosci Methods. 1989;27:35–49. doi: 10.1016/0165-0270(89)90051-4. [DOI] [PubMed] [Google Scholar]
- McNamara JO. Emerging insights into the genesis of epilepsy. Nature. 1999;399:A15–A22. doi: 10.1038/399a015. [DOI] [PubMed] [Google Scholar]
- Mennerick S, Zorumski CF. Postsynaptic modulation of NMDA synaptic currents in rat hippocampal microcultures by paired-pulse stimulation. J Physiol. 1996;490:405–417. doi: 10.1113/jphysiol.1996.sp021154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles R, Wong RK. Single neurones can initiate synchronized population discharge in the hippocampus. Nature. 1983;306:371–373. doi: 10.1038/306371a0. [DOI] [PubMed] [Google Scholar]
- Miles R, Traub RD, Wong RK. Spread of synchronous firing in longitudinal slices from the CA3 region of the hippocampus. J Neurophysiol. 1988;60:1481–1496. doi: 10.1152/jn.1988.60.4.1481. [DOI] [PubMed] [Google Scholar]
- Morgan RJ, Soltesz I. Nonrandom connectivity of the epileptic dentate gyrus predicts a major role for neuronal hubs in seizures. Proc Natl Acad Sci U S A. 2008;105:6179–6184. doi: 10.1073/pnas.0801372105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsubo H, Ochi A, Elliott I, Chuang SH, Rutka JT, Jay V, Aung M, Sobel DF, Snead OC. MEG predicts epileptic zone in lesional extrahippocampal epilepsy: 12 pediatric surgery cases. Epilepsia. 2001;42:1523–1530. doi: 10.1046/j.1528-1157.2001.16701.x. [DOI] [PubMed] [Google Scholar]
- Pastalkova E, Itskov V, Amarasingham A, Buzsáki G. Internally generated cell assembly sequences in the rat hippocampus. Science. 2008;321:1322–1327. doi: 10.1126/science.1159775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlidis P, Madison DV. Synaptic transmission in pair recordings from CA3 pyramidal cells in organotypic culture. J Neurophysiol. 1999;81:2787–2797. doi: 10.1152/jn.1999.81.6.2787. [DOI] [PubMed] [Google Scholar]
- Prince DA, Wilder BJ. Control mechanisms in cortical epileptogenic foci. “Surround” inhibition. Arch Neurol. 1967;16:194–202. doi: 10.1001/archneur.1967.00470200082007. [DOI] [PubMed] [Google Scholar]
- Schevon CA, Trevelyan AJ, Schroeder CE, Goodman RR, McKhann G, Jr, Emerson RG. Spatial characterization of interictal high frequency oscillations in epileptic neocortex. Brain. 2009;132:3047–3059. doi: 10.1093/brain/awp222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz TH, Bonhoeffer T. In vivo optical mapping of epileptic foci and surround inhibition in ferret cerebral cortex. Nat Med. 2001;7:1063–1067. doi: 10.1038/nm0901-1063. [DOI] [PubMed] [Google Scholar]
- Schwartzkroin PA, Prince DA. Penicillin-induced epileptiform activity in the hippocampal in vitro preparation. Ann Neurol. 1977;1:463–469. doi: 10.1002/ana.410010510. [DOI] [PubMed] [Google Scholar]
- Staley KJ, Bains JS, Yee A, Hellier J, Longacher JM. Statistical model relating CA3 burst probability to recovery from burst-induced depression at recurrent collateral synapses. J Neurophysiol. 2001;86:2736–2747. doi: 10.1152/jn.2001.86.6.2736. [DOI] [PubMed] [Google Scholar]
- Stead M, Bower M, Brinkmann BH, Lee K, Marsh WR, Meyer FB, Litt B, Van Gompel J, Worrell GA. Microseizures and the spatiotemporal scales of human partial epilepsy. Brain. 2010;133:2789–2797. doi: 10.1093/brain/awq190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiercz W, Cios KJ, Staley K, Kurgan L, Accurso F, Sagel S. A new synaptic plasticity rule for networks of spiking neurons. IEEE Trans Neural Netw. 2006;17:94–105. doi: 10.1109/TNN.2005.860834. [DOI] [PubMed] [Google Scholar]
- Swiercz W, Cios K, Hellier J, Yee A, Staley K. Effects of synaptic depression and recovery on synchronous network activity. J Clin Neurophysiol. 2007;24:165–174. doi: 10.1097/WNP.0b013e318033756f. [DOI] [PubMed] [Google Scholar]
- Traub RD, Miles R. Neuronal networks of the hippocampus. Cambridge, UK: Cambridge UP; 1991. [Google Scholar]
- Traub RD, Miles R, Wong RK. Models of synchronized hippocampal bursts in the presence of inhibition. I. Single population events. J Neurophysiol. 1987a;58:739–751. doi: 10.1152/jn.1987.58.4.739. [DOI] [PubMed] [Google Scholar]
- Traub RD, Miles R, Wong RK, Schulman LS, Schneiderman JH. Models of synchronized hippocampal bursts in the presence of inhibition. II. Ongoing spontaneous population events. J Neurophysiol. 1987b;58:752–764. doi: 10.1152/jn.1987.58.4.752. [DOI] [PubMed] [Google Scholar]
- Trevelyan AJ, Sussillo D, Watson BO, Yuste R. Modular propagation of epileptiform activity: evidence for an inhibitory veto in neocortex. J Neurosci. 2006;26:12447–12455. doi: 10.1523/JNEUROSCI.2787-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlhaas PJ, Pipa G, Lima B, Melloni L, Neuenschwander S, Nikolić D, Singer W. Neural synchrony in cortical networks: history, concept and current status. Front Integr Neurosci. 2009;3:17. doi: 10.3389/neuro.07.017.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulbert I, Heit G, Madsen J, Karmos G, Halgren E. Laminar analysis of human neocortical interictal spike generation and propagation: current source density and multiunit analysis in vivo. Epilepsia. 2004;45(Suppl 4):48–56. doi: 10.1111/j.0013-9580.2004.04011.x. [DOI] [PubMed] [Google Scholar]
- White A, Williams PA, Hellier JL, Clark S, Dudek FE, Staley KJ. EEG spike activity precedes epilepsy after kainate-induced status epilepticus. Epilepsia. 2010;51:371–383. doi: 10.1111/j.1528-1167.2009.02339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittner L, Miles R. Factors defining a pacemaker region for synchrony in the hippocampus. J Physiol. 2007;584:867–883. doi: 10.1113/jphysiol.2007.138131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worrell GA, Lagerlund TD, Buchhalter JR. Role and limitations of routine and ambulatory scalp electroencephalography in diagnosing and managing seizures. Mayo Clinic Proc. 2002;77:991–998. doi: 10.4065/77.9.991. [DOI] [PubMed] [Google Scholar]
- Ylinen A, Bragin A, Nádasdy Z, Jandó G, Szabó I, Sik A, Buzsáki G. Sharp wave-associated high-frequency oscillation (200 Hz) in the intact hippocampus: network and intracellular mechanisms. J Neurosci. 1995;15:30–46. doi: 10.1523/JNEUROSCI.15-01-00030.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang MM, McArthur JR, Azam L, Bulaj G, Olivera BM, French RJ, Yoshikami D. Synergistic and antagonistic interactions between tetrodotoxin and mu-conotoxin in blocking voltage-gated sodium channels. Channels (Austin) 2009;3:32–38. doi: 10.4161/chan.3.1.7500. [DOI] [PMC free article] [PubMed] [Google Scholar]