

Abstract
Caspases, a family of cysteine proteases, are widely activated in neurons and glia in the injured brain, a response thought to induce apoptosis. However, caspase activation in astrocytes following injury is not strongly associated with apoptosis. The present study investigates the potential role of caspase activation in astrocytes with another characteristic response to neural injury, astrogliosis. Caspase activity and morphological and biochemical indices of astrogliosis and apoptosis were assessed in (i) cultured neonatal rat astrocytes treated with astrogliosis-inducing stimuli (dibutryl cAMP, β-amyloid peptide), and (ii) cultures of adult rat hippocampal astrocytes generated from control and kainate-lesioned rats. The effects of broad spectrum and specific pharmacological caspase inhibitors were assessed on indicators of astrogliosis, including stellate morphology and expression of glutamine synthetase and fibroblast growth factor-2. Reactive neonatal and adult astrocytes demonstrated an increase in total caspase activity with a corresponding increase in the expression of active caspase-3 in the absence of cell death. Broad spectrum caspase inhibition with zVAD significantly attenuated increases in glutamine synthetase and fibroblast growth factor-2 in the reactive astrocytes. In the reactive neonatal astrocyte cultures, specific inhibition of caspases-3 and -11 also attenuated glutamine synthetase and fibroblast growth factor-2 expression, but did not reverse the morphological reactive phenotype. Astrogliosis is observed in all forms of brain injury and despite extensive study, its molecular triggers remain largely unknown. While previous studies have demonstrated active caspases in astrocytes following acute brain injury, here we present evidence functionally implicating the caspases in astrogliosis.
Keywords: caspase, fibroblast growth factor-2, glutamine synthetase, kainate, reactive astrocytes
1. INTRODUCTION
Damage to the brain, be it mechanical trauma, disease, or even normal aging, elicits a robust astrocytic reaction termed astrogliosis (Pekny and Nilsson, 2005). Astrogliosis can be considered an attempt to restore homeostasis in the damaged brain through important functions including glial scar formation, regulation of immune responses, and the modulation of neuronal survival and neurite outgrowth (Sofroniew, 2009). Morphologically, astrogliosis is characterized by changes including hypertrophy, stellation and proliferation, in addition to numerous biochemical changes, which in turn alter neuronal function and viability (Pekny and Nilsson, 2005). Reactive astrocytes express proteins capable of both beneficial and detrimental effects on parameters of neuronal health, outcomes of which are dependent upon factors such as the age of the organism and the type and extent of injury (Sofroniew, 2009). Since astrogliosis can result in such functionally diverse outcomes on neuronal survival, a thorough understanding of the signal transduction pathways regulating astrocyte reactivity may provide insight into normal and pathological astrocytic responses to injury.
Caspase activation is a widespread event following disease or damage to the brain, not only present in neurons and microglia, but also observed in astrocytes (Acarin et al., 2007; Benjelloun et al., 2003; Mouser et al., 2006; Narkilahti et al., 2003; Su et al., 2000; Villapol et al., 2008). Caspases are cysteine proteases classified as either initiator caspases, including caspase-2, -8, -9 and -10, or executioner caspases, including caspase-3, -6 and -7. Initiator caspases proteolytically cleave and activate executioner caspases. In turn, executioner caspases proteolytically cleave and degrade structural proteins, signaling molecules, and DNA repair enzymes (Kumar, 2006). While the caspase family is predominantly associated with the processing of pro-inflammatory cytokines and the execution of apoptosis (Chang and Yang, 2000), various non-apoptotic functions including modulation of cell proliferation, differentiation and cell spreading have been described in a variety of cell types (McLaughlin, 2004; Schwerk and Schulze-Osthoff, 2003). In the brain, the caspase family have also been implicated in neuronal (Fernando et al., 2005) and glial differentiation (Oomman et al., 2006), neuronal cytoskeletal remodeling (Acarin et al., 2007; Rohn et al., 2004), synaptic plasticity (Dash et al., 2000), axon guidance and neurite outgrowth (Campbell and Holt, 2003; Chan and Mattson, 1999) as well as contributing to neuronal survival following preconditioning (Garnier et al., 2003; McLaughlin, 2004; Tanaka et al., 2004).
While some studies attribute caspase activation in astrocytes to glial degeneration (Su et al., 2000), several studies have also demonstrated that astrocytic expression of active caspases is not always associated with cell death following ischemic/excitotoxic damage (Acarin et al., 2007; Duran-Vilaregut et al., 2010; Narkilahti et al., 2003; Villapol et al., 2008; Wagner et al., 2011) and traumatic brain injury (Beer et al., 2000). Considering that gliosis, not cell death, is the principal astrocytic response to injury, it is possible that caspase activation in astrocytes may exert non-apoptotic functions that contribute to astrogliosis.
To investigate a possible non-apoptotic function of caspases in the regulation of astrogliosis, we examined caspase activity and indices of reactivity in rat astrocyte cultures. Experiments were conducted in two different culture paradigms. First, we utilized primary cultures of neonatal rat astrocytes that were exposed to β-amyloid (Aβ), an aggregating peptide that is associated with astrogliosis in both Alzheimer’s disease brain (Rodriguez et al., 2009) and culture models. Treatment of astroglial cultures with Aβ typically does not result in cell death (Kato et al., 1997; Pike et al., 1994) but causes morphological features of gliosis (Abe et al., 1997; Canning et al., 1993; Hu et al., 1998; Pike et al., 1994; Salinero et al., 1997) as well as increased glial fibrillary acid protein (GFAP) immunoreactivity (Pike et al., 1994; Hu et al., 1998) and elevated levels of several cytokines, growth factors, and enzymes (Araujo and Cotman, 1992; Hu et al., 1998; Pike et al., 1994, 1996a). For comparison, we also treated cultures with dibutryl cyclic AMP (dBcAMP), a stimulus that induces some features of astrogliosis (Fedoroff et al., 1984; Le Prince et al., 1991). In our analyses, we focused on two factors that are established markers of astrogliosis in several paradigms, basic fibroblast growth factor (FGF-2) and glutamine synthetase (GS) (Eddleston and Mucke, 1993). Observations from neonatal cultures studies were corroborated using an ex vivo model of astrogliosis induced in adult animals, an approach demonstrated to retain biochemical changes associated with the reactive phenotype in culture even after multiple divisions (Rozovsky et al., 2005; Wu et al., 1998).
2. RESULTS
2.1 In vitro model of astrogliosis
Neonatal astrocyte cultures were treated with either dBcAMP (1 mM), a synthetic analogue of cAMP, or Aβ peptide 25–35 (Aβ, 25 μM), an aggregating polypeptide implicated in Alzheimer’s disease. Exposure to treatments for 48 h induced stellation (Fig. 1A–C) and significantly increased expression of GS (Fig. 1D) and FGF-2 (Fig. 1E). These astrogliosis-related changes were observed as early as 24 h after exposure to dBcAMP and Aβ and persisted for at least 7 d (data not shown).
Figure 1. Reactivity observed in primary astrocytes treated with dB-cAMP or Aβ.

(A) Vehicle-treated control (Ctl) neonatal astrocyte cultures show polygonal morphology. In contrast, stellate morphology is observed in cultures treated with (B) 1 mM dBcAMP (dB) or (C) 25 μM Aβ25–35. Representative western blots demonstrate increased expression of (D) GS and (E) FGF-2 in cultures treated for 48 h with dBcAMP (dB) or Aβ relative to vehicle-treated controls (Ctl).
2.2 Non-apoptotic activation of caspases in neonatal astrocyte cultures
To begin evaluating a potential role of caspases in the observed astrogliosis, we assessed caspase activity in astrocytes cultures treated with dBcAMP or Aβ. In comparison to vehicle-treated controls, cultures exposed for 48 h to 1 mM dBcAMP or 25 μM Aβ showed more than a two-fold increase in total caspase activity (Fig. 2A). This increase in caspase activity was apparent within 24 h and persisted for at least 4 d, although there was no significant change in vehicle-treated control cultures (data not shown). The observed elevation in caspase activity was associated with an up-regulation of the cleaved active fragment of caspase-3, the most common effector caspase in the brain (Fig. 2B). Increases in total caspase activity and cleavage of caspase-3 associated with dBcAMP- and Aβ-induced reactivity were modest compared to marked increases observed following treatment of astrocytes with a toxic concentration of staurosporine (1 μM), a broad kinase inhibitor established to induce caspase activation and apoptosis in numerous cell types (Bertrand et al., 1994; Krohn et al., 1998). Importantly, although both 1 mM dBcAMP and 25 μM Aβ increase caspase activity in astrocytes, this action was not associated with changes in cell viability. To ensure that caspase activation was not associated with delayed cell death, we maintained treatment of astrocyte cultures with dBcAMP and Aβ for 4 days before analysis. Neither dBcAMP nor Aβ induced significant cell death compared to vehicle-treated astrocytes, with no differences observed in the number of cells labeled with the cell death marker, ethidium homodimer (Fig. 3A, B) or the release of the enzyme lactate dehydrogenase (Fig. 3C). The proportion of vehicle-treated cells labeled with ethidium homodimer was always <5% at all time points. As a positive control, we also assessed viability in cultures treated with 1 μM staurosporine.
Figure 2. Increased caspase activity following treatment with reactive stimuli dB-cAMP or Aβ.

Treatment of neonatal astrocytes cultures with 1 mM dB-cAMP (dB) or 25 μM Aβ25–35 for 48 h significantly increased (A) total caspase activity and (B) levels of the active caspase-3 fragment as assessed by western blot (inset: representative western blot). Cultures treated for 24 h with the apoptotic stimulus 1μM staurosporine (STS) demonstrated the greatest increase in total caspase activity and levels of the active caspase-3 fragment. Data show mean levels (+SEM) from ≥ 3 independent experiments. * P < 0.01 relative to vehicle-treated control (Ctl).
Figure 3. Absence of cell death following treatment with reactive stimuli db-cAMP or Aβ.

(A) Viability of neonatal astrocytes cultures treated with vehicle (CTL), 1 mM dBcAMP or 25 μM Aβ25–35 for 4 d or 1 μM staurosporine (STS) for 24 h was assessed by staining with the vital dyes calcein AM (green fluourescence; indicates live cells), and ethidium homodimer (red fluorescence; indicates dead cells). Levels of cell death in treated astrocytes cultures were quantified by (B) counts of cells with positive staining for ethidium homodimer, and (C) measurement of LDH release into the culture medium. Data show mean levels (+SEM) from ≥ 3 independent experiments. * P < 0.001 relative to vehicle-treated control (Ctl).
2.3 Caspase inhibitors modulate reactive responses to Aβ and dBcAMP in neonatal astrocyte cultures
To begin to assess the potential role of the observed increase in caspase activity in regulation of astrogliosis, we evaluated the effect of a broad-spectrum caspase inhibitor, zVAD, on expression of GS and FGF-2 as well as morphological stellation. We observed that 1 h pretreatment of astrocyte cultures with 50 μM zVAD significantly reduced the increases in GS (Fig. 4A,B) and FGF-2 (Fig. 4C,D) induced by 48 h exposure to 1 mM dBcAMP and 25 μM Aβ. In separate experiments, we found that expression of GS and FGF-2 was also reduced by a 48 h exposure to 50 μM zVAD initiated 48 h after the addition of dBcAMP and Aβ (data not shown). In contrast to its effects on GS and FGF-2 expression, zVAD pretreatment did not affect induction of stellate morphology by dBcAMP and Aβ (Fig. 5A, B). As a positive control, we also assessed the effects of pretreatment with 10 nM thrombin, a factor known to reverse stellate morphology of astrocytes (Pindon et al., 1998), and observed an approximately 75% inhibition of astrocyte stellation in dBcAMP and Aβ treated cultures (Fig. 5A, B).
Figure 4. Broad-spectrum caspase inhibition attenuates up-regulation of GS and FGF-2 expression in response to reactive stimuli, dB-cAMP and Aβ, in astrocyte cultures.

Neonatal astrocyte cultures were treated for 48 h with vehicle (Ctl), 1 mM dBcAMP (dB), or 25 μM Aβ25–35 in the absence (−) or presence (+) or 1 h pretreatment with 50 μM zVAD. Following the experiment, cultures were processed for western blot to assess levels of GS (A) and FGF-2 (C). β-Tubulin was assessed as to ensure equal protein loading. Relative protein levels of GS (B) and FGF-2 (D) were determined by densitometric scanning of western blots from ≥ 3 independent experiments. Data show mean values (+SEM) from each condition expressed as a percentage of the vehicle-treated control group. * P < 0.05 relative to matched (−) zVAD condition.
Figure 5. Astrocyte stellation unaffected by broad spectrum capsase inhibition.
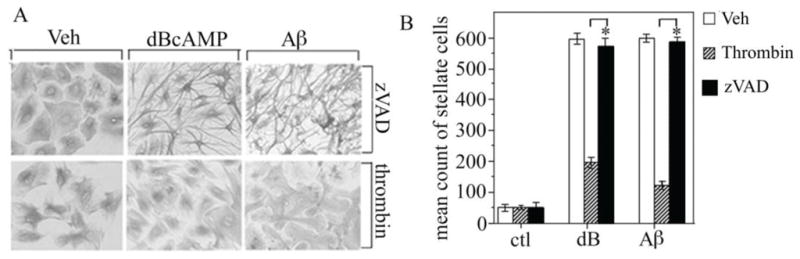
Neonatal astrocytes cultures were pretreated for 1 h with vehicle (veh), 50 μM zVAD, or 10 nM thrombin followed by 48 h exposure to 1 mM dBcAMP (dB) or 25 μM Aβ25–35 and then were processed for GFAP immunocytochemistry. (A) Representative bright field images of GFAP immunostaining show that stellate morphology is attenuated by pretreatment with thrombin but not zVAD. (B) Stellation was quantified from immunostained cultures by determining mean counts of morphologically stellate cells (+SEM) from each condition (N = 3). * P < 0.05 relative to matched condition.
To investigate which members of the caspase family may be contributing to observed effect of the broad spectrum caspase inhibitor, astrocyte cultures were pretreated for 1 h with synthetic peptide inhibitors specific for caspase-1 (YVAD), caspase-3 (DEVD), caspase-6 (AEVD), caspase-8 (LETD), caspase-9 (LEHD) or caspase-11 (WEHD) prior to inducing reactivity with 1 mM dBcAMP. Since the inhibitors can show reduced specificity at higher concentrations (>50 μM), only inhibitory effects on GS and FGF-2 expression observed at lower concentrations (1, 10 and 30 μM) were considered specific. Specific inhibition of caspase-3 with DEVD (Fig. 6A, B) and caspase-11 with WEHD (Fig. 6C, D) significantly attenuated dBcAMP-induced elevations in GS and FGF-2 expression. At higher, less specific concentrations (> 50 μM), AEVD and LEHD significantly attenuated both GS and FGF-2 expression, while YVAD attenuated FGF-2 but not GS expression (Fig. 7).
Figure 6. Specific inhibition of caspases-3 and -11 attenuates GS and FGF-2 expression in reactive astrocytes.

Neonatal astrocytes cultures were pretreated for 1 h with increasing concentration of caspase inhibitors (0, 1, 10, 30, 60, 100 μM) followed by 48 h exposure to 1 mM dBcAMP (dB). For comparison, experiments included conditions with vehicle (Ctl) or 100 μM inhibitor (100) in the absence of dBcAMP. At the conclusion of the experiment, cultures were processed for western blots to assess levels of GS, FGF-2, and β-tubulin (internal control). Representative blots are shown for astrocyte cultures pre-treated with (A) the caspase-3 inhibitor DEVD, and (C) the caspase-11 inhibitor WEHD. Densitometric analysis of blots revealed significant inhibition of both GS and FGF-2 expression in astrocyte cultures treated with (B) 10 – 100 μM DEVD, and (D) 30–100 μM WEHD. Data show mean values (± SEM) relative to dBcAMP-treated cultures in the absence of caspase inhibitor. *P < 0.05 relative to condition lacking inhibitor.
Figure 7. GS and FGF-2 expression in reactive astrocytes unaffected by specific inhibition of caspases-1, -6, -8 and -9.

Neonatal astrocytes cultures were pretreated for 1 h with increasing concentration of caspase inhibitors (Inh; 0, 1, 10, 30, 60, 100 μM) followed by 48 h exposure to 1 mM dBcAMP (dB). For comparison, experiments included conditions with vehicle (Ctl) or 100 μM inhibitor (100) in the absence of dBcAMP. At the conclusion of the experiment, cultures were processed for western blots to assess levels of GS, FGF-2, β-tubulin (internal control). Representative blots for (A) GS and (B) FGF-2 are shown for astrocyte cultures pre-treated with specific inhibitors for caspase-1 (YVAD), caspase-6 (AEVD), caspase-8 (LETD) or caspase-9 (LEHD). At non-specific concentrations (>50μM) AEVD and LEHD significantly attenuated both GS and FGF-2 expression, while YVAD attenuated FGF-2 but not GS expression. Data show mean values (± SEM) relative to dBcAMP-treated cultures in the absence of caspase inhibitor.
2.4 Caspases contribute to astrogliosis in adult astrocyte cultures
To corroborate the relationship between caspases and astrogliosis observed in cultures of neonatal astrocytes, we used a second experimental model of hippocampal astrocytes cultured from kainate-lesioned adult rats and sham-lesioned control rats. We used immunohistochemical analyses to confirm that kainate induced both neuron loss (neuron-specific NeuN antibody) and associated astrogliosis (astrocyte-specific GFAP antibody) in the lesioned animals. As expected, we observed extensive neuron loss confined to the CA3 region of hippocampus (Fig. 8A,B) and robust astrogliosis throughout hippocampus (Fig. 8C,D). Cultures generated from kainate-lesioned and sham-lesioned animals exhibited similar morphological appearance with predominantly polygonal-shaped cells and an absence of significant stellation (Fig. 9A,B). Both kainate-lesioned and sham-lesioned astrocytes cultures were also characterized by low levels of cell death as evidenced by minimal release of lactate dehydrogenase (Fig. 9C).
Figure 8. Kainate lesion results in neuronal loss and astrogliosis.

Adult female rats were lesioned bilaterally by intra-hippocampal injection with kainate then 3 weeks later processed for immunohistochemistry. Labeling with the neuron-specific antibody NeuN shows that the hippocampus CA3 pyramidal neuron layer (arrows) is intact in vehicle-lesioned (Sham) animals (A) but largely degenerated in kainate-lesioned (Kainate) rats (B). Astrocyte staining with GFAP antibody shows that, in comparison to sham-lesioned rats (C), lesioned rats display robust labeling throughout hippocampus (D) that is characterized by robust stellation (magnified inset). Images show representative findings from sham-lesioned (N = 4) and kainate-lesioned (N = 5) animals.
Figure 9. Evidence of non-apoptotic caspase activation in an ex vivo model of astrogliosis.

Astrocytes cultured from adult sham-lesioned (Sham) and kainate-lesioned (Kainate) rats were characterized for morphology, caspase activity and viability. GFAP immunostaining of cultures revealed similar morphological appearance with little stellation in hippocampal astrocyte cultures derived from (A) sham-lesioned (N = 4) and (B) kainate-lesioned (N = 5) rats. (C) Astrocyte cultures from sham-lesioned animals (Sham) and kainate-lesioned animals (Kainate) did not exhibit significant differences in LDH release. For comparison, cultures from sham-lesioned animals treated for 24 h with 1 μM staurosporine (STS) showed a significant increase in LDH release. (D) Total caspase activity in cultures was assessed in the presence (solid bars) and absence (open bars) of the general caspase inhibitor zVAD (50 μM). Caspase activity was significantly elevated both in cultures from kainate-lesioned animals and in cultures of sham-lesioned animals treated for 24 h with 1 μM staurosporine. Data show mean values (+SEM) from 4–5 independent culture preparations. * P < 0.05 relative to Sham condition. (E) Representative western blots show relative levels active caspase-3 expression from sham- and kainate-lesioned astrocyte cultures in the presence (+) and absence (−) of 50 μM zVAD treatment and following treatment with 1μM staurosporine (STS) for 1 h or 24 h. Bots were stripped and reprobed β-tubulin as an internal control for equal protein.
There were significant differences between astrocytes cultures generated from kainate-lesioned and sham-lesioned in terms of both caspase activity and expression of the astrogliosis markers GS and FGF-2. Cultures from kainate-lesioned animals exhibited an approximately two-fold increase in caspase activity (Fig. 9D) and elevated levels of the active, cleaved fragment of caspase-3 (Fig. 9E), relative to cultures from sham-lesioned animals. Levels of both total caspase activity and active caspase-3 were modest in comparison to that observed in astrocyte cultures from sham-lesioned animals treated for 24 h with 1 μM staurosporine. Astrocyte cultures from kainate-lesioned animals were also characterized by increased expression of GS and FGF-2 (Fig. 10), demonstrating a retained reactive phenotype ex vivo. Treatment for 24 h with the broad-spectrum caspase inhibitor zVAD significantly attenuated expression of GS and FGF-2 in cultures from kainate-lesioned rats but not sham-lesioned rats (Fig. 10).
Figure 10. Caspase inhibition attenuates up-regulation of GS and FGF-2 expression in an ex vivo model of astrogliosis.

Astrocyte cultures from sham- and kainate-lesioned rats were treated for 48 h with vehicle (−) or 50 μM general caspase inhibitor zVAD (+) and then processed for western blot. Representative western blots show increased (A) GS and (C) FGF-2 expression in cultures from kainate-lesioned (KA 1, KA 2) rats compared to sham-lesioned controls (Sham 1, Sham 2). Densitometric analysis of western blots revealed zVAD treatment (+zVAD; solid bars) significantly decreased (B) GS and (D) FGF-2 expression in astrocyte cultures from Kainate-lesioned rats (Sham, N = 4; Kainate, N = 5). * P < 0.05 relative to matched condition.
3. DISCUSSION
Following injury, caspase activation in neurons is predominantly associated with neuronal apoptosis. In contrast, in vivo studies have demonstrated poor correlation between caspase activation in astrocytes and cell death (Beer et al., 2000; Narkilahti et al., 2003). In the current study, we demonstrate a functional significance of non-apoptotic activation of caspases, wherein caspase inhibition modulates astrogliotic responses in vitro. Inhibition of caspase activity using a broad-spectrum caspase inhibitor partially attenuated upregulation of two proteins associated with astrogliosis, GS and FGF-2, without altering reactive morphology. Use of subtype-specific caspase inhibitors suggested functional involvement of caspases-3 and -11 in specific aspects of astrogliosis. While these findings suggest a key role of caspase activation in regulating astrogliosis, there are potential limitations to this conclusion. For instance, specific features of astrogliosis, and consequently the signaling pathways underlying astrogliosis, are known to vary across different paradigms (Norton et al., 1992; Wu and Schwartz, 1998). In this context, it is important to note that we observed similar changes in GS, FGF-2, and caspase activation with different inducing stimuli and across two different culture paradigms. The implications of this will be discussed in the context of our broadening understanding of non-apoptotic functions of the caspases in the brain.
3.1 Evidence for functional involvement of caspase-3 and caspase-11 in astrogliosis
In the current study, caspase activation was observed in the absence of cell death in both in vitro and ex vivo models of astrogliosis. Consistent with the increase in total caspase activity, an up-regulation of the cleaved active fragment of caspase-3 was observed. Further, specific inhibition of caspase-3 attenuated biochemical markers of astrogliosis (GS and FGF-2). While nuclear translocation of caspase-3 is indicative of its proteolytic activity and is usually associated with apoptosis (Kamada et al., 2005), previous in vivo studies have demonstrated nuclear caspase-3 activation in non-apoptotic astrocytes following brain injury (Acarin et al., 2007; Beer et al., 2000; Benjelloun et al., 2003; Mouser et al., 2006; Su et al., 2000; Villapol et al., 2008). Caspase-3 has been implicated in the cleavage of structurally important proteins including vimentin (Byun et al., 2001) and GFAP in astrocytes (Mouser et al., 2006). While caspase-3 cleavage of structural proteins may be the result of cytoskeletal dismantling and cell death (Mouser et al., 2006), cleaved GFAP and vimentin have also been found to co-localize with activated caspase-3 in non-apoptotic astrocytes in injury models, indicating a potential non-apoptotic role for caspase-3 in astrocyte cytoskeletal remodeling (Acarin et al., 2007). Interestingly, our findings show that caspase inhibition did not alter reactive morphology and thus argue that caspase activation alone is not sufficient for cytoskeletal remodeling associated with hypertrophy and stellation. We speculate that cytoskeletal changes in astrogliosis reflect contributions from caspase activation and additional signaling pathways. For example, stellation in cultured astrocytes treated with dBcAMP or Aβ is dependent upon Na+ and K+ transport across the plasma membrane (Abe and Saitoh, 2001).
In our in vitro model of astrogliosis, caspase-11 was also implicated in regulating astrogliosis since specific pharmacological inhibition of caspase-11 significantly reduced expression of GS and FGF-2. Caspase-11 is predominantly known as an inflammatory initiator caspase, essential for activation of caspase-1 (Wang et al., 1998). No evidence of caspase-1 involvement in the astrocyte responses to dBcAMP or Aβ was observed in the current study since specific inhibition caspase-1 did not alter expression of the assessed markers of astrogliosis. An alternative role of caspase-11 may be activation of caspase-3, which has been demonstrated under certain pathological conditions (Kang et al., 2000). Further investigation is required to determine if a similar such pathway contributes to our observations.
3.2 Potential mechanisms underlying non-apoptotic caspase activation in astrocytes
Activated caspases are known to regulate several non-apoptotic functions, although the mechanism(s) underlying these actions remain to be fully elucidated. In some systems, caspases mediate their non-apoptotic actions via proteolytic activation of transcription factors. For example, differentiation of erythrocytes involves non-apoptotic caspase-mediated cleavage and activation of GATA-1 (De Maria et al., 1999). Similarly, skeletal muscle differentiation involves a crucial role of caspase-3 activation of Mammalian Sterile Twenty-like kinase (Fernando et al., 2002). Several transcription factors and signaling molecules that are activated during astrogliosis, including CREB and NF-κB (de Freitas et al., 2002; Nikaido et al., 2002), are established caspase substrates (Fran∂cois et al., 2000; Qin et al., 2000) and could be potential down-stream effectors of caspase activation. For instance, NF-κB, while initially thought to be inactivated upon caspase cleavage, is now known to be activated even by low levels of caspase activation, which in turn leads to activation of inflammatory pathways (Lamkanfi et al., 2006). Also, proliferation of reactive astrocytes following a cortical stab wound is associated with a down-regulation of the cell-cycle arrest protein p27kip1 (Koguchi et al., 2002). Caspase-mediated cleavage and inactivation of p27kip1 is essential for proliferation of transformed lymphoid cell lines (Frost et al., 2001). Further investigation is needed to determine key components of this potential mechanism of astrogliosis, including the subcellular localization of active caspases, the identity of relevant transcription factors, and the relationships between these components and regulation of specific factors including GS and FGF-2.
Several non-apoptotic functions of caspases are attributed to sublethal activation (McLaughlin, 2004). Apoptotic or non-apoptotic consequences may occur due to differential cleavage of substrate depending upon the level of caspase activation. In the current study, low levels of caspase activation were observed in astrocytes following dB-cAMP and Aβ apoptosis treatments in the absence of cell death, whereas high levels of caspase activation and apoptosis were observed following staurosporine treatment. Additionally, apoptotic inhibitors including survivin and heat shock protein 25/27 have been found to co-localize with active caspase-3 in the nucleus of reactive astrocytes following brain injury, potentially inhibiting apoptotic actions of caspases (Villapol et al., 2008).
3.3 Conclusions
In summary, these findings support a non-apoptotic role for caspase activation in the regulation of astrogliosis. Growing evidence suggests that caspases exert dual roles in the injured brain: facilitating apoptosis in severely injured neurons while also protecting neurons as a consequence of increased expression of bioactive factors from reactive astrocytes. Similarly, other signaling cascades also perform seemingly opposing functions, such as p38 MAPK, which is commonly activated following neuronal injury and can contribute to both apoptosis and astrogliosis (Che et al., 2001a; Che et al., 2001b). Additional signaling molecules implicated in astrogliosis, including ERK/MAPK (Mandell and VandenBerg, 1999; Mandell et al., 2001) and NF-κB (de Freitas et al., 2002; Perez-Otano et al., 1996) also have diverse functions in the brain, such as differentiation, proliferation, and regulation of apoptosis or cell survival pathways. Although the signaling cascades regulating induction and maintenance of astrogliosis require additional definition, the current findings indicate that caspases are significant contributors to the process.
4. EXPERIMENTAL PROCEDURE
4.1 Materials
Kainate, staurosporine, dBcAMP and other reagents (unless otherwise specified) were purchased from Sigma-Aldrich (St Louis, MO). β-Amyloid 25–35 peptide (Aβ25–35) was purchased from Bachem (Torrence, CA), solubilized in sterile, deionized water to 1 mM to yield aggregated species, as previously described (Pike et al., 1995). Caspase inhibitors (zVAD-fmk, DEVD-fmk, WEHD-fmk, AEVD-fmk, LEHD-fmk and LETD-fmk) were purchased from Enzyme Systems (MP Biochemicals, Irvine, CA) and solubilized in DMSO.
4.2 Astrocyte cultures
Neonatal astrocytes cultures were generated from cerebral cortices of postnatal day 0–1 Sprague-Dawley rat pups using the method of McCarthy and de Vellis (1980) with modifications as described previously (Pike et al., 1996b). In brief, cerebral cortices were dissected and the tissue enzymatically and mechanically dissociated. The resultant cell suspensions were plated into 75 cm2 flasks containing DMEM (Gibco, Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) (Gibco, Invitrogen, Carlsbad, CA) and maintained in a humidified incubator at 37° C with 5% CO2. Media was changed every three days until the cultures reached confluence after 10–14 days in vitro. Once confluent, culture flasks were shaken overnight at 280 RPM in an orbital shaker at 37° C, a procedure that eliminates most non-astroglial cells. The percentage of microglial cells was <5% as determined by postive immunoreactivity with the microglial marker Iba-1. Enriched astroglia were re-plated at 12,000 cells/cm2 onto poly-lysine coated plates in DMEM/10% FBS. The cultures were shifted to serum free medium 1–3 days prior to experimental use.
Adult astrocytes were cultured from hippocampi of 3 month-old adult female Sprague-Dawley rats using the method of Schwartz and Wilson (1992). Dissected hippocampi were enzymatically and mechanically dissociated, then plated into 50 cm2 flasks containing DMEM/20% FBS and placed in a humidified incubator at 37° C with 5% CO2. Media was changed every three days for 10–14 days and at least 15 separate astrocyte colonies were detected. Cultures were then passaged once and grown to confluency. Confluent cultures were shaken overnight at 37°C at 120 RPM on an orbital shaker to eliminate non-astroglial cells. Enriched adult astroglial cultures were re-plated at 12,000 cells/cm2 onto poly-lysine coated plates in DMEM/F12 (Gibco, Invitrogen, Carlsbad, CA) with 20% FBS. The cultures were shifted to serum-free DMEM/F12 1–3 days prior to use in experiments.
4.3 Cell viability
Cell viability was assessed by two methods. First, numbers of non-viable cells were counted following staining with the vital dyes calcein AM (live cells) and ethidium homodimer (dead cells) (Molecular Probes; Invitrogen, Carlsbad, CA). Counts of non-viable cells (negative for calcein AM, positive for ethidium homodimer) were made in four non-overlapping fields per culture well (in a predetermined pattern maintained across all experiments) with each condition represented by 3 separate wells. All experiments were repeated in at least 3 independent culture preparations. Second, viability was also quantified by assaying culture media for levels of the intracellular enzyme lactate dehydrogenase, which is released from dying cells with compromised membranes. Lactate dehydrogenase activity was assessed in culture media via spectrophotometric assay as previously described (Koh and Choi, 1987). Briefly, 0.5 mg/ml β-NADH in potassium phosphate buffer was incubated with the treatment media at room temperature for 30 minutes. Next, the substrate pyruvate (12 mM) was added and the plate read at 340 nm every 8 sec over a 5 min period using a Molecular Devices spectrophotometer and data analyzed using SOFTmax PRO (Molecular Devices, Sunnyvale, CA). Samples were assayed in triplicate and each condition assessed in four independent experiments. For both methods, some culture wells were treated with staurosporine (1 μM) as an internal standard for maximum cell death.
4.4 Western blot
Immunoblotting was performed as previously described (Pike et al., 1996a). In brief, equal amounts of culture lysates were diluted into reducing sample buffer and electrophoresed at constant 100V in 10% or 12% polyacrylamide gels using standard SDS-PAGE methodology. Proteins were transferred onto 0.45 μm PVDF membranes (Millipore; Bedford, MA). Membranes were incubated for 1 h in 3% bovine serum albumin to block non-specific protein binding, then were probed with primary antibodies, including GFAP (1:15000 dilution, DAKO, Carpinteria, CA), GS (1:1000 dilution) and FGF-2 (1:500 dilution, BD Biosciences, San Diego, CA). Caspase-3 was probed with an antibody specific to the cleaved, active fragment (1:50 dilution, EMD Biosciences, Gibbstown, NJ). To confirm equal amount of protein in all conditions, blots were stripped and re-probed with β-tubulin (1:500 dilution, Chemicon, Temecula, CA). For data quantification, images from scanned films were analyzed by NIH image 1.61 and relative protein quantities were normalized to control groups. Results from at least 3 independent experiments were combined for statistical analysis.
4.5 Caspase activity assay
Total caspase activity was assessed in duplicate according to manufacturers’ instructions via fluorometric assay (Calbiochem, Gibbstown, NJ) on cell suspensions collected through mechanical scraping. Briefly, protein levels were assessed in an aliquot of cell suspensions to ensure equal protein content in different treatment condition. After correcting for protein content, cell suspension from each condition was incubated with FITC-conjugated caspase substrate for 45 minutes. Cells were washed three times in wash buffer provided in the kit, then resuspended and loaded onto a 96-well plate and read at emission wavelength of 345 nm and excitation wavelength of 548nm on a fluorometric plate reader (Molecular Devices).
4.6 Kainate lesion
Female Sprague-Dawley (3 months of age, N = 9/group) were anesthetized with sodium pentobarbital (i.p., 50 mg/kg body weight). Kainate (1.5 μl of 0.5 mg/ml kainate in saline) or sterile saline solution (1.5 μl) was injected bilaterally into the hippocampi using a Hamilton syringe and stereotaxis (coordinates: -5.2 bregma, 4.5 lateral to the mid-line, 4.0 mm from the surface of the cortex). Two weeks following kainate lesion, animals were sacrificed by decapitation and the brains quickly harvested. The left hemisphere was immersion fixed for 48 hours in 4% paraformaldehyde/0.1 M PBS for use in immunohistochemical analyses. The right hemisphere was immediately used to generate adult astrocytes cultures in an ex-vivo paradigm of astrogliosis, as described above.
4.7 Immmunochemistry
Fixed hemi-brains were exhaustively sectioned (40 μm) in the horizontal plane using a vibratome, and then subjected to immunohistochemistry using standard avidin-biotin methodology, as previously described (Ramsden et al., 2003). For each brain, every tenth section containing hippocampus was permeabilized in 0.02% Triton-X100 in 0.1 M tris-HCl (pH 7.4), then incubated in 3% bovine serum albumin/0.1M tris followed by either NeuN antibody (1:500 dilution; Chemicon, Temecula CA) or GFAP antibody (1:15,000 dilution; ICN, Irvine, CA) for 24 hrs. Next, sections were incubated with either anti-mouse (for NeuN) or anti-rabbit (for GFAP) biotinylated antibody (Vector Laboratories, Burlingame, CA) for 1 hr followed by avidin-biotin-peroxidase complex (ABC, Vector Laboratories) for 1 hour. Immunostaining was visualized using 3,3′ diaminobenzidine (DAB, Vector Laboratories). Sections were dehydrated and coverslipped with depex mounting medium. For GFAP immunocytochemistry, astroglial cultures were fixed with 2% paraformaldehyde and processed as described above using GFAP antibody (1:15000, DAKO, Carpinteria, CA).
4.8 Statistical analyses
Raw data were statistically assessed using one-way ANOVA followed, when appropriate, by between group comparisons using Fisher Least Squares Difference test. A P value of < 0.05 was considered significant.
Highlights.
Astrogliosis is associated with non-apoptotic activation of caspases
Caspase inhibitors attenuate astrogliosis in neonatal astrocyte cultures
Caspases contribute to astrogliosis in adult astrocyte cultures
Acknowledgments
AMB is supported by the American-Australian Neurological Fellowship. This study was supported in part by NIH grants AG15961 (CJP) and AG026572 (RD Brinton/CJP).
Abbreviations
- Aβ
β-amyloid
- dBcAMP
dibutryl cyclic adenosine monophosphate
- FGF-2
fibroblast growth factor-2
- GFAP
glial fibrillary acidic protein
- GS
glutamine synthetase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abe K, Kato M, Saito H. Human amylin mimicks amyloid [beta] protein-induced reactive gliosis and inhibition of cellular redox activity in cultured astrocytes. Brain Res. 1997;762:285–288. doi: 10.1016/s0006-8993(97)00595-7. [DOI] [PubMed] [Google Scholar]
- Abe K, Saito H. Na+ and K+ dependence of morphological changes of cultured rat cortical astrocytes. Pharmacol Toxicol. 2001;88:319–24. [PubMed] [Google Scholar]
- Abe K, Saito H. The p44/42 mitogen-activated protein kinase cascade is involved in the induction and maintenance of astrocyte stellation mediated by protein kinase C. Neurosci Res. 2000;36:251–7. doi: 10.1016/s0168-0102(99)00134-0. [DOI] [PubMed] [Google Scholar]
- Acarin L, Villapol S, Faiz M, Rohn TT, Castellano B, González B. Caspase-3 activation in astrocytes following postnatal excitotoxic damage correlates with cytoskeletal remodeling but not with cell death or proliferation. Glia. 2007;55:954–965. doi: 10.1002/glia.20518. [DOI] [PubMed] [Google Scholar]
- Araujo DM, Cotman CW. [beta]-Amyloid stimulates glial cells in vitro to produce growth factors that accumulate in senile plaques in Alzheimer’s disease. Brain Res. 1992;569:141–145. doi: 10.1016/0006-8993(92)90380-r. [DOI] [PubMed] [Google Scholar]
- Beer R, Franz G, Srinivasan A, Hayes RL, Pike BR, Newcomb JK, Zhao X, Schmutzhard E, Poewe W, Kampfl A. Temporal Profile and Cell Subtype Distribution of Activated Caspase-3 Following Experimental Traumatic Brain Injury. J Neurochem. 2000;75:1264–1273. doi: 10.1046/j.1471-4159.2000.0751264.x. [DOI] [PubMed] [Google Scholar]
- Benjelloun N, Joly LM, Palmier B, Plotkine M, Charriaut-Marlangue C. Apoptotic mitochondrial pathway in neurones and astrocytes after neonatal hypoxia-ischaemia in the rat brain. Neuropathol Appl Neurobiol. 2003;29:350–360. doi: 10.1046/j.1365-2990.2003.00467.x. [DOI] [PubMed] [Google Scholar]
- Bertrand R, Solary E, O’Connor P, Kohn K, Pommier Y. Induction of a common pathway of apoptosis by staurosporine. Exp Cell Res. 1994;211:314–321. doi: 10.1006/excr.1994.1093. [DOI] [PubMed] [Google Scholar]
- Byun Y, Chen F, Chang R, Trivedi M, Green KJ, Cryns VL. Caspase cleavage of vimentin disrupts intermediate filaments and promotes apoptosis. Cell Death Differ. 2001;8:443–450. doi: 10.1038/sj.cdd.4400840. [DOI] [PubMed] [Google Scholar]
- Campbell D, Holt C. Apoptotic pathway and MAPKs differentially regulate chemotropic responses of retinal growth cones. Neuron. 2003;37:939–52. doi: 10.1016/s0896-6273(03)00158-2. [DOI] [PubMed] [Google Scholar]
- Canning DR, McKeon RJ, DeWitt DA, Perry G, Wujek JR, Frederickson RCA, Silver J. [beta]-Amyloid of Alzheimer’s Disease Induces Reactive Gliosis That Inhibits Axonal Outgrowth. Exp Neurol. 1993;124:289–298. doi: 10.1006/exnr.1993.1199. [DOI] [PubMed] [Google Scholar]
- Chan SL, Mattson MP. Caspase and Calpain Substrates: Roles in Synaptic Plasticity and Cell Death. J Neurosci Res. 1999;58:167–190. [PubMed] [Google Scholar]
- Chang HY, Yang X. Proteases for cell suicide: functions and regulation of caspases. Microbiol Mol Biol Rev. 2000;64(4):821–46. doi: 10.1128/mmbr.64.4.821-846.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Che Y, Piao CS, Han PL, Lee JK. Delayed induction of alpha B-crystallin in activated glia cells of hippocampus in kainic acid-treated mouse brain. J Neurosci Res. 2001a;65:425–31. doi: 10.1002/jnr.1170. [DOI] [PubMed] [Google Scholar]
- Che Y, Yu Y, Han P, Lee J. Delayed induction of p38 MAPKs in reactive astrocytes in the brain of mice after KA-induced seizure. Mol Brain Res. 2001b;94:157–65. doi: 10.1016/s0169-328x(01)00233-9. [DOI] [PubMed] [Google Scholar]
- Dash P, Blum S, Moore A. Caspase activity plays an essential role in long-term memory. Neuroreport. 2000;11:2811–6. doi: 10.1097/00001756-200008210-00040. [DOI] [PubMed] [Google Scholar]
- de Freitas MS, Spohr TC, Benedito AB, Caetano MS, Margulis B, Lopes UG, Moura-Neto V. Neurite outgrowth is impaired on HSP70-positive astrocytes through a mechanism that requires NF-kappaB activation. Brain Res. 2002;958(2):359–70. doi: 10.1016/s0006-8993(02)03682-x. [DOI] [PubMed] [Google Scholar]
- De Maria R, Zeuner A, Eramo A, Domenichelli C, Bonci D, Grignani F, Srinivasula SM, Alnemri ES, Testa U, Peschle C. Negative regulation of erythropoiesis by caspase-mediated cleavage of GATA-1. Nature. 1999;401:489–93. doi: 10.1038/46809. [DOI] [PubMed] [Google Scholar]
- Duran-Vilaregut J, del Valle J, Manich G, Junyent F, Camins A, Pallàs M, Pelegrí C, Vilaplana J. Systemic administration of 3-nitropropionic acid points out a different role for active caspase-3 in neurons and astrocytes. Neurochem Int. 2010;56:443–450. doi: 10.1016/j.neuint.2009.12.001. [DOI] [PubMed] [Google Scholar]
- Eddleston M, Mucke L. Molecular profile of reactive astrocytes--implications for their role in neurologic disease. Neuroscience. 1993;54:15–36. doi: 10.1016/0306-4522(93)90380-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedoroff S, McAuley WAJ, Houkle JD, Devon RM. Astrocyte cell lineage. V Similarity of astrocytes that form in the presence of dBcAMP in cultures to reactive astrocytes in vivo. J Neurosci Res. 1984;12:15–27. doi: 10.1002/jnr.490120103. [DOI] [PubMed] [Google Scholar]
- Fernando P, Kelly JF, Balazsi K, Slack RS, Megeney LA. Caspase 3 activity is required for skeletal muscle differentiation. Proc Natl Acad Sci U S A. 2002;99:11025–30. doi: 10.1073/pnas.162172899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernando P, Brunette S, Megeney L. Neural stem cell differentiation is dependent upon endogenous caspase 3 activity. FASEB J. 2005;19:1671–3. doi: 10.1096/fj.04-2981fje. [DOI] [PubMed] [Google Scholar]
- Fran∂cois F, Godinho MJ, Grimes ML. CREB is cleaved by caspases during neural cell apoptosis. FEBS Lett. 2000;486(3):281–4. doi: 10.1016/s0014-5793(00)02316-4. [DOI] [PubMed] [Google Scholar]
- Frost V, Al-Mehairi S, Sinclair AJ. Exploitation of a non-apoptotic caspase to regulate the abundance of the cdkI p27(KIP1) in transformed lymphoid cells. Oncogene. 2001;20(22):2737–48. doi: 10.1038/sj.onc.1204367. [DOI] [PubMed] [Google Scholar]
- Garnier P, Ying W, Swanson RA. Ischemic preconditioning by caspase cleavage of Poly(ADP-Ribose) Polymerase-1. J Neurosci. 2003;23:7967–7973. doi: 10.1523/JNEUROSCI.23-22-07967.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Akama KT, Krafft GA, Chromy BA, Van Eldik LJ. Amyloid-[beta] peptide activates cultured astrocytes: morphological alterations, cytokine induction and nitric oxide release. Brain Res. 1998;785:195–206. doi: 10.1016/s0006-8993(97)01318-8. [DOI] [PubMed] [Google Scholar]
- Kamada S, Kikkawa U, Tsujimoto Y, Hunter T. Nuclear translocation of caspase-3 is dependent on its proteolytic activation and recognition of a substrate-like protein(s) J Biol Chem. 2005;280(2):857–60. doi: 10.1074/jbc.C400538200. [DOI] [PubMed] [Google Scholar]
- Kang SJ, Wang S, Hara H, Peterson EP, Namura S, Amin-Hanjani S, Huang Z, Srinivasan A, Tomaselli KJ, Thornberry NA, Moskowitz MA, Yuan J. Dual role of caspase-11 in mediating activation of caspase-1 and caspase-3 under pathological conditions. J Cell Biol. 2000;149(3):613–22. doi: 10.1083/jcb.149.3.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, Saito H, Abe K. Nanomolar amyloid β protein-induced inhibition of cellular redox activity in cultured astrocytes. J Neurochem. 1997;68:1889–1895. doi: 10.1046/j.1471-4159.1997.68051889.x. [DOI] [PubMed] [Google Scholar]
- Koguchi K, Nakatsuji Y, Nakayama K, Sakoda S. Modulation of astrocyte proliferation by cyclin-dependent kinase inhibitor p27(Kip1) Glia. 2002;37(2):93–104. doi: 10.1002/glia.10017. [DOI] [PubMed] [Google Scholar]
- Koh JY, Choi DW. Quantitative determination of glutamate mediated cortical neuronal injury in cell culture by lactate dehydrogenase efflux assay. J Neurosci Methods. 1987;20:83–90. doi: 10.1016/0165-0270(87)90041-0. [DOI] [PubMed] [Google Scholar]
- Krohn AJ, Preis E, Prehn JHM. Staurosporine-Induced Apoptosis of Cultured Rat Hippocampal Neurons Involves Caspase-1-Like Proteases as Upstream Initiators and Increased Production of Superoxide as a Main Downstream Effector. J Neurosci. 1998;18:8186–8197. doi: 10.1523/JNEUROSCI.18-20-08186.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S. Caspase function in programmed cell death. Cell Death Differ. 2006;14:32–43. doi: 10.1038/sj.cdd.4402060. [DOI] [PubMed] [Google Scholar]
- Lamkanfi M, Declercq W, Vanden Berghe T, Vandenabeele P. Caspases leave the beaten track: caspase-mediated activation of NF-kappaB. J Cell Biol. 2006;173(2):165–71. doi: 10.1083/jcb.200509092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Prince G, Fages C, Rolland B, Nunez J, Tardy M. DBcAMP effect on the expression of GFAP and of its encoding mRNA in astroglial primary cultures. Glia. 1991;4:322–326. doi: 10.1002/glia.440040310. [DOI] [PubMed] [Google Scholar]
- Mandell JW, VandenBerg SR. ERK/MAP kinase is chronically activated in human reactive astrocytes. Neuroreport. 1999;10:3567–72. doi: 10.1097/00001756-199911260-00019. [DOI] [PubMed] [Google Scholar]
- Mandell JW, Gocan NC, Vandenberg SR. Mechanical trauma induces rapid astroglial activation of ERK/MAP kinase: Evidence for a paracrine signal. Glia. 2001;34:283–95. doi: 10.1002/glia.1062. [DOI] [PubMed] [Google Scholar]
- McCarthy K, de Vellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol. 1980;85:890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin B. The kinder side of killer proteases: Caspase activation contributes to neuroprotection and CNS remodeling. Apoptosis. 2004;9:111–121. doi: 10.1023/B:APPT.0000018793.10779.dc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouser PE, Head E, Ha KH, Rohn TT. Caspase-Mediated Cleavage of Glial Fibrillary Acidic Protein within Degenerating Astrocytes of the Alzheimer’s Disease Brain. Am J Pathol. 2006;168:936–946. doi: 10.2353/ajpath.2006.050798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narkilahti S, Pirttilä TJ, Lukasiuk K, Tuunanen J, Pitkänen A. Expression and activation of caspase 3 following status epilepticus in the rat. Eur J Neurosci. 2003;18:1486–1496. doi: 10.1046/j.1460-9568.2003.02874.x. [DOI] [PubMed] [Google Scholar]
- Nikaido T, Iseki K, Mori T, Takaki H, Yokoya S, Hagino S, Takeda J, Zhang Y, Takeuchi M, Kikuchi S, Wanaka A. Expression of OASIS, a CREB/ATF family transcription factor, in CNS lesion and its transcriptional activity. Molecular brain research. 2002;108(1–2):129–38. doi: 10.1016/s0169-328x(02)00521-1. [DOI] [PubMed] [Google Scholar]
- Norton WT, Aquino DA, Hozumi I, Chiu FC, Brosnan CF. Quantitative aspects of reactive gliosis: a review. Neuroche Res. 1992;17:877–885. doi: 10.1007/BF00993263. [DOI] [PubMed] [Google Scholar]
- Oomman S, Strahlendorf H, Dertien J, Strahlendorf J. Bergmann glia utilize active caspase-3 for differentiation. Brain Res. 2006;1078:19–34. doi: 10.1016/j.brainres.2006.01.041. [DOI] [PubMed] [Google Scholar]
- Pekny M, Nilsson M. Astrocyte activation and reactive gliosis. Glia. 2005;50:427–434. doi: 10.1002/glia.20207. [DOI] [PubMed] [Google Scholar]
- Perez-Otano I, McMillian MK, Chen J, Bing G, Hong JS, Pennypacker KR. Induction of NF-kB-like transcription factors in brain areas susceptible to kainate toxicity. Glia. 1996;16:306–15. doi: 10.1002/(SICI)1098-1136(199604)16:4<306::AID-GLIA3>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Pike CJ, Cummings BJ, Monzavi R, Cotman CW. [beta]-Amyloid-induced changes in cultured astrocytes parallel reactive astrocytosis associated with senile plaques in Alzheimer’s disease. Neuroscience. 1994;63:517–531. doi: 10.1016/0306-4522(94)90547-9. [DOI] [PubMed] [Google Scholar]
- Pike CJ, Walencewiczwasserman AJ, Kosmoski J, Cribbs DH, GGC, Cotman CW. Structure-activity analyses of beta-amyloid peptides - contributions of the beta-25–35 region to aggregation and neurotoxicity. J Neurochem. 1995;64:253–265. doi: 10.1046/j.1471-4159.1995.64010253.x. [DOI] [PubMed] [Google Scholar]
- Pike CJ, Ramezan-Arab N, Miller S, Cotman C. b-Amyloid increases enzyme activity and protein levels of glutamine synthetase in cultured astrocytes. Exp Neurol. 1996a;139:167–171. doi: 10.1006/exnr.1996.0091. [DOI] [PubMed] [Google Scholar]
- Pike CJ, Vaughan PJ, Cunningham DD, Cotman CW. Thrombin attenuates neuronal cell death and modulates astrocyte reactivity induced by beta-amyloid in vitro. J Neurochem. 1996b;66:1374–82. doi: 10.1046/j.1471-4159.1996.66041374.x. [DOI] [PubMed] [Google Scholar]
- Pindon A, Festoff BW, Hantai D. Thrombin-induced reversal of astrocyte stellation is mediated by activation of protein kinase C beta-1. Eur J Biochem. 1998;255:766–74. doi: 10.1046/j.1432-1327.1998.2550766.x. [DOI] [PubMed] [Google Scholar]
- Qin Z, Wang Y, Chasea TN. A caspase-3-like protease is involved in NF-kappaB activation induced by stimulation of N-methyl-D-aspartate receptors in rat striatum. Mol Brain Res. 2000;80(2):111–22. doi: 10.1016/s0169-328x(00)00147-9. [DOI] [PubMed] [Google Scholar]
- Ramsden M, Shin TM, Pike CJ. Androgens modulate neuronal vulnerability to kainate lesion. Neuroscience. 2003;122:573–578. doi: 10.1016/j.neuroscience.2003.08.048. [DOI] [PubMed] [Google Scholar]
- Rohn TT, Cusack SM, Kessinger SR, Oxford JT. Caspase activation independent of cell death is required for proper cell dispersal and correct morphology in PC12 cells. Exp Cell Res. 2004;295:215–225. doi: 10.1016/j.yexcr.2003.12.029. [DOI] [PubMed] [Google Scholar]
- Rozovsky I, Wei M, Morgan TE, Finch CE. Reversible age impairments in neurite outgrowth by manipulations of astrocytic GFAP. Neurobiol Aging. 2005;26:705–15. doi: 10.1016/j.neurobiolaging.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Salinero O, Moreno-Flores MT, Ceballos ML, Wandosell F. β-Amyloid peptide induced cytoskeletal reorganization in cultured astrocytes. J Neurosci Res. 1997;47:216–223. [PubMed] [Google Scholar]
- Schwartz JP, Wilson DJ. Preparation and characterization of type 1 astrocytes cultured from adult rat cortex, cerebellum, and striatum. Glia. 1992;5:75–80. doi: 10.1002/glia.440050111. [DOI] [PubMed] [Google Scholar]
- Schwerk C, Schulze-Osthoff K. Non-apoptotic functions of caspases in cellular proliferation and differentiation. Biochem Pharmacol. 2003;66:1453–1458. doi: 10.1016/s0006-2952(03)00497-0. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009;32:638–647. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su JH, Nichol KE, Sitch T, Sheu P, Chubb C, Miller BL, Tomaselli KJ, Kim RC, Cotman CW. DNA Damage and Activated Caspase-3 Expression in Neurons and Astrocytes: Evidence for Apoptosis in Frontotemporal Dementia. Exp Neurol. 2000;163:9–19. doi: 10.1006/exnr.2000.7340. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Yokota H, Jover T, Cappuccio I, Calderone A, Simionescu M, Bennett MVL, Zukin RS. Ischemic Preconditioning: Neuronal Survival in the Face of Caspase-3 Activation. J Neurosci. 2004;24:2750–2759. doi: 10.1523/JNEUROSCI.5475-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villapol S, Acarin L, Faiz M, Castellano B, Gonzalez B. Survivin and heat shock protein 25/27 colocalize with cleaved caspase-3 in surviving reactive astrocytes following excitotoxicity to the immature brain. Neuroscience. 2008;153:108–119. doi: 10.1016/j.neuroscience.2008.01.054. [DOI] [PubMed] [Google Scholar]
- Wagner DC, Riegelsberger UM, Michalk S, Härtig W, Kranz A, Boltze J. Cleaved caspase-3 expression after experimental stroke exhibits different phenotypes and is predominantly non-apoptotic. Brain Res. 2011;1381:237–242. doi: 10.1016/j.brainres.2011.01.041. [DOI] [PubMed] [Google Scholar]
- Wang S, Miura M, Jung YK, Zhu H, Li E, Yuan J. Murine caspase-11, an ICE-interacting protease, is essential for the activation of ICE. Cell. 1998;92(4):501–9. doi: 10.1016/s0092-8674(00)80943-5. [DOI] [PubMed] [Google Scholar]
- Wu VW, Schwartz JP. Cell culture models of reactive astrocytes: new perspectives. J Neurosci Res. 1998;71:749–56. doi: 10.1002/(SICI)1097-4547(19980315)51:6<675::AID-JNR2>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]