

Ferritins are highly conserved multi-subunit proteins that detoxify and reversibly store intracellular iron(1). Mice lacking the cytosolic ferritin heavy chain (H) exhibit embryonic lethality; mice with a conditional ferritin H deletion exhibit severe tissue damage(2,3). Mitochondrial ferritin (Ftmt) is structurally and functionally homologous to ferritin H chain(4,5). Elevated Ftmt expression in erythroblasts from sideroblastic anemia patients suggests a role for Ftmt in these anemias, a heterogeneous group of inherited and acquired disorders characterized by mitochondrial iron deposition(6). While cytosolic ferritin H and light (L) chains are ubiquitous in mammals, Ftmt mRNA is found largely in the testes(7,8). The lack of an iron responsive element in the Ftmt mRNA, while present in ferritin H and L mRNAs, suggests that Ftmt may function independently of iron metabolism. Studies to date have focused on protective effects of Ftmt overexpression against iron overload and oxidative stress in tissue culture, yeast and flies(9–15). To investigate the in vivo role of Ftmt, we deleted the Ftmt gene in mice and examined baseline hematology, iron metabolism and male fertility phenotypes. In addition, we subjected them to vitamin B6 (pyridoxine) deprivation to induce sideroblast/siderocyte formation. In all cases, we observed no significant defects in mice lacking mitochondrial ferritin.
To construct Ftmt-deficient mice, we replaced the entire open reading frame (ORF) of the intronless Ftmt gene with a neomycin-resistance cassette (Fig. 1A). Southern blotting using probes flanking the region encompassed by the targeting construct indicated successful recombination at the Ftmt locus in embryonic stem (ES) cells (data not shown). We confirmed Ftmt deletion using PCR and primers specific to the flanking regions and the neomycin cassette (Fig. 1B). To measure Ftmt expression, we used reverse transcriptase-polymerase chain reaction (RT-PCR) and primers specific to β-actin and Ftmt; control reactions without reverse transcription were performed to ensure that PCR products did not represent amplification of genomic DNA. As Ftmt is most abundantly expressed in the testis, we measured expression in RNA isolated from testes of wild-type and homozygous littermates; no expression was detected in homozygous mutant animals (Fig. 1C). We could not detect Ftmt expression in bone marrow from either genotype, even after pyridoxine deprivation (data not shown; see below).
Figure 1. Construction and analysis of Ftmt-deficient mice.
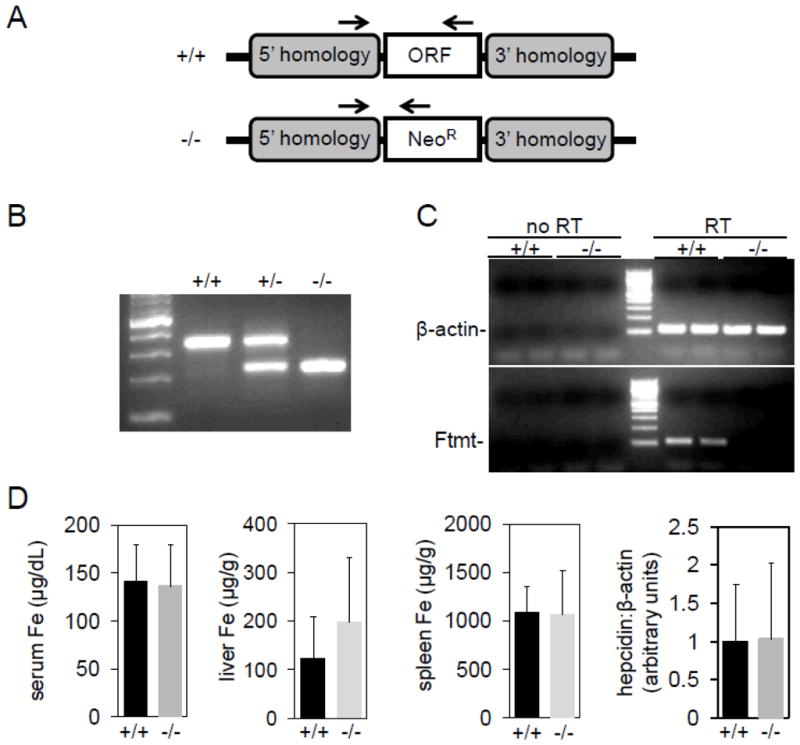
(A) Schematic of wild-type (+/+) and mutant (−/−) Ftmt locus. Arrows indicate site of primers used to genotype mice in (B). (B) Genotyping gel of mice wild-type (+/+), heterozygous (+/−) or homozygous (−/−) for the inactivated Ftmt allele. First lane from left is a DNA size ladder. (C) Gel of RT-PCR using wild-type and homozygous mouse testes RNA and primers specific to β-actin and Ftmt. Reactions with (RT) and without (no RT) a reverse transcriptase step were included to control for amplification of genomic DNA. (D) Analysis of serum iron (Fe), liver Fe, spleen Fe and liver hepcidin levels in wild-type and homozygous animals.
Ftmt−/− animals on the C57BL/6J genetic background at backcross generation greater than N7 were obtained at expected Mendelian frequencies from heterozygous pairings and no gross anatomic or histological abnormalities, including iron staining, were present in the mutant offspring (data not shown). Given the testis-specific expression, we assessed male fertility by pairing mutant and wild type males with two wild type females for 14 days and counted the number of pregnancies and total number of offspring obtained(16); based on this assessment, there was no difference in male fertility between mutants and controls (data not shown).
To determine the effect of Ftmt deletion on baseline murine iron metabolism and hematology, we measured serum, spleen and liver iron concentrations, liver hepcidin expression and red blood cell parameters. There were no significant differences between wild-type and homozygous animals (Fig. 1D, 2A–G). To assess the role of genetic background, we bred the Ftmt deletion onto 129/SvEvTac for five generations (N5); we identified no significant differences between wild-type and homozygous animals on this background as well(data not shown). Given that baseline parameters of iron metabolism differ between these strains, this result suggests that alterations in serum or liver iron levels do not impact the steady state phenotype of Ftmt-deficient mice.
Figure 2. Analysis of Ftmt-deficient mice on a pyridoxine-deficient diet.
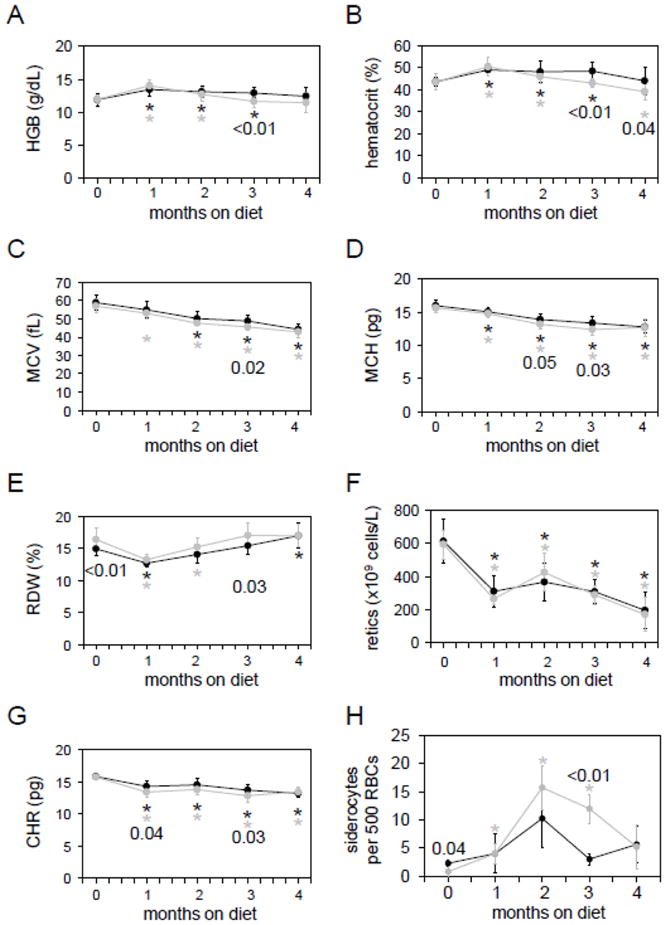
Wild-type (black lines and circles) and homozygous (grey lines and circles) mice were placed on a pyridoxine-deficient diet at one month of age and followed for four months. Parameters measured were (A) hemoglobin level (HGB), (B) hematocrit, (C) mean corpuscular volume (MCV), (D) mean corpuscular hemoglobin level (MCH), (E) red cell distribution width (RDW), (F) reticulocyte count (retics), (G) reticulocyte hemoglobin content (CHR) and (H) number of siderocytes per 500 red blood cells. Six to nine mice were analyzed per time point and genotype. Numbers indicate p values between wild-type and homozygous values at one time point; asterisks indicate value at that time point differs significantly (p<0.05) from initial value at 0 weeks. P values were determined by Student’s two-tailed t-test.
We next placed a cohort of wild-type and homozygous mice on a pyridoxine-deficient diet for four months beginning at one month of age to induce formation of sideroblast or siderocyte (iron-granule positive nucleated or enucleated erythrocytes) (Fig. 2A–2G). Similar to observations made in C57BL/6J x C3H (B6C3F1) mice maintained on a pyridoxine-deficient diet(17), we noted a decreased MCV and hematocrit, along with decreased MCH, reticulocyte counts and CHR. The lack of decrease in HGB levels in our mice, while noted in the study in B6C3F1 mice, may have been due to strain-specific differences. While a small but statistically significant increase in red cell distribution width and an increase in number of siderocytes were noted in the homozygous animals early in the course of the experiment, these differences did not persist. Notably, the number of siderocytes was greater in homozygous than wild-type animals after three months on a pyridoxine-deficient diet; the decrease in siderocyte number thereafter in the homozygous animals may reflect an unknown mechanism of chronic adaptation to the pyridoxine-deficient diet (Fig. 2H). Siderocytes had similar appearance by light microscopy in wild-type and homozygous mutant animals (Fig. 3A–B), as did reticulocyte mitochondria by electron microscopy (Fig. 3C–D). As is true of most mouse sideroblastic anemia models, bone marrow ringed sideroblasts were not present in wild type or mutant animals (data not shown). Overall, there were no pronounced differences in red cell parameters between wild-type and homozygous mice maintained on the pyridoxine-deficient diet.
Figure 3. Microscopic evaluation of Ftmt-deficient erythrocytes.
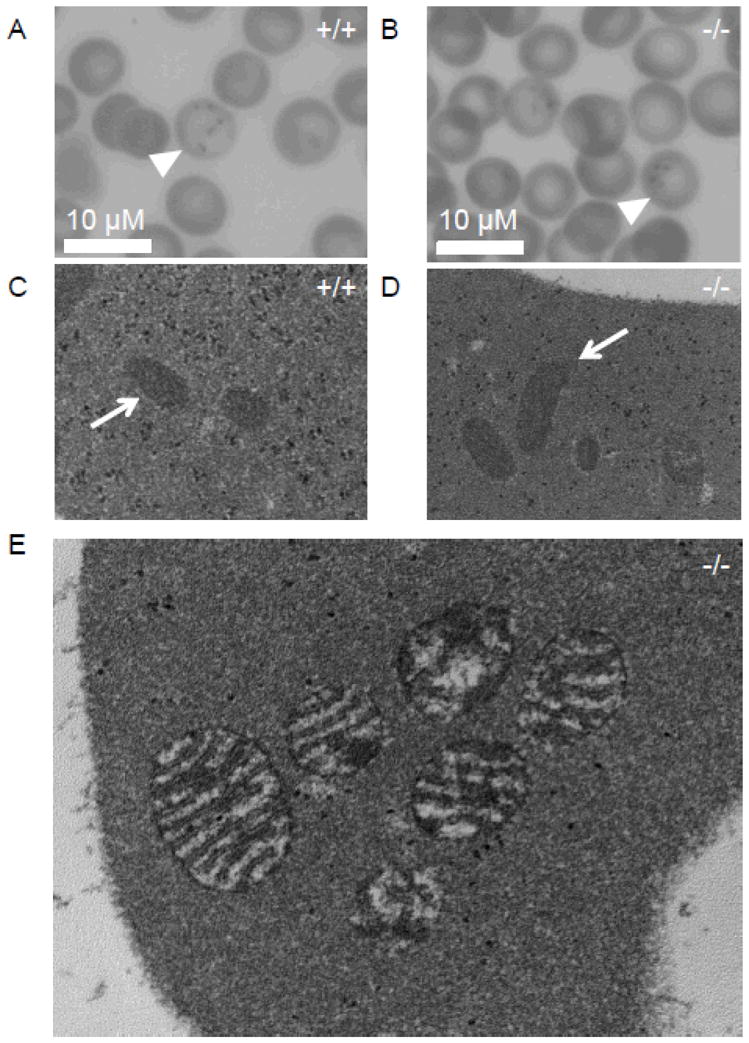
(A,B) Light micrographs of Prussian Blue-stained blood smears from wild-type (+/+) (A) and Ftmt-deficient (−/−) (B) mice; siderocytes are indicated by arrowheads; 40x magnification. (C,D) Electron micrographs of reticulocytes from wild-type (+/+) (C) and Ftmt-deficient mice (−/−) (D); mitochondria are indicated by arrows; 40,000x original magnification. The wild-type and mutant mitochondria are indistinguishable. (E) High-power electron micrograph of a siderocyte from a Ftmt-deficient mouse; 80,000x original magnification. Note the slight enhancement of the mitochondrial membranes and amorphous electron dense deposits detailed in (E), which are similar to previous descriptions of murine siderocytes by electron microscopy(20). All samples were obtained from mice on pyridoxine-deficient diets. All blood samples were stained with acid ferrocyanide prior to processing for electron microscopy.
Our results do not contradict the relatively small body of Ftmt literature. Most, if not all, functional studies to date have relied on overexpression to demonstrate that Ftmt can alter the cellular effects of challenges such as oxidative stress under iron-loaded conditions. We have shown that Ftmt is not essential for viability or male fertility and that Ftmt deletion does not alter several parameters of iron metabolism under baseline conditions, nor does it affect the hematological response to a pyridoxine-deficient diet. Intriguingly, even in the absence of Ftmt, iron-rich deposits can be formed in erythroid cells, suggesting that Ftmt is not essential for siderocyte formation in mice. Furthermore, there may be redundant systems in mice to compensate for the lack of Ftmt function, as is the case with endothelial nitric synthase- or myoglobin-deficient mice in which adaptive mechanisms thoroughly compensate for the respective gene defects(18). One can also speculate that a failure to induce Ftmt expression on a pyridoxine-deficient diet might account for the fact that bone marrow ringed sideroblasts are not typically seen in mice. This demonstrates some of the limitations of using the mouse as a model for sideroblastic anemia—a failure to induce sideroblast formation in mice prevents us from effectively determining if Ftmt plays a role in detoxification of erythroid mitochondrial iron.
Methods
To construct a targeting vector, we cloned 3.2 and 5.8 kb DNA fragments 5′ and 3′ to the single exon encoding the entire Ftmt ORF into pKO Scrambler (Stratagene). The linearized targeting vector was electroporated into J1 (129Sv/Jae derived) ES cells. Homologous recombination was confirmed by Southern blot analysis from both sides of the deleted locus. One ES cell clone with a normal karyotype was injected into C57BL/6 blastocysts. Transmission of the targeted allele (Ftmt−/−) was confirmed by Southern blot analysis. Subsequent genotyping was carried out by PCR analysis using primers within the Ftmt coding sequence or the neomycin resistance cassette and 5′ to the deleted region (Figures 1A, B). The targeted allele was backcrossed onto 129S6/SvEvTac and C57/BL6J for at least 5 generations. The analyses reported here are on C57BL/6J; however many studies were duplicated on 129S6/SvEvTac and yielded similar results. Total RNA isolation, RT-PCR, complete blood counts (CBCs) and serum and tissue iron measurements were performed and hepcidin and β-actin mRNA levels measured by quantitative PCR as previously described(19). For dietary studies, mice were maintained on pyridoxine-deficient (0 ppm) or –replete (15 ppm) diets (Harlan-Teklad TD 01271 and TD 01270) beginning at one month of life. Details of vector construction, probe and primer information and diet compositions are available upon request. Siderocytes were identified by light microscopy of Prussian Blue-stained blood smears. Electron microscopy was performed on acid ferrocyanide-stained samples at the Electron Microscope Facility at Children’s Hospital Boston as previously described(20).
Acknowledgments
M.D.F. was supported by NIH R01DK62474 & R01DK080011 the Wilkes Fund of the Children’s Hospital Department of Pathology, and the Pew Biomedical Scholars Program. T.B.B. is supported by NIH K99 DK084122 C.P. was funded in part by L’Association pour la Recherche sur le Cancer (ARC). Transgenic core services were supported by NIH P30 DK049216 through the Center of Excellence in Molecular Hematology at Children’s Hospital Boston.
Footnotes
The authors declare no conflicts of interest.
References
- 1.Arosio P, Ingrassia R, Cavadini P. Ferritins: a family of molecules for iron storage, antioxidation and more. Biochim Biophys Acta. 2009;1790:589–599. doi: 10.1016/j.bbagen.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 2.Darshan D, Vanoaica L, Richman L, et al. Conditional deletion of ferritin H in mice induces loss of iron storage and liver damage. Hepatology. 2009;50:852–860. doi: 10.1002/hep.23058. [DOI] [PubMed] [Google Scholar]
- 3.Ferreira C, Bucchini D, Martin ME, et al. Early embryonic lethality of H ferritin gene deletion in mice. J Biol Chem. 2000;275:3021–3024. doi: 10.1074/jbc.275.5.3021. [DOI] [PubMed] [Google Scholar]
- 4.Langlois d’Estaintot B, Santambrogio P, Granier T, et al. Crystal structure and biochemical properties of the human mitochondrial ferritin and its mutant Ser144Ala. J Mol Biol. 2004;340:277–293. doi: 10.1016/j.jmb.2004.04.036. [DOI] [PubMed] [Google Scholar]
- 5.Levi S, Corsi B, Bosisio M, et al. A human mitochondrial ferritin encoded by an intronless gene. J Biol Chem. 2001;276:24437–24440. doi: 10.1074/jbc.C100141200. [DOI] [PubMed] [Google Scholar]
- 6.Fleming MD. The genetics of inherited sideroblastic anemias. Semin Hematol. 2002;39:270–281. doi: 10.1053/shem.2002.35637. [DOI] [PubMed] [Google Scholar]
- 7.Santambrogio P, Biasiotto G, Sanvito F, et al. Mitochondrial ferritin expression in adult mouse tissues. J Histochem Cytochem. 2007;55:1129–1137. doi: 10.1369/jhc.7A7273.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Levi S, Arosio P. Mitochondrial ferritin. Int J Biochem Cell Biol. 2004;36:1887–1889. doi: 10.1016/j.biocel.2003.10.020. [DOI] [PubMed] [Google Scholar]
- 9.Lu Z, Nie G, Li Y, et al. Overexpression of mitochondrial ferritin sensitizes cells to oxidative stress via an iron-mediated mechanism. Antioxid Redox Signal. 2009;11:1791–1803. doi: 10.1089/ars.2008.2306. [DOI] [PubMed] [Google Scholar]
- 10.Corsi B, Cozzi A, Arosio P, et al. Human mitochondrial ferritin expressed in HeLa cells incorporates iron and affects cellular iron metabolism. J Biol Chem. 2002;277:22430–22437. doi: 10.1074/jbc.M105372200. [DOI] [PubMed] [Google Scholar]
- 11.Campanella A, Isaya G, O’Neill HA, et al. The expression of human mitochondrial ferritin rescues respiratory function in frataxin-deficient yeast. Hum Mol Genet. 2004;13:2279–2288. doi: 10.1093/hmg/ddh232. [DOI] [PubMed] [Google Scholar]
- 12.Campanella A, Rovelli E, Santambrogio P, et al. Mitochondrial ferritin limits oxidative damage regulating mitochondrial iron availability: hypothesis for a protective role in Friedreich ataxia. Hum Mol Genet. 2009;18:1–11. doi: 10.1093/hmg/ddn308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nie G, Sheftel AD, Kim SF, et al. Overexpression of mitochondrial ferritin causes cytosolic iron depletion and changes cellular iron homeostasis. Blood. 2005;105:2161–2167. doi: 10.1182/blood-2004-07-2722. [DOI] [PubMed] [Google Scholar]
- 14.Zanella I, Derosas M, Corrado M, et al. The effects of frataxin silencing in HeLa cells are rescued by the expression of human mitochondrial ferritin. Biochim Biophys Acta. 2008;1782:90–98. doi: 10.1016/j.bbadis.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 15.Missirlis F, Holmberg S, Georgieva T, et al. Characterization of mitochondrial ferritin in Drosophila. Proc Natl Acad Sci US A. 2006;103:5893–5898. doi: 10.1073/pnas.0601471103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Handel MA, Lessard C, Reinholdt L, et al. Mutagenesis as an unbiased approach to identify novel contraceptive targets. Mol. Cell. Endocrinol. 2006;250:201–205. doi: 10.1016/j.mce.2005.12.046. [DOI] [PubMed] [Google Scholar]
- 17.Tangjarukij C, Navasumrit P, Zelikoff JT, et al. The effects of pyridoxine deficiency and supplementation on hematological profiles, lymphocyte function, and hepatic cytochrome P450 in B6C3F1 mice. J Immunotoxicol. 2009;6:147–160. doi: 10.1080/15476910903083866. [DOI] [PubMed] [Google Scholar]
- 18.Gödecke A, Schrader J. Adaptive mechanisms of the cardiovascular system in transgenic mice--lessons from eNOS and myoglobin knockout mice. Basic Res Cardiol. 2000;95:492–498. doi: 10.1007/s003950070026. [DOI] [PubMed] [Google Scholar]
- 19.Schmidt PJ, Toran PT, Giannetti AM, et al. The transferrin receptor modulates Hfe-dependent regulation of hepcidin expression. Cell Metab. 2008;7:205–214. doi: 10.1016/j.cmet.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pondarre C, Campagna DR, Antiochos B, et al. Abcb7, the gene responsible for X-linked sideroblastic anemia with ataxia, is essential for hematopoiesis. Blood. 2007;109:3567–3569. doi: 10.1182/blood-2006-04-015768. [DOI] [PMC free article] [PubMed] [Google Scholar]