

Abstract
The vertebrate vasculature forms an extensive branched network of blood vessels that supplies tissues with nutrients and oxygen. During vascular development, coordinated control of endothelial cell behaviour at the levels of cell migration, proliferation, polarity, differentiation and cell–cell communication is critical for functional blood vessel morphogenesis. Recent data uncover elaborate transcriptional, post-transcriptional and post-translational mechanisms that fine-tune key signalling pathways (such as the vascular endothelial growth factor and Notch pathways) to control endothelial cell behaviour during blood vessel sprouting (angiogenesis). These emerging frameworks controlling angiogenesis provide unique insights into fundamental biological processes common to other systems, such as tissue branching morphogenesis, mechanotransduction and tubulogenesis.
During embryonic development, endothelial cells (ECs) assemble the tree-like tubular network of blood vessels that eventually permits the transport of fluids, nutrients, circulating cells, hormones and gasses to almost all tissues throughout the vertebrate body (FIG. 1). Once mature, the vasculature consists of an elaborate hierarchical system of arteries, arterioles, capillaries, venules and veins that promotes the circulation of oxygenated blood between the heart, lungs and target tissues. Despite the complexity of the vascular system, virtually all blood vessels that form during development and growth arise by the sprouting of new capillaries from pre-existing vessels, a process termed angiogenesis. Moreover, imbalances in angiogenesis contribute to the pathogenesis of numerous disease states1. For example, insufficient angiogenesis is a principal factor limiting tissue recovery in ischaemic diseases, whereas stimulation of angiogenesis by cancer cells promotes tumour neovascularization, growth and progression to metastasis. Considering the substantial clinical benefits of therapeutically manipulating pathological angiogenesis, the mechanisms controlling this process have formed a major focus for vascular research over the last two decades1.
Figure 1. Development of a functional vasculature from endothelial progenitor cells.

Endothelial progenitors (angioblasts) differentiate from mesodermal cells during early vertebrate development. Once formed, angioblasts may acquire arterial (red) or venous (blue) fates and coalesce to generate the first embryonic blood vessels, the dorsal aorta and cardinal vein. In zebrafish, the coordinated sorting and segregation of arterial and venous angioblasts ensures the assembly of distinct dorsal aorta and cardinal vein vessels. Angioblasts also aggregate to form blood islands, which fuse and remodel in response to haemodynamic stimuli or inherent genetic factors to generate a primitive interlaced network of arterial and venous plexi. Following their vasculogenic assembly, angiogenic remodelling of the dorsal aorta, cardinal vein and vascular plexi creates a complex hierarchical network of arteries, arterioles, capillary beds, venules and veins. Subsequent recruitment of mural cells (pericytes and vascular smooth-muscle cells (vSMCs)) stabilizes nascent vessels and promotes vessel maturation. In addition, the sprouting of lymphatic endothelial cells from venous vessels (lymphangiogenesis) seeds the lymphatic system (indicated by a dotted arrow). Moreover, the emergence of haematopoietic stem cells from arterial ‘haemogenic’ endothelium gives rise to all myeloid and lymphoid blood cell lineages. Vessel diversity is further augmented by tissue-specific specializations that alter key properties, such as permeability, or modify endothelial cells to generate vascular networks with new molecular signatures3,123.
This Review discusses new advances in our understanding of blood vessel morphogenesis, with particular emphasis on emerging mechanisms that control and coordinate EC behaviour during angiogenic sprouting and vascular tube formation. In recent years, a number of seminal studies have begun to unravel the molecular basis of these complex processes in vivo, and they have uncovered remarkable parallels between the signalling pathways that control angiogenesis and the development of epithelial and neuronal tissues. In addition, recent studies reveal intricate post-transcriptional and post-translational mechanisms that direct context-dependent fine-tuning of EC behaviour and allow the integration of cues from the extracellular environment to control angiogenesis. We do not touch on later aspects of vascular development, such as blood vessel maturation, vascular bed-specific specializations or development of the lymphatic vasculature, but refer the reader to excellent reviews covering these topics2–6. We instead limit our discussion to recent developments that alter our view of the early morphogenetic events in angiogenesis. Importantly, a recurring theme throughout this Review is that angiogenesis represents a paradigm for many core biological processes, such as tissue branching, guided cell migration and lumen formation, all of which have wide-reaching roles in the morphogenesis of many other organ systems.
Vascular development and angiogenesis
The de novo formation of embryonic blood vessels (vasculogenesis) involves the differentiation, migration and coalescence of mesoderm-derived endothelial progenitors (angioblasts) to create a primordial vascular network1,2 (FIG. 1). Immediately following vasculogenic blood vessel assembly, ECs undergo specification to either arterial or venous fate in response to a combination of haemodynamic stimuli and underlying genetic factors2,3. For example, Notch-mediated expression of the hairy- and enhancer of split-related with YRPW motif (HEY) basic helix–loop–helix transcription factor genes, HEY1 and HEY2, promotes arterial differentiation7,8. By contrast, the orphan nuclear receptor COUP transcription factor 2 may repress Notch signalling to promote venous differentiation9. Subsequent angiogenic remodelling of these arterial and venous endothelial tissues and the extensive expansion of this primitive network lead to the formation of a complex functional vascular system1,2. Later in development, vessels that arise by angiogenesis may also adopt diverse vascular bed-specific functional specializations in response to local tissue-derived signals2,3 (FIG. 1). In addition, the venous endothelium gives rise to the blind-ended network (that is, closed at one end) of lymphatic vessels that collect and return lymph fluid back to the vasculature4. Thus, ECs have a remarkably plastic capacity to give rise to vessels with diverse functional, morphological and molecular signatures3. Furthermore, a subset of ‘haemogenic’ arterial endothelium generates definitive haematopoietic stem cells that seed the adult haematopoietic system10. Hence, blood vessel morphogenesis not only gives rise to the mature vasculature but also influences the formation and function of a diverse range of vital tissues.
Cellular mechanisms of angiogenesis
Blood vessel formation by angiogenesis is a complex multistep process that critically requires the tight control and coordination of EC behaviour (FIG. 2). In stable vessels, ECs typically form a cobblestone-like monolayer of inactive cells that lines the luminal surface of vascular tubes11. This quiescent phenotype is maintained until ECs detect pro-angiogenic signals that induce fundamental changes in their behaviour. In response to angiogenic cues, ECs loosen their cell–cell junctional contacts, activate pro-teases that degrade the surrounding basement membrane and acquire extensively invasive and motile behaviour to initiate new blood vessel sprouting1,2 (FIG. 2b). For example, disruption of angiopoietin 1 (ANG1) signalling via the TIE2 (also known as TEK) receptor Tyr kinase by the antagonist ANG2 is thought to destabilize vessels and have key roles during early angiogenic vascular remodelling (this is covered in another review12). Importantly, of the ECs that sense angiogenic stimuli, only a small proportion will be selected to lead newly sprouting vessels. These endothelial ‘tip cells’ (TCs) extend numerous dynamic filopodial extensions that sense and respond to attractive or repulsive guidance signals within their immediate microenvironment13,14. Hence, TCs share many morphological and functional similarities with the neuronal growth cones that guide axons15. By contrast, ECs that trail TCs (‘stalk cells’ (SCs)) are less motile but critically support the extension of sprouting vessels, generate the trunk of new capillaries and maintain connectivity with the parental vessel. Furthermore, SCs are thought to establish a vascular lumen in growing vessels2,16,17 (FIG.2c). This subdivision of sprouting ECs into leading TCs and following SCs is strikingly analogous to the hierarchical organization of epithelial TCs and SCs during Drosophila melanogaster tracheal branching morphogenesis18.
Figure 2. Cellular mechanisms of angiogenic sprouting.

a | In the absence of pro-angiogenic stimuli, endothelial cells (ECs) are retained in a quiescent state. In addition, EC homeostasis is maintained by low-level autocrine vascular endothelial growth factor A (VEGFA) signalling124. b | During angiogenesis, high levels of exogenous pro-angiogenic factors (such as VEGFA and VEGFC) and of VEGF receptor 2 (VEGFR2) or VEGFR3 signalling select ‘tip cells’ (TCs; blue) for sprouting. By contrast, Delta-like 4–Notch signalling laterally inhibits TC fate in adjacent ECs. TC sprouting behaviour is facilitated by the vascular endothelial cadherin-mediated loosening of EC–EC junctions, matrix metalloproteinase-mediated degradation of extracellular matrix (ECM) and the detachment of pericytes (purple). c | Invasive TC sprouting is guided by gradients of pro-angiogenic growth factors and various environmental guidance cues, such as semaphorins and ephrins. During sprout elongation, TCs are trailed by endothelial ‘stalk cells’ (SCs; yellow), which maintain connectivity with parental vessels and initiate partitioning-defective 3 (PAR3)-mediated vascular lumen morphogenesis. Expression of VEGFR1 and activation of Notch, Roundabout homologue 4 and WNT signalling in SCs repress TC behaviour to maintain the hierarchical organization of sprouting ECs. However, TCs and SCs may also shuffle and exchange positions during angiogenic sprouting. Upon contact with other vessels, TC behaviour is repressed and vessels fuse by the process of anastomosis, which is assisted by associated myeloid cells. d | Nascent perfused vessels are subsequently stabilized by the platelet-derived growth factor B-mediated recruitment of supporting pericytes, the strengthening of EC–EC contacts and the deposition of an ECM to re-establish a quiescent endothelial phenotype. ANG2, angiopoietin 2.
Once initiated, EC sprouting continues in a highly directional manner until TCs connect with adjacent vessels and undergo anastomosis, which leads to the fusion of the contacting vessels (FIG. 2c). On contact with other ECs, TCs lose their motile phenotype, generate tight EC–EC junctions and fuse with recipient vessels to form a continuous unobstructed (or patent) lumen, which allows blood flow. Although our understanding of the anastomotic process is rather limited, it is clear that other cell types may also influence vessel fusion. In particular, certain macrophage populations may act as cellular chaperones that promote vascular anastomosis19. Furthermore, the recruitment of other accessory cells is a critical factor in the subsequent maturation of this nascent vasculature. Factors such as platelet-derived growth factor B (PDGFB) and transforming growth factor-β1 (TGFβ1) recruit mural cells (pericytes and vascular smooth muscle cells) to the developing vasculature, which stabilizes vessel walls2,5,6 (BOX 1). Furthermore, deposition of the basement membrane at the abluminal surface and strengthening of cell–cell junctions suppress EC sprouting behaviour and re-establish a mature quiescent phenotype (FIG. 2d). Subsequent rounds of angiogenesis then allow further expansion of the vasculature. In addition, the intussusceptive insertion of pillars of tissue into blood vessel lumina promotes the splitting of vessels and permits additional remodelling of pre-existing vascular networks20. However, the molecular mechanisms of intussusceptive angiogenesis remain unclear. Simultaneous pruning of superfluous vessels allows overall remodelling of this actively growing network into a mature vascular bed.
Box 1. Endothelial cell–mural cell interactions.
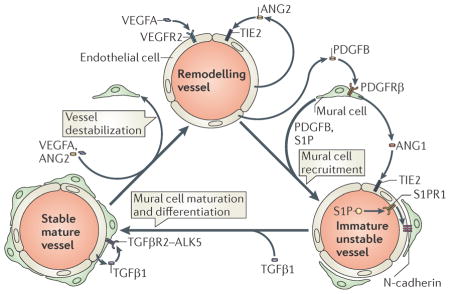
The recruitment of mural cells (pericytes and vascular smooth muscle cells (vSMCs)) to nascent blood vessels plays an essential part in the stabilization and maturation of new vascular networks. Whereas pericytes primarily associate with small-calibre capillaries, vSMCs ensheath larger arteries and veins. Initially, platelet-derived growth factor B (PDGFB) is released from endothelial cells (ECs) undergoing angiogenic remodelling (see the figure). Extracellular matrix-bound PDGFB acts as a chemoattractant for co-migrating pericytes that express the receptor Tyr kinase PDGF receptor β (PDGFRβ)5,6,112,113. Recruited pericytes are incorporated into the wall of immature vessels and establish direct cell–cell contacts with ECs. EC–mural cell contacts may be strengthened by the bioactive lipid spingosine 1 phosphate (S1P) through signalling via its EC expressed guanine nucleotide binding protein (G protein) coupled receptor, S1P receptor 1 (S1PR1; also known as EDG1)5,6,114,115. Secretion of S1P by platelets, haematopoietic cells and ECs may promote S1PR1 mediated trafficking of neural cadherin (N-cadherin) to areas of EC–pericyte contact and the assembly of adherens junctions116. Furthermore, angiopoietin 1 (ANG1) release from mural cells activates the TIE2 receptor in ECs117, which is thought to promote EC survival, maintain EC quiescence and mediate mural cell attachment5,6,12. However, a recent study using conditional Ang1 out mice questions some of these previous findings118. In addition, work in mice suggests that ephrin B2–ephrin receptor (EPH) Tyr kinase interactions may also have key roles in mural cell motility and adhesion to nascent vessels119. Subsequent differentiation of pericytes into vSMCs is poorly understood but can be induced, in part, by the release of EC-derived transforming growth factor β1 (TGFβ1). Activation of activin receptor like kinase 5 (ALK5; also known as TGFβR1) (which forms a heteromeric complex with TGFβ receptor type 2 (TGFβR2)) by TGFβ1 in mural cells may then promote vSMC differentiation120 to generate vSMC-ensheathed quiescent mature vessels5,6. In mice, Notch 3 signalling may also modulate the subsequent maturation of arterial vSMCs121. Importantly, this whole process can be reversed in response to pro angiogenic signals, such as vascular endothelial growth factor A (VEGFA) or the TIE2 antagonist ANG2, which promote mural cell detachment and vessel destabilization to allow further rounds of vascular remodelling12,122. VEGFR2, VEGF receptor 2.
VEGF stimulates angiogenic sprouting
Several general signalling pathways control EC behaviour during angiogenic sprouting, including TIE2 and Notch signalling (FIG. 3a), as does signalling via known axon guidance receptors that are expressed in ECs (FIG. 3b). However, the secreted growth factor vascular endothelial growth factor A (VEGFA; also known as vascular permeability factor (VPF)) is the principal master regulator of new blood vessel sprouting during development, growth and disease21–23 (FIG. 3a). VEGFA is the best-characterized member of a family of homodimeric glycoproteins that includes placental growth factor (PLGF), VEGFB, VEGFC and VEGFD21–23. During angiogenesis, VEGFA binds its cognate receptor Tyr kinase, VEGF receptor-2 (VEGFR2; also known as KDR and FLK1), and activates multiple downstream pathways via signalling intermediates, such as mitogen-activated protein kinases (MAPKs), phosphoinositide 3-kinases (PI3Ks), AKT, phospholipase Cγ and small GTPases21–23. As a result, VEGF signalling promotes EC proliferation, filopodial extension, degradation of the extracellular matrix and chemotaxis2,14. Hence, VEGFA signalling induces the motile and invasive behaviour that drives TC sprouting and activates the angiogenic switch14 (FIG. 4a). Expression of VEGFA is, chiefly, positively regulated by hypoxia, thus the angiogenic response is rapidly initiated in response to tissue oxygen deficiency during development, growth and disease24. Consequently, heterozygous Vegfa+/− mice manifest severe vascular defects and display haploinsufficient lethality25,26. Similarly, early embryonic lethality and deficient vascular assembly are observed in Vegfr2-null mice27.
Figure 3. Key signalling pathways that control angiogenesis.

a | General signalling pathways that control endothelial cell (EC) behaviour. Mammalian vascular endothelial growth factors (VEGFs) bind homodimers and heterodimers of three VEGF receptors (VEGFRs). Signalling via VEGFR2, VEGFR3 or VEGFR2 VEGFR3 heterodimers is pro-angiogenic. Proteolytic processing of VEGFC and VEGFD is required to permit their interaction with VEGFR2. VEGFA binding to VEGFR1 and the secreted VEGFR1 extracellular domain (soluble VEGFR1 (sVEGFR1)) acts as a sink for VEGFA that limits its availability to activate VEGFR2. The interaction of TIE2 receptor with matrix-associated angiopoietin 1 (ANG1) at EC–extracellular matrix (ECM) junctions induces migration125. By contrast, at EC–EC junctions ANG1–TIE2 interactions promote quiescence upon trans-complex formation with TIE2 on adjacent cells. These complexes include vascular endothelial protein Tyr phosphatase (VE-PTP; also known as PTPRB) and activate distinct signalling pathways from those at cell–matrix contacts125. ANG2 antagonizes ANG1 activity on TIE2 to destabilize vessels and aid angiogenic remodelling. Homophilic VE-cadherin interactions maintain EC–EC junctions. Delta-like 4 (DLL4)-mediated activation of Notch receptors represses angiogenic cell behaviour and promotes vessel stability upon the proteolytic release of the Notch intracellular domain (NICD). In certain contexts, Jagged 1 competes with DLL4 for Notch to decrease DLL4–Notch-mediated signalling. b | Known axon guidance receptors expressed in ECs. Roundabout homologue 4 (ROBO4)–uncoordinated 5 homologue B (UNC5B) interactions promote UNC5B-mediated inhibition of VEGFR signalling, block angiogenesis and maintain vessel integrity. Activation of ROBO4 by Slit 2 may also block VEGFR signalling but is controversial. Activation of UNC5B by netrins may also disrupt angiogenesis. Secreted class III semaphorins (such as semaphorin 3E (SEMA3E)) promote EC repulsion to perturb angiogenesis upon binding their receptor, plexin D1. By contrast, neuropilin 1 (NRP1) or NRP2 augment angiogenic EC behaviour on binding VEGFA or VEGFC and/or on interaction with VEGFR2 or VEGFR3. Activation of ephrin receptor B4 (EPHB4) upon interaction with its membrane-associated ligand, ephrin B2, may promote EC–EC repulsion or attraction in various cellular contexts and is essential for angiogenesis. Reverse EPHB4–ephrin B2 signalling may also play key parts in vascular development. Importantly, association of ephrin B2 with VEGFR2 or VEGFR3 promotes membrane internalization of the VEGFR and enhances angiogenic signalling. PLGF, placental growth factor.
Figure 4. Molecular mechanisms of endothelial tip cell selection.

a | Sprouting endothelial cells are hierarchically organized into leading ‘tip cells’ (TCs) and trailing ‘stalk cells’ (SCs) that exhibit very distinct and specialized cell behaviours. TC formation is induced by vascular endothelial growth factor (VEGF) signalling, whereas Delta-like 4 (DLL4)–Notch signalling represses VEGF receptor (VEGFR) signalling and TC fate in SCs. b | VEGFA and VEGFC signalling via VEGFR2 and VEGFR3 induce the invasive and motile TC behaviour that drives angiogenesis. VEGFR2 activation induces DLL4 expression in TCs, which activates Notch on adjacent SCs. VEGF-mediated disruption of a repressive translocation ETS leukaemia (TEL) and carboxy-terminal-binding protein (CtBP) complex that binds theDLL4 promoter may partially account for the ability of VEGF to induceDLL4 expression. Notch signalling in SCs downregulates the expression of VEGFR3 (encoded by FLT4) and upregulates the expression of VEGFR1 (encoded by FLT1) and soluble VEGFR1 (sVEGFR1), which represses VEGFR2 function and blocks TC behaviour. Notch activation also inducesDLL4 expression in SCs to propagate the DLL4 Notch-mediated lateral inhibition of VEGFR2 and VEGFR3 along developing vessels. Notch-induced Notch-regulated ankyrin repeat-containing protein (NRARP) expression enhances WNT signalling in SCs, which maintains EC–EC junctions, promotes proliferation and may augment DLL4 expression via β-catenin. NRARP also promotes feedback inhibition of Notch signalling. Notch signalling in TCs is blocked by Jagged 1, which is expressed in SCs and impedes DLL4–Notch interactions on TCs when Notch is glycosylated (Gl) by Fringe family glycosyltransferases in the Golgi apparatus. Deacetylation of the Notch intracellular domain (NICD) by sirtuin 1 (SIRT1) may also negatively influence TC Notch signalling. CLDN5, claudin 5; LRP, low-density lipoprotein receptor-related protein.
Alternative splicing of the VEGFA transcript gives rise to a number of protein variants that have divergent functions and bioavailabilities during blood vessel morphogenesis1,2,28. For example, the heparan sulphate-binding isoform VEGFA165 (VEGFA164 in mice) is matrix associated and forms gradients that promote the extension of filopodia, the directional migration of ECs and the branching of blood vessels29. By contrast, VEGFA121 (VEGFA120 in mice) is unable to bind heparan sulphate, is freely diffusible and influences EC proliferation but not migration29. Recent evidence indicates that the VEGFR2 signalling output elicited by matrix-associated VEGFA is also distinct to that induced by soluble isoforms30. Moreover, VEGFA splice variants with altered carboxyl termini (b isoforms) possess greatly reduced abilities to activate VEGFR2 (REFS 28,31). Hence, tight spatial regulation of the production of distinct VEGFA isoforms is a key control point during blood vessel morphogenesis.
In addition to VEGFR2, VEGFA binds to the related receptor Tyr kinase VEGFR1 (encoded by FLT1) (FIG. 3a), which has a higher affinity for VEGFA than VEGFR2 but possesses weak Tyr kinase activity. Hence, VEGFR1 is considered to be a decoy receptor that counteracts pro-angiogenic signalling32. Furthermore, alternative splicing of VEGFR1 generates a secreted and catalytically inactive isoform (soluble VEGFR1 (sVEGFR1) that acts as a sink for free VEGFA. Consequently, Flt1 knockout is associated with EC overgrowth, abnormal angiogenesis and embryonic lethality in mice32–34. Similarly, flt1 knockdown enhances angiogenic EC behaviour in zebrafish35. In addition, Flt1 expression in non-EC cell types may also directly influence angiogenesis. For example, recent data indicate that WNT signalling-induced Flt1 expression in retinal myeloid cells suppresses angiogenesis in the mouse retina36. Other VEGF family members, such as PLGF and VEGFB, selectively bind VEGFR1; however, their functional role during vascular development is unclear.
VEGFC is another emerging determinant of vascular development. VEGFC binding to its receptor, VEGFR3 (encoded by FLT4) (FIG. 3a), is well-known to play key parts during lymphangiogenesis4. However, VEGFR3 signalling also positively regulates angiogenesis37. In particular, VEGFR3 is highly expressed in TCs and is required for EC sprouting in mice and zebrafish37,38. Furthermore, recent evidence indicates that VEGFC promotes the assembly of VEGFR2–VEGFR3 heterodimers, which are enriched in TCs and positively influence angiogenic sprouting39. Hence, the correct spatiotemporal expression of VEGFR3 is increasingly considered to be an important determinant of TC function, as is discussed below.
Mechanisms of TC and SC selection
Angiogenesis requires the hierarchical organization of sprouting cells into leading TCs and trailing SCs. This coordination of cell migratory behaviour is a key aspect of tissue branching morphogenesis in many systems18. But why are only some ECs selected as TCs in response to VEGFA signalling? In recent years, our understanding of the molecular mechanisms of TC selection has benefited greatly from the development of in vivo models of blood vessel sprouting. In particular, studies of TC and SC organization and behaviour during retinal angiogenesis in mice, and intersegmental vessel (ISV) sprouting in zebrafish, have allowed dissection of the pathways involved at the level of single cells in vivo. These studies reveal remarkable conservation of the mechanisms controlling angiogenesis between vertebrate species.
Notch signalling controls TC and SC fate
Notch signalling is an evolutionarily conserved pathway that has well-established roles in the determination of cell fate in all metazoans40. Ligand binding induces Notch receptor cleavage (by ADAM10 (a disintegrin and metalloprotease 10), ADAM17 and presenilins) and the release of an intracellular fragment (known as the Notch intra-cellular domain (NICD)) (FIG. 3a), which functions as a key transcriptional regulator during cell-fate specification. ECs express multiple Notch receptors (NOTCH1, NOTCH3 and NOTCH4) and transmembrane Notch ligands (Delta-like 1 (DLL1), DLL4, Jagged 1 and Jagged 2) (FIG. 3a), and recent studies demonstrate that Notch signalling plays a critical part in TC and SC fate decisions during angiogenesis40,41. Studies in mouse and zebrafish reveal that expression of Dll4 and dll4, respectively, in TCs activates Notch in adjacent SCs to laterally inhibit TC fate and maintain the hierarchical organization of sprouting ECs38,42–47 (FIG. 4a). VEGFA–VEGFR2 signalling promotes DLL4 expression in TCs (FIG. 4b). Hence, ECs experiencing the highest level of VEGF signalling will be selected as TCs, whereas in adjacent cells TC fate is repressed. Consequently, reduced DLL4–Notch signalling in vivo is accompanied by excessive TC formation, expanded expression of TC-associated genes (such as FLT4, uncoordinated 5 homologue B (UNC5B) and PDGFB), increased EC proliferation, uncontrolled sprouting and disordered vessel branching38,42–45. Furthermore, elegant mosaic studies in zebrafish reveal that ECs expressing a constitutively active form of Notch are excluded from the TC position of sprouting vessels38. In addition, disruption of Notch cleavage on the EC-specific knockout of ADAM10 also promotes enhanced EC sprouting48. The role of DLL4–Notch signalling in suppressing TC specification is not only critical for physiological blood vessel formation but also controls tumour angiogenesis. Tumours are extensively vascularized on antibody-mediated reduction of DLL4–Notch interactions but vessels are hypoperfused and poorly functional, resulting in tumour hypoxia and decreased growth46,47. Hence, DLL4 is considered to be a promising target for anticancer therapeutics. However, recent evidence suggesting that chronic DLL4 blockade induces vascular tumours may hamper this effort49.
DLL4–Notch signalling efficiently suppresses TC fate by negatively regulating VEGFR signalling. In particular, Notch activation inhibits VEGFR2 function and blocks FLT4 expression in SCs and consequently renders them less responsive to VEGF-mediated TC-inducing signals38,44–46,50 (FIG. 4b). For example, flt4 expression in zebrafish is normally restricted to endothelial TCs but is expanded throughout the SC domain in the absence of Notch signalling as a consequence of increased TC formation38. Consequently, ectopic flt4 expression drives the hyper-sprouting phenotype observed in Notch signalling-deficient zebrafish embryos37,38,45. However, a recent study in zebrafish proposes that Dll4 suppresses Vegfc-induced Vegfr3 function but does not influence the expression of Vegfr3’s encoding gene, flt4, suggesting additional roles for other Notch ligands in the transcriptional control of flt4 (REF. 51). By contrast, Notch signalling positively regulates FLT1 expression52 (FIG. 4b). In this context, VEGFR1 is likely to act as a negative determinant of TC behaviour by acting as a decoy receptor for VEGFA to further prevent VEGFA-mediated TC specification. Consistent with this concept, knockdown of flt1 in zebrafish promotes increased TC formation and vessel hyperbranching35. Interestingly, recent data indicate that ECs can rapidly shift between the TC and SC positions during angiogenic sprouting in vitro and during the sprouting of blood vessels in the head of zebrafish53. Hence, TC and SC fates may be somewhat plastic rather than rigidly fixed. Consequently, sprouting ECs continually shuffle and compete for the TC position by dynamically regulating their levels of VEGFR2 versus FLT1 expression53. Furthermore, sFLT1 expression in ECs adjacent to TCs was recently shown to act as a spatial cue that guides new angiogenic sprouts away from the parental vessel54. Hence, in addition to controlling VEGF signalling, the DLL4–Notch pathway may indirectly influence local guidance of sprouting vessels.
Control of TC specification by DLL4–Notch signalling shares remarkable similarities with the molecular mechanisms of epithelial tracheal branching in D. melanogaster. Similar to the role of VEGF signalling in endothelial TC formation, tracheal TCs are specified by the fibroblast growth factor (FGF) ligand Branchless (BNL). Furthermore, in this system, Notch signalling via the ligand Delta limits excessive epithelial TC formation by repressing expression of the BNL receptor, breathless (btl), in SCs. Hence, the Notch-mediated lateral inhibition of receptor Tyr kinase expression controls the correct specification of TC numbers, position and behaviour during tissue branching morphogenesis in multiple systems.
Whereas DLL4-mediated Notch signalling negatively regulates TC selection, SC-restricted expression of another Notch ligand, Jagged 1, promotes TC formation and angiogenesis55 (FIG. 4b). In sprouting ECs, the extracellular domain of Notch is post-translationally glycosylated by the Fringe family of glycosyltransferases. This sugar modification enhances Notch signalling via DLL4 but represses signalling via Jagged 1. Hence, Jagged 1–Notch interactions do not induce productive Notch signalling. According to this model, SC-restricted Jagged 1 competes with DLL4 for binding to Notch receptors on TCs and effectively suppresses TC Notch signalling. Consequently, TC formation and vascular sprouting is severely disrupted in the retinal vessels of EC-specific Jagged 1-knockout mice. By contrast, TC formation is enhanced upon the EC-specific Jagged 1 gain-of-function. These findings indicate that distinct Notch ligands have opposing roles in the control of TC selection and angiogenic sprouting, which may have important implications for tissue branching morphogenesis in other systems.
Emerging players in TC and SC fate decisions
The remarkable similarity between angiogenesis and the branching of the D. melanogaster trachea raises the exciting possibility that other known determinants of epithelial sprouting may control blood vessel morphogenesis. Indeed, a recent study highlights this evolutionary conservation by identifying a critical role for the ETS-related factor translocation ETS leukaemia (TEL; also known as ETV6) in angiogenic sprouting56. TEL is a mammalian orthologue of the D. melanogaster transcriptional repressor YAN, which is negatively regulated by BTL signalling and is important for trachea branching57,58. In ECs, TEL interacts with the co-repressor C-terminal-binding protein (CtBP) to repress the expression of sprouting antagonists (such as DLL4 and Sprouty homologue 4 (SPRY4)) before VEGF-driven TC formation and angiogenesis56 (FIG. 4b). Consequently, EC sprouting in in vitro assays and during zebrafish angiogenesis is disrupted upon TEL or tel knockdown, respectively, which correlates with increased DLL4 expression. These findings indicate that further research exploiting lessons learnt in other systems will have a direct impact on our understanding of blood vessel morphogenesis.
As discussed above, the acquisition of TC and SC fates and behaviours involves the tight spatiotemporal control of EC-type-specific gene expression. Consequently, recent comparative transcriptome analyses of TCs versus SCs and wild-type versus Dll4-deficient mouse retinal ECs are yielding a wealth of knowledge regarding the molecular control of angiogenic sprouting59,60. In particular, these studies indicate key roles for CXC chemokine receptor 4 (CXCR4) in TC behaviour59 and the APJ endogenous ligand (APELIN) guanine-nucleotide-binding protein (G protein)-coupled receptor APJ in SC function60. Hence, further functional analyses of TC and SC enriched genes identified in these and other studies should yield additional critical insight.
Recent studies of angiogenesis are also shedding light on fundamental mechanisms that control Notch signalling in different cellular contexts. For example, studies using mice overexpressing β-catenin in ECs show that the WNT–β-catenin pathway positively regulates Dll4 expression in the vasculature61 (FIG. 4b). Consequently, β-catenin gain-of-function induces endothelial Notch signalling and disrupts EC branching, although the role of this pathway in TC selection is unclear. In parallel, DLL4–Notch interactions can feed back to activate WNT signalling in SCs, which maintains EC–EC connections62. Notch signalling promotes vascular stability by inducing expression of the WNT regulator gene Notch-regulated ankyrin repeat-containing protein (NRARP). NRARP limits Notch activity and stimulates lymphoid enhancer-binding factor 1 (LEF1)- and β-catenin-dependent WNT signalling to stabilize SCs and prevent EC retraction (FIG. 4b). As a result, loss of Nrarp in mice or nrarpa and/or nrarpb in zebrafish causes ectopic vessel regression and reduced WNT-mediated vascular stability62. Hence, functional interplay between Notch and WNT signalling is critical for vascular development.
Strikingly, a recent study of the NAD+-dependent deacetylase sirtuin 1 (SIRT1) reveals that acetylation of the NICD decreases its turnover, thereby enhancing Notch signalling63 (FIG. 4b). SIRT1 deacetylates NICD in ECs to oppose its acetylation-mediated stabilization. Consequently, knockdown of Sirt1 in mice (which enhances DLL4-mediated Notch signalling) and sirt1 in zebrafish disrupts EC sprouting and perturbs vessel branching. Interestingly, enriched expression of SIRT1 in SCs suggests that the repression of Notch signalling might have key roles in SC biology in addition to its known role in TC selection. Most importantly, these studies define SIRT1 as a potential general regulator of Notch signalling, which may have important implications for cell fate decisions in multiple tissues.
Vascular lumen morphogenesis
During angiogenic sprouting, all functional blood vessels need to form a patent vascular lumen to establish blood flow. Whereas the molecular pathways controlling epithelial lumen morphogenesis are under intense investigation64, progress towards defining the mechanisms of vascular tube formation has been slow. Indeed, until a few years ago our understanding of blood vessel lumen formation relied almost exclusively on in vitro research exploiting three-dimensional tubulogenesis assays. Although in vitro studies have revealed a wealth of knowledge regarding potential players in this process (including integrins, CDC42, SRC, RAC1, p21-activated kinase 2 (PAK2), PAK4, RAF, partitioning defective 3 (PAR3; also known as PARD3), PAR6 and atypical protein kinase C)16, it has been difficult to confirm these observations in vivo. Recent studies in mice and zebrafish are shedding new light on the cellular and molecular mechanisms of vascular lumen morphogenesis, confirming many previous in vitro observations and adding important new insights. In particular, it is now clear that lumen formation in most multicellular vessels involves the transition through a number of discrete phases, as discussed below.
Establishing EC polarity
Prior to tube formation, blood vessels consist of multicellular rods of ECs that are interconnected by uniform EC–EC junctions and have yet to establish apicobasal polarity (FIG. 5a). PAR3 is a critical determinant of cell polarity that is known to influence epithelial lumen formation and has been implicated in vascular tube formation in vitro16. The first phase of blood vessel lumen morphogenesis appears to involve PAR3-mediated acquisition of EC apicobasal polarity and the lateral redistribution of junctional proteins (including zonula occludens 1 (ZO1), claudin 5, CD99 and vascular endothelial cadherin (VE-cadherin)) from the apical EC surface to the vascular cord periphery65,66 (FIG. 5a). Importantly, β1 integrin–matrix interactions at the basal EC surface establish EC polarity in vivo by regulating PAR3 expression and localization to the basal EC surface65. In addition, recent evidence indicates that RAS-interacting protein 1 (RASIP1) and its binding partner RHOA GTPase-activating protein 29 (ARHGAP29) also influence lumen morphogenesis in mouse66. Vascular-specific expression of RASIP1 regulates RHO GTPase activity and also shifts junctional proteins laterally by activating integrins (including β1 integrin) and controlling PAR3 localization. Hence, correct acquisition of apicobasal polarity and spatial redistribution of EC–EC junctions initiate lumen formation.
Figure 5. Molecular mechanisms of lumen morphogenesis.

a | Prior to lumen morphogenesis, blood vessels consist of coalesced cords of endothelial cells (ECs) that lack apicobasal polarity. β1 integrin–matrix interactions and RAS interacting protein 1 (RASIP1) establish EC apicobasal polarity in a partitioning defective 3 (PAR3)-mediated manner to promote the lateral redistribution of junctional components from the apical surface to the periphery of EC–EC contacts. b | Once EC apicobasal polarity is established, lumen formation is triggered, at least in part, by vascular endothelial cadherin (VE-cadherin)-mediated redistribution of CD34 and podocalyxin (PODXL) to the apical surface.β1 integrin may also promote the redistribution of PODXL to EC apical membranes. Subsequently, protein kinase C (PKC)-mediated phosphorylation and redistribution of moesin to PODXL-enriched apical EC membranes promotes the deposition of filamentous actin (F-actin). Furthermore, PODXL may initiate lumen formation by inducing the electrostatic repulsion of EC–EC apical surfaces. c | Lumenal expansion proceeds by a variety of mechanisms. For example, vascular endothelial growth factor receptor 2 (VEGFR2) signalling and activation of RHO-associated coiled-coil kinase (ROCK) may promote the association of non-muscle myosin II with apical F-actin to drive actomyosin-mediated cell shape changes. By contrast, RASIP1 may repress actomyosin contractility to fine-tune this response. Alternatively, or more likely in addition, directed exocytic vacuole trafficking and fusion of these vacuoles with the apical surface may also drive lumen expansion.
ZO1, zonula occludens 1; CLDN5, claudin 5.
Recruiting key proteins to the site of lumen formation
Once EC polarity is established, CD34 and podocalyxin (PODXL) (glycoproteins that are expressed on vascular endothelium) are recruited to apical membranes, which are initially at contact points between adjacent ECs65–67 (FIG. 5b). Both β1 integrin and VE-cadherin (but not RASIP1) regulate the accumulation of PODXL at the apical surface, which may be important for lumen formation in vivo65–67. Following its redistribution, PODXL recruits other apical proteins (moesin and filamentous actin (F-actin)) as the vascular lumen begins to form67. Similar to what is seen with PODXL and VE-cadherin, the apical accumulation of moesin may control lumen morphogenesis and appears to function by recruiting F-actin67,68. Hence, the acquisition of apicobasal polarity seems to function, in part, to sequester key components of the tubulogenesis machinery at sites of lumen formation.
Lumenal expansion
Expansion of the blood vessel lumen probably occurs by a number of mechanisms (FIG. 5c). For example, VEGFA signalling promotes the recruitment of non-muscle myosin II to the apical surface, suggesting that actomyosin contractility may induce shape changes that help to form the lumen67. Furthermore, the fact that RASIP1 suppresses actomyosin contractility may suggest that a fine balance is required for effective tube formation66. By contrast, blood vessels lacking the beta1 integrin gene do not form lumina but accumulate large numbers of intracellular vesicles, suggesting that vesicular trafficking may also contribute to luminal expansion65. In addition, interpretation of live-imaging studies in zebrafish ISV tube formation suggests that EC lumina assemble by the coalescence and fusion of intracellular vacuoles17. However, this is disputed by recent studies indicating that the ISV lumen mainly forms extracellularly69, consistent with studies in mice65–67. Importantly, the role of β1 integrin in tubulogenesis is restricted to the arterial endothelium, and venous vessels must use alternative mechanisms65. Consistent with these observations, the first embryonic arterial and venous vessels in zebrafish form lumina by very distinct mechanisms70. Thus, it appears likely that a range of tubulogenesis mechanisms are used in different cellular contexts, indicating that we have only just scratched the surface of this complex process.
Axon guidance signals and angiogenesis
Endothelial TCs share many of the morphological and functional features of axonal growth cones15. For example, both migrating TCs and growth cones represent motile guidance structures that extend dynamic filopodial protrusions and respond to both attractive and repulsive cues. Importantly, recent evidence suggests that ECs and neurons have also recruited various common mechanisms to control directional migration and patterning of the complex vascular and neuronal networks.
Netrins in angiogenesis
Netrins are a family of secreted guidance molecules that bind the extracellular matrix and can have either attractive or repulsive effects on axon guidance71. For example, netrin binding to members of the deleted in colorectal cancer (DCC) family of receptors is attractive, whereas binding to members of the UNC5 receptor family promotes axon repulsion15,72. Of the netrin receptors, UNC5B expression is vascular-specific and restricted to arterial ECs and TCs in sprouting capillaries45,73 (FIG. 3b). Importantly, the vasculature of Unc5b-knockout mice is excessively branched and TCs lacking Unc5b display uncontrolled filopodial extensions73. Hence, similar to the role of UNC5 receptors in neurons, UNC5B normally represses the motile behaviour of ECs. Consequently, stimulation of UNC5B with netrin 1 promotes retraction of TC filopodia and blocks neovessel sprouting73,74. By contrast, netrins may also have pro-angiogenic roles, as local delivery of netrin 1 or netrin 4 promotes therapeutic angiogenesis75. These apparently contradictory observations that netrin 1 acts as both an anti- and a pro-angiogenic factor may be explained by recent evidence indicating that UNC5B is an EC dependence receptor that stimulates apoptosis in the absence of ligand76. Consequently, netrin 1 may enhance EC sprouting by inhibiting the pro-apoptotic effect of unbound UNC5B76. However, which endogenous netrins trigger UNC5B signalling in vivo remains unclear considering that netrin 1-knockout mice display no vascular defects77.
ROBO receptors in angiogenesis
The Slit proteins form another family of secreted molecules important for guidance, and they repel axon growth cones upon binding Roundabout (ROBO) receptors. Of the four known mammalian ROBO genes (ROBO1, ROBO2, ROBO3 and ROBO4), ROBO4 (also known as Magic roundabout) is selectively expressed in ECs78 (FIG. 3b). Studies of ROBO4 in mice and Robo4 in zebrafish indicate that this protein maintains vascular stability and inhibits angiogenesis by counteracting VEGF-mediated signalling79–83. Considering that ROBO4 is enriched in endothelial SCs80, these findings may suggest that ROBO4–Slit signalling negatively regulates VEGF signalling to repress TC behaviour in SCs. However, whether the interaction of ROBO4 with Slit ligands regulates ROBO4 function in vivo is controversial. For example, Slit 2 was shown to co-immunoprecipitate with ROBO4 (REF. 84); however, Slit 2 binding to ROBO4 is not observed in Biacore binding assays85. Furthermore, structural analysis suggests that ROBO4 lacks residues that are essential for ROBO–Slit binding15. Importantly, recent data show that ROBO4 unexpectedly binds to and activates UNC5B in ECs82. These findings indicate that ROBO4 function may not be regulated by Slit 2 but might involve the interaction of two unrelated guidance receptors.
Semaphorins in angiogenesis
The secreted or membrane-anchored semaphorin family of guidance molecules also has key roles during axon guidance and vascular patterning86 (FIG. 3b). Membrane-associated semaphorins influence cell motility by binding the plexin family of transmembrane receptors, whereas the secreted class III semaphorins (from semaphorin 3A (SEMA3A) to SEMA3G) typically interact with the neuropilin (NRP) family of transmembrane co-receptors15. One exception is SEMA3E, which promotes signalling via the EC-enriched receptor, plexin D1 (REF. 87). EC-specific knockdown of plexin D1 in mice disrupts vascular patterning, enhances EC sprouting and is associated with neonatal lethality88. Furthermore, precocious ISV sprouting is observed in the zebrafish plexin D1 mutant, out of bounds (obd)89. Hence, semaphorin–plexin D1 signalling plays an evolutionarily conserved role as a repulsive guidance cue during angiogenesis. However, recent evidence showing that SEMA3E–plexin D1 signalling negatively regulates DLL4–Notch signalling may indicate an additional role for this pathway during TC fate decisions in mouse retinal vessels90.
NRP co-receptors in angiogenesis
NRP co-receptors (NRP1 and NRP2) play key parts in axon guidance upon binding class III semaphorins15,91. In addition, NRPs modulate angiogenic sprouting upon binding VEGFA165 (VEGFA164 in mice), but not VEGFA121 (VEGFA120 in mice)2,91,92 (FIG. 3b). NRPs form complexes with various VEGFRs and enhance VEGFR-mediated signalling when bound to VEGFA. Consequently, NRPs are essential for vascular development and Nrp1-knockout mice display disrupted EC remodelling and vessel branching during development93,94. However, vascular defects in Nrp1-null mice are, overall, relatively mild, suggesting that NRP-mediated VEGF signalling may not have key roles during TC specification and sprouting.
EPHs in angiogenesis
Ephrin receptors (EPHs) are transmembrane receptor Tyr kinases that bind, and are activated by, cell surface proteins called ephrins95. EPH–ephrin forward and reverse signalling have long been known to be important for both neuronal and vascular development; however, the cellular and molecular mechanisms underlying EPH–ephrin function in the vasculature in vivo are only just emerging. Ephrin B2 and its cognate receptor EPHB4 have key roles during blood vessel morphogenesis and have attracted the most attention owing to their strikingly restricted expression in arteries and veins, respectively95 (FIG. 3b). For example, recent data suggests that ephrin B2 may directly interact with VEGFRs to regulate VEGF signalling96,97, as is discussed later in this Review. In addition, a recent study in zebrafish indicates that ephrin b2a–Ephb4a interactions regulate segregation and sorting of arterial and venous EC precursor cells into discrete primary arterial and venous vessels during early development70. In this process, Vegfa-induced expression of ephrin b2a in arterial precursors blocks the ventral migration of these cells to exclude them from the primary venous vessel. By contrast, ephb4a expression in venous precursors permits ventral migration of venous progenitors and promotes exclusion from the first embryonic artery. Hence, ephrin b2- and Ephb4-mediated directional control of EC migration drives arterial and venous segregation and the generation of separate parallel vessels. Furthermore, these results are supported by previous observations that ephrin B2–EPHB4 signalling regulates the calibre of arterial and venous vessels in mice98. However, further research is required to determine whether this pathway functions as the chief mechanism for regulating primary vessel formation or, alternatively, acts more as a checkpoint to suppress arterial and venous vessel mixing.
These findings clearly demonstrate that ECs respond to multiple neuronal signals during vascular development. Similarly, a growing body of evidence suggests that vascular guidance cues may also influence neuronal development. For example, recent data indicate that VEGFR3 signalling controls neurogenesis in mice99. Furthermore, the release of neural-derived signals that influence neurovascular interactions (such as thrombospondin type I domain-containing 7A (THSD7A)) may also play key parts in the regulation of angiogenesis100. Hence, future studies investigating the functional overlap between vascular and neuronal pathways will shed important light on the mechanisms controlling the development of both systems.
Fine-tuning angiogenesis
Vascular research over the past 20 years has succeeded in identifying many key molecular pathways that control angiogenesis. However, recent studies are additionally starting to define elegant post-transcriptional and post-translational mechanisms that adjust responses mediated by known angiogenic pathways. Hence, current research is revealing a remarkable level of complexity in the control of pro-angiogenic signalling, as discussed below.
Post-transcriptional control of angiogenesis
MicroRNAs (miRNAs) are a large family of small (~22 base pairs) non-coding single-stranded RNAs that are considered to be key post-transcriptional regulators of gene expression101. miRNAs function by binding complementary sequences within target mRNAs, which blocks the translation, or promotes the degradation, of the bound mRNA. Recent data implicate certain miRNAs as crucial determinants of EC behaviour during angiogenesis101 (FIG. 6a). For example, modulation of VEGF signalling during development by the EC-restricted miRNA miR-126 is essential for normal angiogenesis in mice and zebrafish102–104. miR-126 is located in intron 7 of the endothelial gene epidermal growth factor-like 7 (EGFL7). Expression of miR-126 positively influences VEGF-induced signalling by post-transcriptionally repressing SPRED1 (Sprouty-related EVH1 domain-containing 1) and PI3K regulatory subunit 2 (PIK3R2; also known as p85β), which are negative regulators of MAPK and PI3K signalling, respectively102–104. Consequently, enhanced SPRED1 and PIK3R2 expression upon miR-126 knockdown blocks VEGF-induced MAPK and PI3K signalling. As a result, angiogenesis and vascular integrity are compromised in mice or zebrafish deficient for miR-126, resulting in fragile and leaky vessels that often haemorrhage.
Figure 6. Fine-tuning angiogenic signals.

a | Post-transcriptional modification of key angiogenic signalling pathways by microRNAs (miRNAs) modulates angiogenesis. Expression of miR-126 de-represses phosphoinositide 3-kinase (PI3K) and/or RAF1 signalling to promote vascular endothelial growth factor (VEGF)-induced angiogenesis. Blood flow also influences VEGF signalling by promoting Krüppel-like factor 2 (KLF2)-induced expression of miR-126 during angiogenic sprouting. Furthermore, endothelial cell expression of miR-132 enhances angiogenic signalling controlled by RAS small GTPases by reducing the expression of RAS-specific GTPase-activating protein p120 (p120RASGAP), a negative regulator of RAS. By contrast, miR-92a blocks angiogenesis by repressing pro-angiogenic α5 integrin protein expression. miR-92a may also activate Notch signalling to block VEGF-induced angiogenesis by reducing the expression of sirtuin 1 (SIRT1), which deacetylates the Notch intracellular domain to destabilize it. b | Post-translational modulation of VEGF receptor 2 (VEGFR2) or VEGFR3 membrane trafficking determines the duration and magnitude of VEGFA and VEGFC signalling. Synectin and ephrin B2-mediated membrane internalization protects phosphorylated active VEGFR from inactivation by cell-surface Tyr phosphatases. Furthermore, transport of VEGFR to intracellular endocytic compartments enhances pro-angiogenic signalling. Internalized VEGFR is subsequently either degraded or recycled to the cell surface for another round of activation. Hence, αvβ3 integrin and RAB4A-mediated recycling of VEGFR stabilizes receptor protein expression and enhances VEGF signalling. PIK3R2, PI3K regulatory subunit 2; SPRED1, Sprouty-related EVH1 domain-containing 1; ERK, extracellular signal-regulated kinase.
Strikingly, recent data in zebrafish indicate that modulation of the miR-126–Spred1 axis may even allow integration of mechanical cues from the extracellular environment to control blood vessel morphogenesis105 (FIG. 6a). In response to blood flow, the mechanosensitive transcription factor Krüppel-like factor 2a (Klf2a) promotes miR-126 expression in sprouting vessels of the aortic arches. Consequently, miR-126-mediated augmentation of VEGF signalling is essential for flow-induced aortic arch vessel angiogenesis. Hence, these findings shed new light on the mechanosensitive mechanisms that integrate blood flow and VEGFR signalling during remodelling of the developing vasculature.
Other vascular miRNAs seem to have critical roles during pathological angiogenesis106,107. Expression of miR-132 in ECs promotes angiogenesis by targeting RAS-specific GAP p120 (p120RASGAP), a known negative regulator of RAS activity106 (FIG. 6a). Hence, miR-132 activates RAS to promote EC proliferation, tube formation and angiogenesis in vivo. Consequently, miR-132 expression in sprouting ECs acts as an angiogenic switch that promotes tumour neovascularization and growth. By contrast, EC expression of miR-92a appears to block new blood vessel sprouting by repressing the expression of pro-angiogenic proteins, such as a5 integrin107. Consequently, inhibition of miR-92a enhances therapeutic angiogenesis and neovascularization of ischaemic tissues in mice. Interestingly, a potential target of miR-92a is SIRT1, suggesting that miR-92a may suppress SIRT1-mediated deacetylation of NICD and inhibit angiogenesis by stabilising anti-angiogenic Notch signalling63. However a direct link between miR-92a and Notch signalling has not been reported.
Post-translational control of VEGFR signalling
The internalization of ligand-bound VEGFR-2 into endosomal compartments is known to prolong the magnitude and duration of downstream pro-angiogenic signalling in vitro108. Importantly, a number of recent studies are confirming that tight spatial control of VEGFR trafficking is also a critical control point during angiogenesis in vivo (FIG. 6b). For example, synectin–myosin VI-dependent trafficking of VEGFR2-containing endosomes shuttles VEGF-activated receptors away from the plasma membrane109. This spatial redistribution of VEGFR2 blocks the dephosphorylation of active receptors by protein Tyr phosphatase 1B (PTP1B; also known as PTPN1) at the plasma membrane. Hence, synectin–myosin VI-mediated maintenance of VEGFR2 signalling influences many VEGF-regulated processes in vivo. Following these observations, two recent studies also identified a key role for the neuronal guidance molecule ephrin B2 in this process96,97. Expression of ephrin B2 in ECs unexpectedly promotes the internalization of VEGFRs (VEGFR2 and VEGFR3) and their signalling from endosomes, which is essential for the extension of TC filopodia. Consequently, loss of ephrin B2 and ephrin b2a expression in mice and zebrafish, respectively, disrupts TC formation, blocks EC sprouting and perturbs angiogenesis96,97.
In addition to receptor internalization, enhanced recycling of endocytosed VEGFR2 appears to play important parts in angiogenesis in vivo (FIG. 6b). In particular, recent data implicate αvβ3 integrin in the RAB4A-mediated recycling of VEGFR2 to the plasma membrane of ECs110. Consequently, pharmacological augmentation of this mechanism blocks VEGF-induced lysosomal degradation of VEGFR2 and enhances EC migration and angiogenesis in vitro and in vivo. Moreover, RAB-mediated membrane trafficking of other signalling components (such as RHOA and SYX (also known as PLEKHG5)) may also have key roles in angiogenic sprouting111. As new determinants of VEGFR internalization and recycling will probably emerge over the next few years, a key question will be whether (and how) these mechanisms are modulated in cell-type-specific manners during angiogenesis. For example, can these pro-angiogenic mechanisms be selectively recruited by TCs during sprouting?
Perspectives
In recent years, our understanding of the molecular mechanisms that control and coordinate diverse EC behaviours during blood vessel morphogenesis has expanded remarkably. In particular, the development of advanced vertebrate model systems has allowed the field to address fundamental vascular questions in vivo at a spatiotemporal resolution never before possible. Studies exploiting these tools have greatly enriched our knowledge of key aspects of blood vessel assembly, such as the mechanisms of TC selection and vascular lumen morphogenesis. However, our understanding of many processes is still in its infancy. For example, the molecular machinery that terminates EC sprouting and stimulates vessel anastomosis remains largely a mystery. Moreover, the cell-type-specific mechanisms regulating venous vascular tube formation remain unknown. Importantly, glaring gaps still exist in our knowledge of the sprouting process itself. For example, are endothelial SCs also specified during angiogenesis or do they just passively trail migrating TCs? Likewise, the plasticity of the TC and SC fates needs to be investigated further. Furthermore, the precise spatiotemporal dynamics of TC and SC Notch signalling during EC sprouting is unclear. As recent studies have demonstrated, blood vessel morphogenesis represents a paradigm for many fundamental processes common to other biological systems3,18. Hence, future studies addressing the questions raised above should reveal important mechanistic insights that can be readily translated into other systems.
The therapeutic targeting of blood vessel morphogenesis remains a predominant strategy for tackling the diverse range of diseases that are driven by pathological angiogenesis1. Considering the critical role of the DLL4–Notch–VEGF signalling axis in angiogenic sprouting38,42–47, much of the current clinical research is focused on modulating this pathway1. However, it is clear that there is still vast therapeutic demand and scope for identifying new determinants of blood vessel formation. In particular, a better molecular understanding of currently under-explored processes, such as lumen morphogenesis and SC formation, will pave the way for novel targets that may ultimately translate into more effective therapies.
Acknowledgments
We apologize to those authors whose work could not be referenced directly because of space limitations. S.P.H. is a Wellcome Trust research career development fellow. Vascular work in the laboratory of D.Y.R.S. is supported in part by grants from the US National Institutes of Health (HL54737) and the Packard Foundation to D.Y.R.S.
Glossary
- Ischaemic diseases
Disease states that are characterized by a restriction in the blood supply to an organ or tissue, such as ischaemic heart disease, stroke or ischaemic colitis
- Metastasis
The spread of cancer cells from the site of a primary tumour to distant parts of the body
- Basement membrane
The sheet-like layer of laminin-and collagen-rich fibres that separates endothelial or epithelial cells from adjacent tissues
- Filopodial extensions
Thin cytoplasmic protrusions containing bundled actin filaments that dynamically extend from the leading edge of migrating cells and explore the surrounding microenvironment
- Neuronal growth cones
Specialized guidance structures located at the distal end of developing axons that sense extrinsic guidance cues to direct the movement of axons
- Vascular lumen
The open and unobstructed (or patent) space within a hollow vascular tube that is lined by endothelial cells and through which blood flows
- Anastomosis
The union of two hollow structures so as to interconnect and establish continuity between both structures
- Abluminal surface
The surface away from the lumen (to distinguish it from the luminal surface, which is the one adjacent to the lumen)
- Intussusceptive angiogenesis
Blood vessel formation by the splitting of existing vessels. Intussusception involves the insertion of a tissue pillar into the vascular lumen to split a single parent vessel into two daughter vessels
- Hypoxia
A deficiency in the supply of oxygen to an organ or tissue
- Haploinsufficient
The term applied to a gene of a diploid organism if a single copy of that gene is insufficient to support a wild-type phenotype
- Heparan sulphate
A linear polysaccharide present on the surface of cells or in extracellular matrices, which function by binding wide varieties of proteins (such as growth factors) to regulate key biological processes
- Decoy receptor
Generally considered to be a non-signalling receptor that binds a ligand and reduces the interaction of the bound ligand with its signalling receptor
- Lymphangiogenesis
The formation of new lymphatic vessels by sprouting from pre-existing vessels
- Lateral inhibition
In developmental biology, this term refers to the ability of a cell to inhibit the differentiation of its immediate neighbours through cell–cell interactions
Footnotes
Competing interests statement
The authors declare no competing financial interests.
Contributor Information
Shane P. Herbert, Email: S.P.Herbert@leeds.ac.uk.
Didier Y.R. Stainier, Email: Didier.Stainier@ucsf.edu.
References
- 1.Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adams RH, Alitalo K. Molecular regulation of angiogenesis and lymphangiogenesis. Nature Rev Mol Cell Biol. 2007;8:464–478. doi: 10.1038/nrm2183. [DOI] [PubMed] [Google Scholar]
- 3.Rocha SF, Adams RH. Molecular differentiation and specialization of vascular beds. Angiogenesis. 2009;12:139–147. doi: 10.1007/s10456-009-9132-x. [DOI] [PubMed] [Google Scholar]
- 4.Tammela T, Alitalo K. Lymphangiogenesis: molecular mechanisms and future promise. Cell. 2010;140:460–476. doi: 10.1016/j.cell.2010.01.045. [DOI] [PubMed] [Google Scholar]
- 5.Gaengel K, Genove G, Armulik A, Betsholtz C. Endothelial-mural cell signaling in vascular development and angiogenesis. Arterioscler Thromb Vasc Biol. 2009;29:630–638. doi: 10.1161/ATVBAHA.107.161521. [DOI] [PubMed] [Google Scholar]
- 6.Jain RK. Molecular regulation of vessel maturation. Nature Med. 2003;9:685–693. doi: 10.1038/nm0603-685. [DOI] [PubMed] [Google Scholar]
- 7.Fischer A, Schumacher N, Maier M, Sendtner M, Gessler M. The Notch target genes Hey1 and Hey2 are required for embryonic vascular development. Genes Dev. 2004;18:901–911. doi: 10.1101/gad.291004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhong TP, Childs S, Leu JP, Fishman MC. Gridlock signalling pathway fashions the first embryonic artery. Nature. 2001;414:216–220. doi: 10.1038/35102599. [DOI] [PubMed] [Google Scholar]
- 9.You LR, et al. Suppression of Notch signalling by the COUP-TFII transcription factor regulates vein identity. Nature. 2005;435:98–104. doi: 10.1038/nature03511. [DOI] [PubMed] [Google Scholar]
- 10.Bertrand JY, Traver D. Hematopoietic cell development in the zebrafish embryo. Curr Opin Hematol. 2009;16:243–248. doi: 10.1097/MOH.0b013e32832c05e4. [DOI] [PubMed] [Google Scholar]
- 11.Mazzone M, et al. Heterozygous deficiency of PHD2 restores tumor oxygenation and inhibits metastasis via endothelial normalization. Cell. 2009;136:839–851. doi: 10.1016/j.cell.2009.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Augustin HG, Koh GY, Thurston G, Alitalo K. Control of vascular morphogenesis and homeostasis through the angiopoietin-Tie system. Nature Rev Mol Cell Biol. 2009;10:165–177. doi: 10.1038/nrm2639. [DOI] [PubMed] [Google Scholar]
- 13.De Smet F, Segura I, De Bock K, Hohensinner PJ, Carmeliet P. Mechanisms of vessel branching: filopodia on endothelial tip cells lead the way. Arterioscler Thromb Vasc Biol. 2009;29:639–649. doi: 10.1161/ATVBAHA.109.185165. [DOI] [PubMed] [Google Scholar]
- 14.Gerhardt H, et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J Cell Biol. 2003;161:1163–1177. doi: 10.1083/jcb.200302047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adams RH, Eichmann A. Axon guidance molecules in vascular patterning. Cold Spring Harb Perspect Biol. 2010;2:a001875. doi: 10.1101/cshperspect.a001875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iruela-Arispe ML, Davis GE. Cellular and molecular mechanisms of vascular lumen formation. Dev Cell. 2009;16:222–231. doi: 10.1016/j.devcel.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kamei M, et al. Endothelial tubes assemble from intracellular vacuoles in vivo. Nature. 2006;442:453–456. doi: 10.1038/nature04923. [DOI] [PubMed] [Google Scholar]
- 18.Affolter M, Zeller R, Caussinus E. Tissue remodelling through branching morphogenesis. Nature Rev Mol Cell Biol. 2009;10:831–842. doi: 10.1038/nrm2797. [DOI] [PubMed] [Google Scholar]
- 19.Fantin A, et al. Tissue macrophages act as cellular chaperones for vascular anastomosis downstream of VEGF-mediated endothelial tip cell induction. Blood. 2010;116:829–840. doi: 10.1182/blood-2009-12-257832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Makanya AN, Hlushchuk R, Djonov VG. Intussusceptive angiogenesis and its role in vascular morphogenesis, patterning, and remodeling. Angiogenesis. 2009;12:113–123. doi: 10.1007/s10456-009-9129-5. [DOI] [PubMed] [Google Scholar]
- 21.Lohela M, Bry M, Tammela T, Alitalo K. VEGFs and receptors involved in angiogenesis versus lymphangiogenesis. Curr Opin Cell Biol. 2009;21:154–165. doi: 10.1016/j.ceb.2008.12.012. [DOI] [PubMed] [Google Scholar]
- 22.Coultas L, Chawengsaksophak K, Rossant J. Endothelial cells and VEGF in vascular development. Nature. 2005;438:937–945. doi: 10.1038/nature04479. [DOI] [PubMed] [Google Scholar]
- 23.Shibuya M. Differential roles of vascular endothelial growth factor receptor-1 and receptor-2 in angiogenesis. J Biochem Mol Biol. 2006;39:469–478. doi: 10.5483/bmbrep.2006.39.5.469. [DOI] [PubMed] [Google Scholar]
- 24.Pugh CW, Ratcliffe PJ. Regulation of angiogenesis by hypoxia: role of the HIF system. Nature Med. 2003;9:677–684. doi: 10.1038/nm0603-677. [DOI] [PubMed] [Google Scholar]
- 25.Carmeliet P, et al. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;380:435–439. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- 26.Ferrara N, et al. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 1996;380:439–442. doi: 10.1038/380439a0. [DOI] [PubMed] [Google Scholar]
- 27.Shalaby F, et al. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature. 1995;376:62–66. doi: 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- 28.Ladomery MR, Harper SJ, Bates DO. Alternative splicing in angiogenesis: the vascular endothelial growth factor paradigm. Cancer Lett. 2007;249:133–142. doi: 10.1016/j.canlet.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 29.Ruhrberg C, et al. Spatially restricted patterning cues provided by heparin-binding VEGF-A control blood vessel branching morphogenesis. Genes Dev. 2002;16:2684–2698. doi: 10.1101/gad.242002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen TT, et al. Anchorage of VEGF to the extracellular matrix conveys differential signaling responses to endothelial cells. J Cell Biol. 2010;188:595–609. doi: 10.1083/jcb.200906044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kawamura H, Li X, Harper SJ, Bates DO, Claesson-Welsh L. Vascular endothelial growth factor (VEGF)-A165b is a weak in vitro agonist for VEGF receptor-2 due to lack of coreceptor binding and deficient regulation of kinase activity. Cancer Res. 2008;68:4683–4692. doi: 10.1158/0008-5472.CAN-07-6577. [DOI] [PubMed] [Google Scholar]
- 32.Hiratsuka S, et al. Membrane fixation of vascular endothelial growth factor receptor 1 ligand-binding domain is important for vasculogenesis and angiogenesis in mice. Mol Cell Biol. 2005;25:346–354. doi: 10.1128/MCB.25.1.346-354.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fong GH, Rossant J, Gertsenstein M, Breitman ML. Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature. 1995;376:66–70. doi: 10.1038/376066a0. [DOI] [PubMed] [Google Scholar]
- 34.Fong GH, Zhang L, Bryce DM, Peng J. Increased hemangioblast commitment, not vascular disorganization, is the primary defect in flt-1 knockout mice. Development. 1999;126:3015–3025. doi: 10.1242/dev.126.13.3015. [DOI] [PubMed] [Google Scholar]
- 35.Krueger J, et al. Flt1 acts as a negative regulator of tip cell formation and branching morphogenesis in the zebrafish embryo. Development. 2011;138:2111–2120. doi: 10.1242/dev.063933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stefater JA, III, et al. Regulation of angiogenesis by a non-canonical Wnt-Flt1 pathway in myeloid cells. Nature. 2011;474:511–515. doi: 10.1038/nature10085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tammela T, et al. Blocking VEGFR-3 suppresses angiogenic sprouting and vascular network formation. Nature. 2008;454:656–660. doi: 10.1038/nature07083. [DOI] [PubMed] [Google Scholar]
- 38.Siekmann AF, Lawson ND. Notch signalling limits angiogenic cell behaviour in developing zebrafish arteries. Nature. 2007;445:781–784. doi: 10.1038/nature05577. References 37 and 38 are the first papers to identify VEGFR3 as a key determinant of endothelial TC behaviour and define a critical role for Notch signalling in the negative regulation of VEGFR3 expression in endothelial SCs. [DOI] [PubMed] [Google Scholar]
- 39.Nilsson I, et al. VEGF receptor 2/-3 heterodimers detected in situ by proximity ligation on angiogenic sprouts. EMBO J. 2010;29:1377–1388. doi: 10.1038/emboj.2010.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roca C, Adams RH. Regulation of vascular morphogenesis by Notch signaling. Genes Dev. 2007;21:2511–2524. doi: 10.1101/gad.1589207. [DOI] [PubMed] [Google Scholar]
- 41.Phng LK, Gerhardt H. Angiogenesis: a team effort coordinated by notch. Dev Cell. 2009;16:196–208. doi: 10.1016/j.devcel.2009.01.015. [DOI] [PubMed] [Google Scholar]
- 42.Hellstrom M, et al. Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature. 2007;445:776–780. doi: 10.1038/nature05571. [DOI] [PubMed] [Google Scholar]
- 43.Leslie JD, et al. Endothelial signalling by the Notch ligand Delta-like 4 restricts angiogenesis. Development. 2007;134:839–844. doi: 10.1242/dev.003244. [DOI] [PubMed] [Google Scholar]
- 44.Lobov IB, et al. Delta-like ligand 4 (Dll4) is induced by VEGF as a negative regulator of angiogenic sprouting. Proc Natl Acad Sci USA. 2007;104:3219–3224. doi: 10.1073/pnas.0611206104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Suchting S, et al. The Notch ligand Delta-like 4 negatively regulates endothelial tip cell formation and vessel branching. Proc Natl Acad Sci USA. 2007;104:3225–3230. doi: 10.1073/pnas.0611177104. References 38 and 42–45 are the first to describe the important role of DLL4–Notch signalling in the selection of endothelial TCs during blood vessel sprouting in vivo. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Noguera-Troise I, et al. Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature. 2006;444:1032–1037. doi: 10.1038/nature05355. [DOI] [PubMed] [Google Scholar]
- 47.Ridgway J, et al. Inhibition of Dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature. 2006;444:1083–1087. doi: 10.1038/nature05313. References 46 and 47 report that blocking DLL4–Notch signalling inhibits tumour growth by promoting excessive non-productive angiogenesis that results in poor tumour perfusion and hypoxia. [DOI] [PubMed] [Google Scholar]
- 48.Glomski K, et al. Deletion of Adam10 in endothelial cells leads to defects in organ-specific vascular structures. Blood. 2011;118:1163–1174. doi: 10.1182/blood-2011-04-348557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yan M, et al. Chronic DLL4 blockade induces vascular neoplasms. Nature. 2010;463:E6–E7. doi: 10.1038/nature08751. [DOI] [PubMed] [Google Scholar]
- 50.Sainson RC, et al. Cell-autonomous notch signaling regulates endothelial cell branching and proliferation during vascular tubulogenesis. FASEB J. 2005;19:1027–1029. doi: 10.1096/fj.04-3172fje. [DOI] [PubMed] [Google Scholar]
- 51.Hogan BM, et al. Vegfc/Flt4 signalling is suppressed by Dll4 in developing zebrafish intersegmental arteries. Development. 2009;136:4001–4009. doi: 10.1242/dev.039990. [DOI] [PubMed] [Google Scholar]
- 52.Funahashi Y, et al. Notch regulates the angiogenic response via induction of VEGFR-1. J Angiogenes Res. 2010;2:3. doi: 10.1186/2040-2384-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jakobsson L, et al. Endothelial cells dynamically compete for the tip cell position during angiogenic sprouting. Nature Cell Biol. 2010;12:943–953. doi: 10.1038/ncb2103. [DOI] [PubMed] [Google Scholar]
- 54.Chappell JC, Taylor SM, Ferrara N, Bautch VL. Local guidance of emerging vessel sprouts requires soluble Flt-1. Dev Cell. 2009;17:377–386. doi: 10.1016/j.devcel.2009.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Benedito R, et al. The notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell. 2009;137:1124–1135. doi: 10.1016/j.cell.2009.03.025. Shows an unexpected pro-angiogenic role for the Notch ligand Jagged 1, which antagonises EC DLL4–Notch signalling to regulate new blood vessel sprouting in mice. [DOI] [PubMed] [Google Scholar]
- 56.Roukens MG, et al. Control of endothelial sprouting by a Tel–CtBP complex. Nature Cell Biol. 2010;12:933–942. doi: 10.1038/ncb2096. Identifies a new role for the transcriptional repressor complex TEL–CtBP in the regulation of VEGFA-mediated DLL4 expression during angiogenesis. [DOI] [PubMed] [Google Scholar]
- 57.Rebay I, Rubin GM. Yan functions as a general inhibitor of differentiation and is negatively regulated by activation of the Ras1/MAPK pathway. Cell. 1995;81:857–866. doi: 10.1016/0092-8674(95)90006-3. [DOI] [PubMed] [Google Scholar]
- 58.Hacohen N, Kramer S, Sutherland D, Hiromi Y, Krasnow M. Asprouty encodes a novel antagonist of FGF signaling that patterns apical branching of the Drosophila airways. Cell. 1998;92:253–263. doi: 10.1016/s0092-8674(00)80919-8. [DOI] [PubMed] [Google Scholar]
- 59.Strasser GA, Kaminker JS, Tessier-Lavigne M. Microarray analysis of retinal endothelial tip cells identifies CXCR4 as a mediator of tip cell morphology and branching. Blood. 2010;115:5102–5110. doi: 10.1182/blood-2009-07-230284. [DOI] [PubMed] [Google Scholar]
- 60.del Toro R, et al. Identification and functional analysis of endothelial tip cell-enriched genes. Blood. 2010;116:4025–4033. doi: 10.1182/blood-2010-02-270819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Corada M, et al. The Wnt/β-catenin pathway modulates vascular remodeling and specification by upregulating Dll4/Notch signaling. Dev Cell. 2010;18:938–949. doi: 10.1016/j.devcel.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Phng LK, et al. Nrarp coordinates endothelial Notch and Wnt signaling to control vessel density in angiogenesis. Dev Cell. 2009;16:70–82. doi: 10.1016/j.devcel.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Guarani V, et al. Acetylation-dependent regulation of endothelial Notch signalling by the SIRT1 deacetylase. Nature. 2011;473:234–238. doi: 10.1038/nature09917. Reveals that acetylation of the NICD increases its stability, and that this is counteracted by the SIRT1 deacetylase to allow fine-tuning of Notch signalling in sprouting ECs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bryant DM, Mostov KE. From cells to organs: building polarized tissue. Nature Rev Mol Cell Biol. 2008;9:887–901. doi: 10.1038/nrm2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zovein AC, et al. β1 integrin establishes endothelial cell polarity and arteriolar lumen formation via a Par3-dependent mechanism. Dev Cell. 2010;18:39–51. doi: 10.1016/j.devcel.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xu K, et al. Blood vessel tubulogenesis requires Rasip1 regulation of GTPase signaling. Dev Cell. 2011;20:526–539. doi: 10.1016/j.devcel.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Strilic B, et al. The molecular basis of vascular lumen formation in the developing mouse aorta. Dev Cell. 2009;17:505–515. doi: 10.1016/j.devcel.2009.08.011. [DOI] [PubMed] [Google Scholar]
- 68.Wang Y, et al. Moesin1 and VE-cadherin are required in endothelial cells during in vivo tubulogenesis. Development. 2010;137:3119–3128. doi: 10.1242/dev.048785. References 65–68 are the first to define molecular mechanisms that control vascular lumen morphogenesis in vivo and reveal key roles for signalling mediated by integrins, small GTPases, PAR3, VEGFA and VE-cadherin. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Blum Y, et al. Complex cell rearrangements during intersegmental vessel sprouting and vessel fusion in the zebrafish embryo. Dev Biol. 2008;316:312–322. doi: 10.1016/j.ydbio.2008.01.038. [DOI] [PubMed] [Google Scholar]
- 70.Herbert SP, et al. Arterial–venous segregation by selective cell sprouting: an alternative mode of blood vessel formation. Science. 2009;326:294–298. doi: 10.1126/science.1178577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tessier-Lavigne M, Goodman CS. The molecular biology of axon guidance. Science. 1996;274:1123–1133. doi: 10.1126/science.274.5290.1123. [DOI] [PubMed] [Google Scholar]
- 72.Carmeliet P, Tessier-Lavigne M. Common mechanisms of nerve and blood vessel wiring. Nature. 2005;436:193–200. doi: 10.1038/nature03875. [DOI] [PubMed] [Google Scholar]
- 73.Lu X, et al. The netrin receptor UNC5B mediates guidance events controlling morphogenesis of the vascular system. Nature. 2004;432:179–186. doi: 10.1038/nature03080. [DOI] [PubMed] [Google Scholar]
- 74.Larrivee B, et al. Activation of the UNC5B receptor by Netrin-1 inhibits sprouting angiogenesis. Genes Dev. 2007;21:2433–2447. doi: 10.1101/gad.437807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wilson BD, et al. Netrins promote developmental and therapeutic angiogenesis. Science. 2006;313:640–644. doi: 10.1126/science.1124704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Castets M, et al. Inhibition of endothelial cell apoptosis by netrin-1 during angiogenesis. Dev Cell. 2009;16:614–620. doi: 10.1016/j.devcel.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 77.Serafini T, et al. Netrin-1 is required for commissural axon guidance in the developing vertebrate nervous system. Cell. 1996;87:1001–1014. doi: 10.1016/s0092-8674(00)81795-x. [DOI] [PubMed] [Google Scholar]
- 78.Huminiecki L, Gorn M, Suchting S, Poulsom R, Bicknell R. Magic roundabout is a new member of the roundabout receptor family that is endothelial specific and expressed at sites of active angiogenesis. Genomics. 2002;79:547–552. doi: 10.1006/geno.2002.6745. [DOI] [PubMed] [Google Scholar]
- 79.Bedell VM, et al. roundabout4 is essential for angiogenesis in vivo. Proc Natl Acad Sci USA. 2005;102:6373–6378. doi: 10.1073/pnas.0408318102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jones CA, et al. Robo4 stabilizes the vascular network by inhibiting pathologic angiogenesis and endothelial hyperpermeability. Nature Med. 2008;14:448–453. doi: 10.1038/nm1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jones CA, et al. Slit2–Robo4 signalling promotes vascular stability by blocking Arf6 activity. Nature Cell Biol. 2009;11:1325–1331. doi: 10.1038/ncb1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Koch AW, et al. Robo4 maintains vessel integrity and inhibits angiogenesis by interacting with UNC5B. Dev Cell. 2011;20:33–46. doi: 10.1016/j.devcel.2010.12.001. Reports that the interaction of two unrelated axon guidance receptors, ROBO4 and UNC5B, negatively regulates VEGFA signalling in ECs to inhibit angiogenesis. [DOI] [PubMed] [Google Scholar]
- 83.Marlow R, et al. Vascular Robo4 restricts proangiogenic VEGF signaling in breast. Proc Natl Acad Sci USA. 2010;107:10520–10525. doi: 10.1073/pnas.1001896107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Park KW, et al. Robo4 is a vascular-specific receptor that inhibits endothelial migration. Dev Biol. 2003;261:251–267. doi: 10.1016/s0012-1606(03)00258-6. [DOI] [PubMed] [Google Scholar]
- 85.Suchting S, Heal P, Tahtis K, Stewart LM, Bicknell R. Soluble Robo4 receptor inhibits in vivo angiogenesis and endothelial cell migration. FASEB J. 2005;19:121–123. doi: 10.1096/fj.04-1991fje. [DOI] [PubMed] [Google Scholar]
- 86.Yazdani U, Terman JR. The semaphorins. Genome Biol. 2006;7:211. doi: 10.1186/gb-2006-7-3-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gu C, et al. Semaphorin 3E and plexin-D1 control vascular pattern independently of neuropilins. Science. 2005;307:265–268. doi: 10.1126/science.1105416. [DOI] [PubMed] [Google Scholar]
- 88.Gitler AD, Lu MM, Epstein JA. PlexinD1 and semaphorin signaling are required in endothelial cells for cardiovascular development. Dev Cell. 2004;7:107–116. doi: 10.1016/j.devcel.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 89.Torres-Vazquez J, et al. Semaphorin-plexin signaling guides patterning of the developing vasculature. Dev Cell. 2004;7:117–123. doi: 10.1016/j.devcel.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 90.Kim J, Oh WJ, Gaiano N, Yoshida Y, Gu C. Semaphorin 3E–Plexin-D1 signaling regulates VEGF function in developmental angiogenesis via a feedback mechanism. Genes Dev. 2011;25:1399–1411. doi: 10.1101/gad.2042011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Neufeld G, et al. The neuropilins: multifunctional semaphorin and VEGF receptors that modulate axon guidance and angiogenesis. Trends Cardiovasc Med. 2002;12:13–19. doi: 10.1016/s1050-1738(01)00140-2. [DOI] [PubMed] [Google Scholar]
- 92.Pan Q, et al. Blocking neuropilin-1 function has an additive effect with anti-VEGF to inhibit tumor growth. Cancer Cell. 2007;11:53–67. doi: 10.1016/j.ccr.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 93.Gerhardt H, et al. Neuropilin-1 is required for endothelial tip cell guidance in the developing central nervous system. Dev Dyn. 2004;231:503–509. doi: 10.1002/dvdy.20148. [DOI] [PubMed] [Google Scholar]
- 94.Kawasaki T, et al. A requirement for neuropilin-1 in embryonic vessel formation. Development. 1999;126:4895–4902. doi: 10.1242/dev.126.21.4895. [DOI] [PubMed] [Google Scholar]
- 95.Kuijper S, Turner CJ, Adams RH. Regulation of angiogenesis by Eph–ephrin interactions. Trends Cardiovasc Med. 2007;17:145–151. doi: 10.1016/j.tcm.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 96.Sawamiphak S, et al. Ephrin-B2 regulates VEGFR2 function in developmental and tumour angiogenesis. Nature. 2010;465:487–491. doi: 10.1038/nature08995. [DOI] [PubMed] [Google Scholar]
- 97.Wang Y, et al. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature. 2010;465:483–486. doi: 10.1038/nature09002. References 96 and 97 show that ephrin B2 unexpectedly positively regulates angiogenesis upon its interaction with VEGFR2 and VEGFR3, which promotes receptor internalisation and signalling from intracellular endosomal compartments. [DOI] [PubMed] [Google Scholar]
- 98.Kim YH, et al. Artery and vein size is balanced by Notch and ephrin B2/EphB4 during angiogenesis. Development. 2008;135:3755–3764. doi: 10.1242/dev.022475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Calvo CF, et al. Vascular endothelial growth factor receptor 3 directly regulates murine neurogenesis. Genes Dev. 2011;25:831–844. doi: 10.1101/gad.615311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wang CH, et al. Zebrafish Thsd7a is a neural protein required for angiogenic patterning during development. Dev Dyn. 2011;240:1412–1421. doi: 10.1002/dvdy.22641. [DOI] [PubMed] [Google Scholar]
- 101.Small EM, Olson EN. Pervasive roles of microRNAs in cardiovascular biology. Nature. 2011;469:336–342. doi: 10.1038/nature09783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Fish JE, et al. miR-126 regulates angiogenic signaling and vascular integrity. Dev Cell. 2008;15:272–284. doi: 10.1016/j.devcel.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kuhnert F, et al. Attribution of vascular phenotypes of the murine Egfl7 locus to the microRNA miR-126. Development. 2008;135:3989–3993. doi: 10.1242/dev.029736. [DOI] [PubMed] [Google Scholar]
- 104.Wang S, et al. The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev Cell. 2008;15:261–271. doi: 10.1016/j.devcel.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Nicoli S, et al. MicroRNA-mediated integration of haemodynamics and Vegf signalling during angiogenesis. Nature. 2010;464:1196–1200. doi: 10.1038/nature08889. Provides evidence that blood flow positively influences angiogenesis by promoting the Klf2a-mediated expression of a pro-angiogenic miRNA, miR-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Anand S, et al. MicroRNA-132-mediated loss of p120RasGAP activates the endothelium to facilitate pathological angiogenesis. Nature Med. 2010;16:909–914. doi: 10.1038/nm.2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bonauer A, et al. MicroRNA-92a controls angiogenesis and functional recovery of ischemic tissues in mice. Science. 2009;324:1710–1713. doi: 10.1126/science.1174381. [DOI] [PubMed] [Google Scholar]
- 108.Lampugnani MG, Orsenigo F, Gagliani MC, Tacchetti C, Dejana E. Vascular endothelial cadherin controls VEGFR-2 internalization and signaling from intracellular compartments. J Cell Biol. 2006;174:593–604. doi: 10.1083/jcb.200602080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lanahan AA, et al. VEGF receptor 2 endocytic trafficking regulates arterial morphogenesis. Dev Cell. 2010;18:713–724. doi: 10.1016/j.devcel.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Reynolds AR, et al. Stimulation of tumor growth and angiogenesis by low concentrations of RGD-mimetic integrin inhibitors. Nature Med. 2009;15:392–400. doi: 10.1038/nm.1941. [DOI] [PubMed] [Google Scholar]
- 111.Wu C, et al. Rab13-dependent trafficking of RhoA is required for directional migration and angiogenesis. J Biol Chem. 2011;286:23511–23520. doi: 10.1074/jbc.M111.245209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lindahl P, Johansson BR, Leveen P, Betsholtz C. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science. 1997;277:242–245. doi: 10.1126/science.277.5323.242. [DOI] [PubMed] [Google Scholar]
- 113.Lindblom P, et al. Endothelial PDGF-B retention is required for proper investment of pericytes in the microvessel wall. Genes Dev. 2003;17:1835–1840. doi: 10.1101/gad.266803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Liu Y, et al. Edg-1, the G protein-coupled receptor for sphingosine-1-phosphate, is essential for vascular maturation. J Clin Invest. 2000;106:951–961. doi: 10.1172/JCI10905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Allende ML, Yamashita T, Proia RL. G-protein-coupled receptor S1P1 acts within endothelial cells to regulate vascular maturation. Blood. 2003;102:3665–3667. doi: 10.1182/blood-2003-02-0460. [DOI] [PubMed] [Google Scholar]
- 116.Paik JH, et al. Sphingosine 1-phosphate receptor regulation of N-cadherin mediates vascular stabilization. Genes Dev. 2004;18:2392–2403. doi: 10.1101/gad.1227804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Suri C, et al. Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell. 1996;87:1171–1180. doi: 10.1016/s0092-8674(00)81813-9. [DOI] [PubMed] [Google Scholar]
- 118.Jeansson M, et al. Angiopoietin-1 is essential in mouse vasculature during development and in response to injury. J Clin Invest. 121:2278–2289. doi: 10.1172/JCI46322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Foo SS, et al. Ephrin-B2 controls cell motility and adhesion during blood-vessel-wall assembly. Cell. 2006;124:161–173. doi: 10.1016/j.cell.2005.10.034. [DOI] [PubMed] [Google Scholar]
- 120.Chen S, Lechleider RJ. Transforming growth factor-β-induced differentiation of smooth muscle from a neural crest stem cell line. Circ Res. 2004;94:1195–1202. doi: 10.1161/01.RES.0000126897.41658.81. [DOI] [PubMed] [Google Scholar]
- 121.Domenga V, et al. Notch3 is required for arterial identity and maturation of vascular smooth muscle cells. Genes Dev. 2004;18:2730–2735. doi: 10.1101/gad.308904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Maisonpierre PC, et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science. 1997;277:55–60. doi: 10.1126/science.277.5322.55. [DOI] [PubMed] [Google Scholar]
- 123.Red-Horse K, Ueno H, Weissman IL, Krasnow MA. Coronary arteries form by developmental reprogramming of venous cells. Nature. 2010;464:549–553. doi: 10.1038/nature08873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lee S, et al. Autocrine VEGF signaling is required for vascular homeostasis. Cell. 2007;130:691–703. doi: 10.1016/j.cell.2007.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Saharinen P, et al. Angiopoietins assemble distinct Tie2 signalling complexes in endothelial cell–cell and cell–matrix contacts. Nature Cell Biol. 2008;10:527–537. doi: 10.1038/ncb1715. [DOI] [PubMed] [Google Scholar]