

Abstract
Members of the rodent Ly49 receptor family control natural killer (NK) cell responsiveness and demonstrate allele specificity for major histocompatibility complex class I (MHC-I) ligands. For example, the rat Ly49i2 inhibitory NK cell receptor binds RT1-A1c, but not other rat MHC class Ia or b molecules. RT1-A1c preferentially binds peptides with proline at the second or P2 position, which defines it as a HLA-B7 supertype MHC-I molecule. Previously, our laboratory showed that mutations within the MHC-I supertype-defining B-pocket of RT1-A1c could lead to alterations in P2 anchor residues of the peptide repertoire bound by RT1-A1c and loss of recognition by Ly49i2. Although suggestive of peptide involvement, it was unclear whether the peptide P2 anchor residue, or alteration of the RT1-A1c primary sequence influenced Ly49i2 recognition. Therefore, we directly investigated the role of the P2 anchor residue of RT1-A1c-bound peptides in Ly49i2 recognition. First, fluorescent multimers generated by refolding soluble recombinant RT1-A1c with individual synthetic peptides differing only at the P2 anchor residue were examined for binding to Ly49i2 NK cell transfectants. Secondly, cytotoxicity by Ly49i2-expressing NK cells towards RMA-S target cells expressing RT1-A1c bound with peptides that only differ at the P2 anchor residue was evaluated. Our results demonstrate that Ly49i2 recognizes RT1-A1c bound with peptides that have Pro or Val at P2, while little or no recognition is observed when RT1-A1c is complexed with peptide bearing Gln at P2. Thus, the identity of the P2 peptide anchor residue is an integral component of MHC-I recognition by Ly49i2.
Keywords: Rodents, Natural Killer Cells, Cell Surface Molecules, MHC, Cytotoxicity
INTRODUCTION
Natural killer cells are large granular lymphocytes that play an important role in innate immunity against tumors and viruses (1, 2). They are defined by their ability to kill tumor and virally infected cells and release cytokines without the need for prior sensitization. Protection from NK cell-mediated lysis is dependent on a balance of activating and inhibitory receptors (3, 4). Normally inhibitory receptors dominate when NK cells encounter healthy cells, thereby maintaining self-tolerance. Inhibition of NK cell activation is regulated by several NK receptors which include those that recognize self major histocompatibility complex class I (MHC-I)3 and MHC-I-like molecules (3). In pathological conditions, such as during viral infection or cell transformation, down-regulation of MHC-I to avoid T-cell detection would disrupt inhibitory receptor interactions and allow NK cell activation by activating NK receptors. The MHC-I-specific NK cell receptors include the rodent Ly49 and the human killer immunoglobulin-like receptors (KIR) (5). Although structurally distinct, Ly49 and KIR are polygenic, and perform functionally equivalent roles in NK cells (5).
Determination of Ly49-MHC-I co-crystal structures and mutagenesis of solvent exposed MHC-I residues indicated that Ly49 interact with a highly conserved MHC-I region, that combines below the base of the peptide binding groove and portions of the α3 domain and β2-microglobulin (β2m), referred to as site 2 in Ly49-MHC-I co-crystals (6–8). This raised the puzzling question of how Ly49 could distinguish between different MHC-I ligands, based on the mostly non-polymorphic MHC-I surface at site 2.
The α1 and α2 domains forming the peptide-binding groove of MHC-I molecules are extremely polymorphic. Despite extensive polymorphism, MHC-I molecules from humans can be grouped into about nine supertypes based on their distinct preferences for binding peptides with specific anchor residues (9, 10). In addition, MHC-I from other species have been found to bind peptide anchor motifs similar to human supertypes (9). The rat inhibitory receptor Ly49i2 is allele specific; for example, of two classical rat MHC-I molecules, RT1-A1c and RT1-Aa, it only recognizes the former and not the latter (11). RT1-A1c fits the HLA-B7 supertype, based on its ability to bind peptides with proline at P2 and a hydrophobic residue at the C-terminus (12). Previously, our laboratory demonstrated that mutation of RT1-A1c amino acids forming the B-pocket where the P2 residue of bound peptide binds, and not other residues in the rest of the peptide binding groove, disrupted Ly49i2 recognition (13). The B-pocket mutations also resulted in a quantitative shift in the frequency of the RT1-A1c bound peptides with proline at the P2 position. In this report, we directly compare the influence of different P2 residues of bound peptides in Ly49i2 recognition of RT1-A1c. Our results indicate a requirement for specific P2 residues bound in RT1-A1c for recognition by the MHC-I allele specific Ly49i2 NK inhibitory receptor. Thus, Ly49i2 is peptide selective in MHC-I recognition and bound peptide affects ligand identity.
MATERIALS AND METHODS
Cell lines, peptides, and Abs
RNK-16, a spontaneous F344 rat strain NK cell leukemia and RNK-16 transfected to express Ly49i2 (RNK.49i2) have been described (13, 14). The TAP-2–deficient RMA-S mouse T lymphoma (15) was obtained from Dr. W. Jefferies (University of British Columbia, Vancouver, Canada). Three 9-mer peptides, NPRKVTAYL (P2P), NQRKVTAYL (P2Q), and NVRKVTAYL (P2V) were commercially synthesized (GenScript) for peptide loading of RT1-A1c. Purified STOK2 (rat IgG2a; anti-Ly49i2) (11) was purchased from BD Biosciences. Tissue culture supernatant of the YR5/12 hybridoma (rat IgG2b; anti-RT1-A1c) (16) was purchased from Serotec. Purified 4D11 (rat IgG2a; anti-Ly49G) and F4/80 (rat IgG2b; anti-F4/80) were produced from hybridomas in this laboratory and used as isotype controls, Fluorescein (FITC)-coupled and R-phycoerythrin (PE)-coupled mouse anti-rat IgG were purchased from Jackson ImmunoResearch Laboratories.
Expression, refolding, and purification of RT1-A1c
The cDNA encoding the extracellular region of RT1-A1c mature alpha chain (residues 1–277) was ligated to a sequence that encoded a C-terminal BirA recognition tag (17) and was inserted into the bacterial expression vector pET21a (Novagen). The cDNA encoding the mature rat β2m cloned from a PVG rat cDNA library was also subcloned into pET21a. Sequences were confirmed by automated DNA sequencing using an ABI 3730 DNA Analyzer (Applied Biosystems). These plasmid DNAs were then transformed into Escherichia coli strain BL21(DE3) for protein expression. The bacteria cultured in LB broth supplemented with 50 μg/ml ampicillin were induced with 0.5 mM isopropyl-1-thio-β-D-galactopyranoside (IPTG) and further supplemented with 20 μg/mL biotin for 4 hrs. Bacteria were pelleted and then treated with 1 mg/ml lysozyme for 1 hr at RT in TE (0.1 M Tris pH 8.0, 0.2 mM EDTA) supplemented with 1% Triton X-100 and then lysed by sonication. Pellets containing the inclusion bodies were washed once with TE without Triton X-100 and dissolved overnight at 4°C in 8M Urea. Protein concentrations were determined by using absorbance at 280 nm and molar extinction coefficients of 67760 and 16740 for RT1-A1c and β2m, respectively.
The refolding of soluble RT1-A1c was achieved by rapid dilution of a mixture of 0.5 μmole RT1-A1c heavy chain and 1 μmole rat β2m into 400 mL arginine buffer (0.4 M L-arginine-HCl, 0.1 M Tris, pH 8, 2 mM EDTA, 5 mM reduced glutathione, and 0.5 mM oxidized glutathione) in the presence or absence of 10 mg of peptide at 4°C. With peptide present, this resulted in a molar ratio of 1:2:20 of heavy chain to β2m to peptide. The mixture was allowed to stir for 72 hrs and then concentrated by ultrafiltration. The concentrated proteins were dialyzed against 100 mM NaCl, 100 mM Tris, pH 8.5, and then purified by size exclusion chromatography on a HiLoad Superdex-75 26/60 prep grade column (GE Healthcare).
Epitope and thermal stability analysis
Refolding of RT1-A1c to acquire the YR5/12 recognized epitope was assessed by flow cytometry. Latex beads (5 μm diameter; Interfacial Dynamics Corp) were coated with avidin and used to capture equal amounts of refolded RT1-A1c by its biotin tag. Beads were incubated with YR5/12 for 1 hr at 4°C, washed twice with 1% BSA in PBS, incubated with FITC-coupled mouse anti-rat IgG for 30 mins before flow cytometric analysis. Flow cytometric acquisition was completed using a Becton Dickinson FACSCalibur and CellQuest software. Analyses were performed using FCS Express (De Novo Software).
In thermal stability studies, refolded RT1-A1c was again captured onto avidin-coated latex beads. Half of the beads were incubated at 37°C for 1 hr while the other half was left at 4°C for the same amount of time. After incubation, both sets of beads were stained in parallel with YR5/12 followed by FITC-coupled secondary antibody and analyzed by flow cytometry as described above.
Protein analysis
Proteins were analyzed by glycine SDS-PAGE (18). About 2.5 μg of protein were boiled in 1x SDS-PAGE sample buffer and run on a 15% polyacrylamide minigel. Proteins were visualized by staining the gel with Gelcode Blue (Pierce). To visualize the presence of peptide in the refolded RT1-A1c, tricine SDS-PAGE was used as previously described (19, 20). Fifteen μg of protein were boiled in 1x tricine sample buffer and then run on a 16.5% tricine SDS polyacrylamide minigel at 100 V for 3.75 hrs. Proteins were visualized by first fixing in 5% glutaraldehyde solution for 1 hr and then staining with Gelcode Blue. All proteins were quantified using the BCA protein assay kit (Pierce).
Peptide elution and amino acid quantification
Five nmoles of each purified rat MHC-I-peptide complex (in 100 mM Tris, pH 8.5, 100 mM NaCl) were diluted to 100 μl and 10 μl of glacial acetic acid (10% final) was added to acidify the solution and elute the peptides. Peptides were then filtered through 5 kDa MWCO Biomax Ultrafree 0.5 ml centrifugal ultrafiltration membranes (Millipore). The retentate was reduced to about 25 μl then another 100 μl of 10% glacial acetic acid was added and the filtration was repeated twice more. The filtrate was then recovered for amino acid quantification. Peptides were hydrolyzed under vacuum in 6 M HCl, 0.1% phenol for 1 hr at 160°C. Amino acids were then quantified using a Beckman 6300 amino acid analyzer with post-column ninhydrin detection. Efficiency was calculated by the addition of 12 nmole of norleucine just before the hydrolysis step. Asn, Leu, and Val were considered the most stable amino acids after hydrolysis and were used to calculate the amount of peptide in the original sample.
Production and use of RT1-A1c multimers
RT1-A1c was multimerized by mixing 40.5 μg of monomers in five additions with 11.9 μg of Extravidin-PE conjugate (Sigma) in 5-min intervals to prepare 100 μg of multimers. To assess the interaction of RT1-A1c with Ly49i2, 5×105 RNK.49i2 or RNK-16 cells were incubated with 0, 0.125, 0.25, 0.5, 1, 5, or 10 μg of multimers in 50 μl of RNK medium (RPMI 1640 supplemented with 10% fetal calf serum, penicillin, streptomycin, L-Gln, and 50 μM 2-mercaptoethanol) for 1 hr at 4°C. As a negative control, RNK.49i2 and RNK-16 were also incubated with 5 μg of Extravidin-PE conjugate. The cells were then pelleted and washed twice before flow cytometric analysis as described above. Ly49i2 expression was assessed in parallel by antibody staining with STOK2 followed by secondary staining with PE-coupled mouse anti-rat IgG.
Generation of RNK-16 effector cells expressing chimeric Ly49Wi2 receptors
A chimeric receptor consisting of the intracellular and transmembrane domains of Ly49W and the extracellular domain of Ly49i2 (Ly49Wi2) was created from the respective encoding cDNA using two rounds of PCR amplification. The first round consisted of amplification of the intracellular- and transmembrane-encoding domains of Ly49W using the 5′ primer GGCCTCGAGACCATGAGTGAGCAGGAGGTCAC which adds a XhoI restriction site and 3′ primer CATGATTATACTGCAAACAGTTTGTCACAAGCACTGAGACAATTAC which included a priming sequence for the 5′ end of the Ly49i2 ectodomain for the second round of PCR. The amplified product was separated by gel electrophoresis and the band corresponding to the product was purified. This was used as the 5′ primer for the second round of PCR with cDNA encoding Ly49i2 and 3′ primer GGCTCTAGATCACAGAGCAGACTGTGACC that adds a XbaI restriction site to the 3′ end of the final construct. The final amplified product was separated by gel electrophoresis and the band corresponding to the correct product was excised and purified. This was then digested by restriction enzymes and inserted into the BSRαEN vector (provided by Dr. A. Shaw, Washington University, St. Louis, MO) at the XhoI-XbaI sites to create BSR.Ly49Wi2. The identity of the insert was verified by DNA sequencing. RNK-16 cells were stably transfected with the construct as previously described (21). Positive expression of Ly49Wi2 was verified by surface staining with STOK2 (anti-Ly49i2) antibody by flow cytometry.
Generation of RMA-S cells expressing RT1-A1c
The cDNA encoding RT1-A1c was previously cloned from a cDNA library of the PVG strain rat (22) and inserted into the expression vector pCI-neo (Promega). Stable transfectants were generated by resuspending RMA-S cells (2×106) in 0.4 ml of RPMI and electroporating 20 μg of plasmid in 4 mm-gap cuvettes at 180 mV and 960 μF with a Genepulser (BioRad). Transfected cells were transferred into 96-well microtiter plates and grown in the presence of 0.5 mg/ml G418. Cells surviving G418 selection were clonally expanded and incubated overnight at 26°C to enhance RT1-A1c cell surface expression. Expression of RT1-A1c was assessed by flow cytometry using the YR5/12 mAb followed by FITC-conjugated mouse anti-rat IgG secondary mAb.
Peptide loading and cytotoxicity assays
RMA-S cells transfected with RT1-A1c were cultured overnight at 26°C and the surface MHC-I were loaded with specific peptide by incubation in the presence of 10 μM of peptides, similar to what has been previously described (15). These target cells were subsequently labeled at 37°C with 100–150 μCi of Na51CrO4 (51Cr; Mandel) for 1 hr. The cells were then extensively washed and 1 × 104 target cells were incubated for 4 hrs at 37°C with varying effector:target ratios with Ly49Wi2-expressing RNK-16 cells in 96-well V-bottom microtitre plates. Following incubation, plates were centrifuged for 5 min. Supernatants (25 μl) were transferred to 96-well plates and 100 μl of scintillant (OptiPhase ‘SuperMix’, PerkinElmer) was added. The plates were analyzed in a beta counter (MicroBeta, PerkinElmer). Percent specific lysis was calculated as [(experimental release) − (spontaneous release)/(maximal release) − (spontaneous release)] × 100. All cytotoxicity assays were performed a minimum of three separate times.
RESULTS
Design of peptides
Major histocompatibility complex class Ia molecules of several mammalian species including primates and rodents can be classified into approximately nine supertypes based on their preference for defined peptide anchor residues bound within specific pockets of the MHC-I peptide binding groove (9, 10). The rat MHC class Ia molecule, RT1-A1c, can be classified into the HLA-B7 supertype based on its preference for binding nonamer peptides with proline as the second residue from the N-terminus at the P2 anchor position (Fig. 1A) (12). The P2 residue of bound peptide is typically held in the B-pocket and RT1-A1c shares the B-pocket amino acid residues Y7, N63, I66, Y67, and Y99 seen with other molecules of the HLA-B7 supertype (9). Previous work from our laboratory showed that site-directed mutations of polymorphic residues D9 and S24 in the B-pocket, but not polymorphic residues in other pockets of the peptide binding groove of RT1-A1c, resulted in loss of recognition by the rat NK receptor, Ly49i2 (13). Sequencing of acid-eluted peptides from the D9A and S24A mutants demonstrated that the proportion of bound peptides bearing proline at the P2 anchor position was reduced relative to peptides eluted from wild-type RT1-A1c (13). In the case of mutant D9A, a significant increase in valine was observed at the P2 anchor position in bound peptides. Valine is a previously reported secondary P2 anchor residue for RT1-A1c and other HLA-B7 supertype members (Fig. 1A) (12, 23, 24). As for the S24A mutant, a significant increase in glutamine as the P2 anchor with a yield equivalent to proline was found at this position in bound peptides (13). Glutamine is neither a previously reported P2 anchor residue for RT1-A1c, nor an accepted HLA-B7 supertype P2 residue (9, 23). These results raised the interesting possibility that the identity of the P2 anchor residue of bound peptides may be critical for Ly49i2 recognition of RT1-A1c, where substitution from a proline to a non-B7 supertype P2 residue or possibly to a secondary B7 supertype P2 residue in bound peptides, may be sufficient to disrupt recognition. We first explored this concept by employing fluorescent RT1-A1c multimers loaded with peptides that were otherwise identical except for containing different residues at the P2 position. Peptides were designed based on the “ideal RT1-A1c binding peptide” previously identified by Stevens et al. (12, 24). Their studies involved peptide elution and Edman degradation sequencing with the most abundant residue found at each peptide position used to design a nonamer, NPRKVTAYL, that showed high binding affinity (Fig. 1B), and thus received the designation “ideal” (12). Their results also showed that proline was the predominant anchor residue found at P2 in natural peptides eluted from RT1-A1c. To test the importance of bound peptide P2 anchor residue identity in Ly49i2 recognition of RT1-A1c, we synthesized peptides for binding to RT1-A1c that shared the “ideal RT1-A1c binding peptide” sequence, with the exception that they each differed in the amino acid Pro, Gln, or Val at the P2 anchor position: NPRKVTAYL (P2P), NQRKVTAYL (P2Q), and NVRKVTAYL (P2V) (Fig. 1B). The Gln and Val were chosen as alternative P2 anchor residues, as they were increased in peptides bound to the mutant RT1-A1c molecules S24A and D9A that were not recognized by Ly49i2 (13). Furthermore, Val but not Gln is a previously reported secondary P2 anchor residue for HLA-B7 supertype MHC molecules, including RT1-A1c (Fig. 1B).
FIGURE 1. RT1-A1c has a preference for binding B7-supertype peptides.
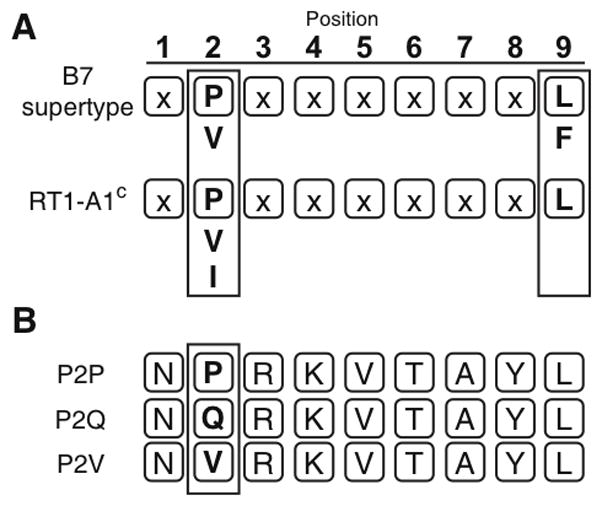
A. The HLA-B7 supertype (top) and RT1-A1c (bottom) peptide anchor residue binding preferences are shown (boxed). Both HLA-B7 and RT1-A1c have preference for proline at the P2 position and have leucine as a preferred amino acid residue at the C-terminus. Secondary anchor residues appear below primary anchor residues. The B7 supertype has a more variable amino acid binding at the C-terminus (A,L,I,V,M,F,W,Y), but only the binding preference for the prototypical member, HLA-B7, is shown for simplicity. B. Three nonamer peptides were used in this study. P2P is the “ideal” peptide sequence previously reported (12). P2Q and P2V have the same sequence, except glutamine or valine has been substituted at the P2 position.
Expression and purification of soluble RT1-A1c monomers
A fusion protein of the extracellular domain of RT1-A1c with a C-terminal BirA recognition sequence (17) was over-expressed in E. coli. Using identical conditions, RT1-A1c and rat β2m were refolded in the presence of either P2P, P2Q, P2V or no peptide (see Materials and Methods). Refolded proteins were purified by size exclusion chromatography (Fig. 2A). Refolding of RT1-A1c in the presence of each of the three peptides produced nearly the same elution profile with a prominent peak at about 158 ml retention volume (~47 kDa) corresponding to the peptide-RT1-A1c-β2m complex. By contrast, folding of RT1-A1c in the absence of peptide produced a much smaller peak at about 154 ml corresponding to the RT1-A1c-β2m complex and a much higher peak at the void volume corresponding to protein aggregation. This is consistent with what has been reported previously for folding of recombinant MHC-I molecules (24, 25), and demonstrates that RT1-A1c requires peptide to refold efficiently. The 47 kDa fractions were pooled, concentrated, and analyzed by SDS-PAGE (Fig. 2B). The results show two bands, one consistent with heavy chain RT1-A1c at about 34 kDa and a second consistent with β2m at 12 kDa after refolding with each of the peptides, P2P, P2Q, and P2V. This confirms that the peak that eluted at about 158 ml was indeed refolded MHC-I complexes. In addition, the pooled fractions of RT1-A1c refolded in the absence of peptide also separated into heavy chain and β2m indicating that empty RT1-A1c complexes were produced.
FIGURE 2. RT1-A1c refolds with peptides containing Pro, Gln, or Val at the P2 anchor position.

A. The ectodomain of RT1-A1c tagged with a BirA recognition sequence on the C-terminus and rat β2m were expressed as inclusion bodies in E. coli. The inclusion bodies were solubilized and refolded in the presence of synthetic peptides differing at P2. Peptides used were P2P (NPRKVTAYL), P2Q (NQRKVTAYL), P2V (NVRKVTAYL), or no peptide. Folded MHC-I molecules were concentrated and dialyzed into 100 mM NaCl, 100 mM Tris, pH 8.5 before purification by size exclusion chromatography. The chromatographs are shown with molecular mass standards indicated by the arrows above. The highlighted (shaded) fractions corresponding to refolded RT1-A1c were pooled and concentrated. B. Purified RT1-A1c was analyzed by SDS-PAGE. Approximately 2.5 μg of protein was run on 15% SDS-PAGE and visualized by Gelcode Blue stain.
Purified Soluble RT1-A1c monomers show conformational integrity, thermal stability, and contain peptides
As a prerequisite to employing the soluble RT1-A1c-peptide complexes in receptor binding studies, we assessed them for conformational integrity, thermal stability, and for peptide content following acid elution. Equivalent quantities of RT1-A1c refolded with P2P, P2Q, P2V or no peptide were captured on avidin-coated beads and incubated at 4 or 37°C for 1 hr before staining with a RT1-A1c-specific mAb, YR5/12. Similar staining levels for RT1-A1c refolded with P2P, P2Q and P2V were observed, indicating that all three RT1-A1c monomers folded to equal levels and displayed a serological epitope recognized by YR5/12 (Fig. 3A) Staining by the YR5/12 antibody was only slightly diminished for RT1-A1c bound with P2P, P2Q, or P2V after incubation at 37°C compared with incubation at 4°C, and antibody staining was equivalent to RT1-A1c bound with each of the three peptides at the same temperature (Fig. 3A). Thus, RT1-A1c refolded in the presence of any of the three peptides showed equal thermal stability. In contrast, the RT1-A1c refolded in the absence of peptide showed substantially reduced thermal stability, as shown by the significantly diminished YR5/12 staining after incubation at 37°C (Fig. 3A). Therefore, all three peptides had an equal ability to stabilize against thermal disruption of the RT1-A1c complex.
FIGURE 3. Conformational integrity, thermal stability and peptide content of refolded RT1-A1c.

A. Conformational integrity and thermal stability analysis. RT1-A1c refolded with P2P, P2Q, P2V, or no peptide was captured onto avidin-coated 5 μm latex beads and incubated for 1 hr at 37°C (grey histograms) or 4°C (open histograms) followed by FACS analysis with the RT1-A1c specific Ab, YR5/12. Avidin beads with no protein captured were stained with antibodies in parallel (black histograms). B. The presence of P2P, P2Q, or P2V peptide in the refolded and purified RT1-A1c was analyzed by tricine SDS-PAGE. Approximately 15 μg of purified RT1-A1c was denatured in sample buffer and run on a 16.5% Tricine-SDS polyacrylamide gel. C. Peptide amino acid quantification. Five nmoles of each refolded RT1-A1c–peptide complex were treated with acid to separate the peptide as described in the Materials and Methods. Eluted peptides were subjected to acid hydrolysis and the amino acids quantified.
We next confirmed the presence and equivalent loading of peptide in refolded RT1-A1c. The RT1-A1c refolded with P2P, P2Q, P2V, or no peptide was denatured with sample buffer and separated into its components by tricine-SDS PAGE. A band corresponding to peptide is present in all the refolded RT1-A1c complexes, except for the one refolded without peptide (Fig. 3B).
Peptides eluted with mild acid from equal amounts (5 nmol) of the P2P, P2Q and P2V-RT1-A1c complexes, respectively, were hydrolyzed and quantified for amino acid content (Fig. 3C). The results show a roughly 1:1 ratio of peptide to MHC in the three different peptide-RT1-A1c complexes. The internal relationships of the amino acid quantification show a 1:1:1 molar ratio of Asn, Leu, and Val for P2P and P2Q and a 1:1:2 molar relationship for P2V, which accurately reflect the amino acid representation in the peptide sequences. The results show slightly higher values for the yield of amino acids compared to theoretical input, however this fractional difference can be accounted for by small deviations in both protein and amino acid residue quantifications. Nonetheless, the results demonstrate that the refolded peptide-RT1-A1c complexes were similarly loaded and stabilized with the three different peptides, P2P, P2Q, and P2V.
Binding of RT1-A1c multimers to Ly49i2 is determined by the identity of the P2 residue bound in the supertype defining B-pocket
The Ly49i2 inhibitory NK cell receptor of the PVG rat strain is allele specific in its recognition of rat MHC-I ligands. Using a rat NK leukemia cell line, RNK-16, stably transfected to express Ly49i2, our laboratory previously showed that recognition of RT1-A1c is dependent on the composition of MHC-I residues of the B-pocket, which determines the P2 anchor residue accepted and in turn defines the MHC-I supertype (13). With the refolded RT1-A1c bound with P2P, P2Q, or P2V and RNK.49i2 transfectants, we could now directly determine whether the identity of the P2 peptide residue influences Ly49i2 recognition of RT1-A1c. First, the RNK.49i2 transfectant (RNK.49i2) expresses significant levels of Ly49i2, while the parental RNK-16 cells are negative, as measured by staining with the Ly49i2 specific antibody, STOK2 (Fig. 4A). Next we captured the individual P2P, P2Q, or P2V peptide-RT1-A1c complexes onto avidin-PE conjugates through their biotin tags to form multimeric staining reagents (26). We then incubated the multimers with RNK-16 or RNK.49i2 cells. Phycoerythrin-labeled RT1-A1c multimers bearing the P2P or P2V peptide bound RNK.49i2 but not RNK-16 cells (Fig. 4B). In contrast, RT1-A1c folded without peptide shows little or no binding to RNK.49i2 cells, demonstrating a need for the presence of peptide for Ly49i2 recognition, and is consistent with what has been reported previously for Ly49A recognition of H-2Dd (27, 28). Interestingly, RT1-A1c multimers refolded with P2Q peptide also showed little or no binding to RNK.49i2 cells, indicating that the presence of Gln at P2 could not support RT1-A1c recognition by Ly49i2 (Fig. 4B). An increasing titration of RT1-A1c multimers presenting P2Q peptide showed a slight increase in staining of RNK.49i2 cells, but not much more than RT1-A1c bearing no peptide, and significantly less than RT1-A1c presenting P2P or P2V peptides which showed dose dependent increases in RNK.49i2 binding (Fig. 4C). Our findings demonstrate that Ly49i2 can discriminate between RT1-A1c peptide complexes due to differences in bound peptides. Specifically, our results indicate that RT1-A1c bound with peptides that contain the HLA-B7 supertype P2 anchor residue, Pro, or Val an acceptable secondary supertype residue, are supportive for Ly49i2 recognition of RT1-A1c. In contrast, Gln, which is not a HLA-B7 supertype P2 peptide residue, is insufficient in supporting Ly49i2 recognition of RT1-A1c. It is possible that when bound with Pro or Val anchor residues, RT1-A1c complexes assume specific conformation(s) conducive to Ly49i2 recognition that are not assumed when Gln is the P2 anchor. A strong preference or requirement for Pro or Val bound in the supertype defining B-pocket of RT1-A1c may be a component of NK cell surveillance for expression of an MHC-I supertype.
FIGURE 4. RT1-A1c bound with P2 anchor residues Pro or Val, but not Gln, bind to Ly49i2 expressing cells.

A. RNK-16 and RNK-16 cells stably transfected with Ly49i2 (RNK.49i2) were stained with STOK2 (anti-Ly49i2) Ab (open histograms) or isotype control (grey histograms) B. RNK-16 and RNK.49i2 cells were stained with PE-labeled RT1-A1c multimers bearing the peptides P2P, P2Q, or P2V, or no peptide (open histograms) or Extravidin-PE alone (grey histograms). C. Increasing amounts of PE-labeled RT1-A1c multimers were used to stain RNK.49i2 cells. The extent of staining was determined by FACS analysis and plotted as MFI vs. amount of multimers. RT1-A1c was refolded with P2P, P2Q, and P2V and purified three separate times with similar cell-staining results.
The P2 residue of RT1-A1c bound peptide affects cytolytic activation of RNK cells by Ly49i2
We next wanted to examine the consequences of the altered Ly49i2 recognition of RT1-A1c on cell-mediated cytolytic function. We employed the TAP-2 deficient mouse T lymphoma cell line, RMA-S, as target cells which allowed us to control the peptide exogenously loaded into the MHC-I on the cell surface (15). RMA-S cells were stably transfected with RT1-A1c and cell surface expression was confirmed by staining with the YR5/12 mAb after incubating cells with peptide and analyzing by flow cytometry (Fig. 5A). The behavior of RT1-A1c on the transfected RMA-S (RMA-S.A1c) cells in the presence or absence of peptide was similar to what has been described for endogenous H-2Kb on RMA-S (15). Specifically, in the absence of exogenously added peptide, RMA-S.A1c display low cell surface levels of RT1-A1c after incubation at 37°C, indicating thermal instability of the peptide-deficient MHC-I (Fig. 5A). However, in the presence of any of the three peptides, P2P, P2Q, or P2V, RT1-A1c is able to stably express to similar high levels on the RMA-S.A1c cell surface for at least 4 hrs (Fig. 5A). For cytotoxicity experiments, we used the rat NK cell line to assess ligand recognition by Ly49i2. Since RNK-16 does not recognize RMA-S cells, we could not assess receptor functionality by transfecting Ly49i2 as an inhibitory receptor (data not shown). Instead, we recombinantly converted Ly49i2 into an activating receptor by fusing its extracellular domain to the transmembrane and intracellular domains of the activating mouse Ly49W receptor. The chimeric activating receptor, Ly49Wi2, would contain the Ly49i2 ectodomain and therefore retain the recognition properties of Ly49i2, but transmit an activating rather than an inhibitory signal upon ligand engagement. Our laboratory successfully used this strategy previously to convert mouse inhibitory receptors into activating receptors that retain the ligand specificity of the original inhibitory receptor (29). RNK-16 cells were stably transfected with Ly49Wi2 (RNK.49Wi2) and expression was confirmed by staining with anti-Ly49i2 mAb, STOK2 (Fig. 5B). We next assessed the role of the P2 anchor residue on cytolytic activity mediated through Ly49Wi2. RMA-S.A1c cells were incubated overnight at 26°C to maximize surface expression of empty RT1-A1c molecules. The cells were then incubated in the presence of P2P, P2V, P2Q or no peptide and then used as targets in cytotoxicity assays with RNK.49Wi2 cells (Fig. 5C). No recognition or cytolysis was observed when RMA-S.A1c cells were incubated without peptide. However, when RT1-A1c expressed by the RMA-S transfectants presented either P2P or P2V, RNK.49Wi2 cells were able to recognize the MHC-I and mediate substantial lysis of targets. In contrast, when RT1-A1c presented P2Q, which contains the non-B7 supertype P2 residue, a significantly depressed level of cytolysis was mediated by the RNK.49Wi2 cells. Thus, the pattern of RT1-A1c-peptide complex recognition by Ly49i2 observed with soluble multimers was also found in target cell lysis, where RT1-A1c bound with peptides bearing supertype P2 anchor residues, Pro or Val, are efficient Ly49i2 ligands, whereas RT1-A1c bound with a non-supertype P2 anchor residue is not. These results indicate that the identity of the P2 anchor residue of bound peptide plays an important role in Ly49i2 recognition of its RT1-A1c ligand.
Figure 5. The P2 anchor residue affects cytolytic activity by Ly49Wi2-expressing RNK-16 cells.

A. RMA-S cells stably transfected with RT1-A1c (RMA-S.A1c) were cultured overnight at 26°C and then incubated in the presence of 10 μM P2P, P2Q, P2V or no peptide at 37°C for 4 hrs. Cells were then stained with YR5/12 (black histograms) or isotype control (grey histograms) followed by secondary staining with FITC conjugated anti-rat IgG. Cells were analyzed by flow cytometry. The MFI are indicated on the top right corner of each histogram. B. RNK-16 cells were stably transfected with the chimeric recptor Ly49Wi2 (RNK.49Wi2) which retains the recognition domain of Ly49i2, but possesses the activation domain of Ly49W. Expression was confirmed by staining cells with STOK2. C. RNK.49Wi2 cells were used as effector cells in a 4-hr cytotoxicity assay with RMA-S.A1c target cells incubated with P2P, P2Q, P2V, or no peptide.
DISCUSSION
RT1-A1c preferentially binds peptides with a proline residue at P2 which defines it as a HLA-B7 supertype (9, 10). Previously, our laboratory showed that point mutations in the B-pocket of RT1-A1c resulted in a loss of protection from NK cell lysis mediated by Ly49i2 (13). These mutations were accompanied by alterations in the P2 anchor residues of the RT1-A1c bound peptides. This raised the possibility that bound peptide could influence MHC recognition by Ly49i2 and the hypothesis that Ly49i2 will only recognize RT1-A1c that is bound with peptides bearing the HLA-B7 supertype-defining P2 residue Pro, or possibly the secondary HLA-B7 supertype residue Val, but not P2 anchor residues that are not normally associated with the HLA-B7 supertype (13). Requirements for Ly49i2 recognition of MHC-I could include MHC-I bound with: 1) no peptide; 2) any peptide; 3) a peptide with an acceptable B7 supertype P2 anchor; or 4) specifically only Pro at P2. Using three synthesized nonamer peptides that differed only at the P2 residues, we were able to refold each with RT1-A1c and show that each peptide was loaded into the RT1-A1c to an equivalent extent. Furthermore, all three RT1-A1c-peptide complexes were recognized by the RT1-A1c specific mAb YR5/12 and had equivalent thermal stability. The refolded RT1-A1c complexes, in the form of PE-labeled multimers were used to stain Ly49i2 transfected cells. The RT1-A1c complexed with peptides having Pro or Val at P2 were able to bind Ly49i2, while RT1-A1c complexed with peptides that had Gln at P2 was greatly diminished in its ability to bind Ly49i2, to the point of being nearly equivalent to the negligible Ly49i2 binding of peptide-empty RT1-A1c. When this interaction was tested functionally through cell-mediated lysis, a similar pattern was observed. RMA-S cells displaying RT1-A1c peptides with Pro or Val at P2 were able to be killed by RNK-16 cells expressing the chimeric Ly49Wi2 receptor. However, when the peptide presented Gln at P2, cytolysis mediated through the Ly49Wi2 receptor was substantially diminished. This indicates that the identity of the P2 residue bound in the B-pocket of RT1-A1c indeed influences the binding of Ly49i2 and both the prototypic Pro or secondary Val HLA-B7 supertype P2 anchor residues support Ly49i2 recognition of RT1-A1c. Although our analysis was limited to three different peptides, due to limitations in the choice of P2 anchor residues that can be expected to stabilize RT1-A1c, it shows the dramatic differences in Ly49i2 recognition that can result from just a single amino acid difference in the bound peptide at an anchor position, which is not explained by a difference in MHC stability or level of peptide occupancy. Previously, we suggested that Ly49i2 recognition of RT1-A1c may be influenced by the supertype family of the MHC-I (13, 30). Our demonstration of the importance of the identity of P2 residues, Pro or Val, which are anchor residues of the HLA-B7 supertype, for Ly49i2 recognition are consistent with this possibility. Ly49 recognition of MHC-I based on supertype families may be a possible solution for NK cells to maintain innate self-recognition of MHC-I, despite rapid MHC-I evolution.
The role of MHC-I bound peptide in mouse Ly49 recognition has been approached by others. For example, the interaction of mouse Ly49A with its MHC-I ligand, H-2Dd, was shown to be peptide dependent but non-discriminating of H-2Dd-bound peptide (27, 31) and it was possible that the contribution of bound peptide was simply to stabilize the MHC-I (28). Consistent with our results, it was previously noted that “peptide empty” H-2Dd could not be recognized by Ly49A, suggesting that the peptide confers a particular MHC-I conformation needed for Ly49A recognition (28). However other studies have shown that mouse Ly49C and Ly49I display selectivity in bound peptide for MHC-I recognition, independent of MHC-I stabilization (8, 32–34). In light of our findings demonstrating the importance of peptide anchor residue identity in rat Ly49i2 recognition, if we consider the identity of peptide anchor residues, a possible explanation emerges for the peptide selectivity of Ly49C recognition of H-2Kb. The peptide motif bound by H-2Kb has two primary anchor residues, at P5 and P8, and an auxiliary anchor residue at P3 (23, 35, 36). From the pattern of Ly49C recognition of H-2Kb bound with peptides (8, 32), Ly49C interaction may depend on the identity of the P3 auxiliary anchor residue. A similar relationship may exist for Ly49I recognition of H-2Kd, except in this case, from the pattern of peptide selectivity (33, 34), and location of anchor residues, the identity of the P5 auxiliary anchor residue may determine receptor interaction. Thus, the identity of peptide anchor residues, whether primary or auxiliary, may significantly influence the extent of interaction with, or specificity for, MHC-I ligands by multiple Ly49 receptors.
The influence of different peptides on Ly49 recognition of MHC-I could be viewed as unexpected, given that the Ly49A/H-2Dd and Ly49C/H-2Kb co-crystal structures revealed that Ly49 receptors bind at the highly conserved region known as site 2 which involves the base of the peptide binding groove, the α3 domain, and β2m, and therefore makes no direct contact with MHC-I bound peptide (6, 8). Previous results from our laboratory suggested that Ly49i2 also interacts with the highly conserved site 2 of RT1-A1c, and like Ly49A and Ly49C, would have no opportunity for direct contact with peptide (13). Therefore, Ly49i2 detection of differences in bound peptide occurs indirectly. In our present study, RT1-A1c was able to bind Ly49i2 when bound with peptides containing Pro or Val at P2. These were the preferred residues found at P2 as determined by peptide elution studies using both randomly-generated and naturally-derived peptides bound to RT1-A1c (12, 24). RT1-A1c was not able to bind to Ly49i2 when the P2 residue of the bound peptide was Gln. Interestingly, and consistent with these results, the yield of peptides with Gln at P2 increased with the RT1-A1c S24A B-pocket mutant which was not recognized by Ly49i2 (13). Also, RT1-Aa is a rat MHC class Ia allele product that binds peptides with a Gln P2 anchor, and RT1-Aa is also not recognized by Ly49i2 (11, 24). Lack of recognition of RT1-A1c with a Gln at P2 of the bound peptide may be due to conformational differences imposed on the B-pocket. Our data indicate that Pro or Val in the P2 position of peptides, bound in the RT1-A1c B-pocket, are permissive for Ly49i2 recognition, while Gln is not. Pro and Val are compact, fitting readily into the RT1-A1c B-pocket, which is relatively confined due to Tyr67 minimizing the B-pocket volume, as indicated in the RT1-A1c crystal structure (37). From the crystal structure of RT1-Aa bound with the B6 peptide (AQFSASASR), Gln in the P2 position binds in the B-pocket of RT1-Aa which is lined with hydrophobic residues to interact with its methylene groups as well as accommodation of the Gln carboxamide through hydrogen bonding (37). When the P2 Gln-containing peptide binds RT1-A1c, the larger size and polar group in Gln, as opposed to Pro or Val, may require a large displacement of the residues within the B pocket, or possibly it does not fit in the pocket and points into solvent, perhaps resulting in more dependence on the auxiliary P3 Arg anchor for MHC binding. In both cases, we would expect changes in the B-pocket conformation relative to that with Pro or Val bound either by fitting a larger than preferred residue or remaining vacant. Differences in B-pocket conformation may be transmitted through the floor of the peptide-binding groove, particularly the β1 strand, influencing the conformation of solvent exposed RT1-A1c residues extending below the groove such as Arg6 and Phe8, which have been demonstrated to play a role in Ly49i2 interaction (30),and/or possibly influencing contacts with associated β2m and therefore its orientation. RT1-A1c has three β1 strand residues, Arg6, Phe8, and Asp9, that have distinct van der Waals interactions with β2m residues Phe56, Ser57, and Lys58 which are not found in RT1-Aa interactions with β2m. The only polymorphic residue in the β1 strand differing between RT1-A1c and Aa is residue 9 (Asp and Tyr, respectively). The Asp9 on the β1 strand interfaces the peptide-binding groove B-pocket and the β2m binding site on RT1-A1c. In RT1-Aa, Tyr9 does not contact β2m. Interestingly, we have previously mutated Asp9 to Ala in A1c, and this disrupted Ly49i2 recognition, despite this mutant maintaining the correct peptide P2 anchor residue, Pro or Val in bound peptides (13). This suggests that Asp9 has a role in Ly49i2 recognition independent of its influence on RT1-A1c binding of peptides with specific P2 anchor residues. This may involve facilitating an RT1-A1c conformation conducive to direct interaction of Ly49i2 with the RT1-A1c heavy chain, and/or maintaining β2m in an associated orientation that promotes interaction with Ly49i2, perhaps in conjunction with A1c residues Arg6 and Phe8. Further to the possible role of β2m in Ly49i2 recognition, the conformation of the S4 strand of rodent β2m, which includes residues Phe56, Ser57, and Lys58 that contact the heavy chain β1 strand, is flexible and can be dramatically different between different rodent MHC-I allele products, resulting in different β2m contacts with the heavy chain (38). By contrast, the human β2m S4 strand has a single conformation (38). Also, the orientation of β2m and the MHC-I heavy chain α1/α2 peptide binding domains can differ significantly between rodent MHC-I molecules, compared to human (39). Such variation in the nature of β2m orientation and association with the heavy chain in rodents may provide identity and specificity for Ly49 interactions, not employed in humans. Thus, rodent β2m may be a transmitter of structural changes in the peptide binding groove, e.g. from the B-pocket bound with different P2 peptide residues, via MHC-I β1 strand residues, that confers specificity to Ly49 interactions. In the case of the peptides we have examined, P2P and P2V bound in the RT1-A1c B-pocket, through conformational effects communicated through the β1 strain, may be conducive to association of β2m to the heavy chain in such an orientation, conformation or affinity that it promotes Ly49i2 interaction. In contrast, it is possible that P2Q would not support the appropriate β2m association/conformation that starts with a distortion of the B-pocket that in turn affects the β1 strand and the nature of its interaction with β2m. Another example of the importance of polymorphic β1 strand residues in controlling MHC-I recognition is indicated by a single mutation of Asp9 to Val in the mouse H-2Ld molecule. In this case, antigen specific T cells could no longer recognize an Ld-presented peptide, yet at the same time the stability of β2m association with Ld was improved (40). Thus, the interface between the bound peptide, the floor of the peptide groove and β2m has significant influence on MHC-I complex conformation, and likely how the MHC-I is perceived by multiple receptor types.
A recent report has indicated that residues 211–231 encompassing the L3 loop on mouse Ly49 determines the allele specificity for H-2Kb (41). In mouse Ly49C, part of this L3 loop forms an α-helix which is not seen in mouse Ly49A or G. Exchange of the Ly49C L3/α3 loop into Ly49A led to a gain in recognition of H-2Kb (41). Interestingly, the L3 loop interacts with MHC-I on the platform below the peptide binding groove in an area proximal to the A-, B-, and D-pockets. It is therefore possible that the L3 loop of mouse Ly49, and equivalent sequences in rat Ly49 such as Ly49i2, could indirectly discriminate between the shapes of these pockets through their conformational effects on neighboring MHC-I heavy chain solvent exposed residues. Crystal structures of RT1-A1c bound with peptides differing in P2 anchor residues, that either do or do not support Ly49i2 binding, e.g. with Pro vs Gln at the P2 position, may be informative in this regard, and further enhanced if in conjunction with a co-crystal of Ly49i2 and RT1-A1c bound with a peptide bearing Pro at the P2 position.
With the potential involvement of β2m in rat Ly49i2 recognition of MHC-I molecules and the possibility that RT1-A1c-bound peptides influence this interaction, it is worth pointing out that FCS, which contains bovine β2m, was used in our Ly49i2 tetramer binding and cytotoxicity assays. Exogenous β2m (e.g. bovine) has a potential to exchange with endogenous β2m (e.g. rodent) bound to MHC-I molecules on the cell surface (42) and therefore some percentage of the RT1-A1c in our assays contain bovine β2m, rather than rat β2m, which could influence receptor recognition. It has been shown that mouse Ly49 will not recognize mouse MHC-I when associated with human β2m (7). This is because human β2m lacks S4 strand residues that are involved in rodent specific recognition by Ly49 (43). Bovine β2m, like human β2m, lack the rodent β2m specific S4 strand residues and thus if associated with RT1-A1c, would likely not be recognized by Ly49i2. However, despite the presence of exogenous bovine β2m from FCS in our assays, we still saw significant recognition by rat Ly49i2 of rat MHC-I complexed with peptides in both multimer binding and cytotoxicity assays. In the case of the Ly49i2 multimer assays, the recombinant RT1-A1c-complexes were produced with rat β2m, and the binding assays were carried out at 4°C, a temperature that allows very little β2m exchange (44). As for the cytotoxicity experiments, there would be greater β2m exchange, however, here rat Ly49 recognition is also robustly observed and with the same relative patterns with the different bound peptides found in the Ly49 multimer assays. This may be analogous to mouse Ly49A recognition, where despite human β2m replacing ~35% of the mouse β2m on mouse MHC-I, Ly49A recognition was entirely unaffected and completely blockable by antibody to mouse β2m (7).
Based on KIR2DL1/HLA-Cw4 and KIR2DL2/HLA-Cw3 co-crystal structures, KIR make direct contact with the α1 and α2 helices at the top of the peptide-binding groove and particularly for KIR2DL2, also make direct contact with positions 7 and 8 of the bound peptide (45, 46). For KIR2DL2 and KIR2DL3, their binding to HLA-C and inhibition of NK cell function has been shown to be peptide dependent, and the strength of binding/inhibition is strongly influenced by the identity of residues 7 and 8 of HLA-C-bound peptide (47). Thus, peptide influences on KIR recognition are through direct peptide contact with KIR, via peptide residues 7 and 8, which are not anchor residues and instead extend up and out of the peptide binding groove. In the case of rodents, as we have mentioned, β2m can directly contact Ly49 receptors and may confer specificity to MHC-I/Ly49 interactions as a result of MHC bound peptide-dependent conformational changes imposed on β2m via the floor of the peptide binding groove. The S4 strand of rodent β2m contacting the heavy chain is flexible and can be dramatically different when bound to different rodent MHC-I allele products (38) and could potentially be further influenced by the nature of the peptide bound in the MHC-I groove. These differences may affect the association and orientation of β2m bound to MHC-I. Such variation may provide identity and specificity for rodent Ly49 interactions. Since the S4 strand of human β2m has only a single conformation, irrespective of the MHC-I allele or nature of bound peptide (38), it would not be conducive to MHC-I ligand discrimination by Ly49 receptors in humans. The purpose of just a single β2m conformation in humans is unclear, however it could have been a contributor to the evolution of an independent receptor system, the KIRs, to detect and distinguish between MHC-I ligands, by a means that was not reliant on β2m conformation or orientation. Furthermore, by interacting with MHC-I ligands on top of the peptide-binding groove, KIR could directly interact with residue(s) of the MHC-I bound peptide, perhaps providing greater discrimination of peptides, than sensing them indirectly as occurs with Ly49. It has also been demonstrated that HLA-C bound with peptides that induce weak KIR binding antagonize NK cell inhibition by HLA-C-peptide complexes that mediate strong KIR binding (47). It remains to be determined whether rat RT1-A1c-peptide complexes that weakly interact with Ly49i2 also antagonize NK cell inhibition by strongly interacting RT1-A1c-peptde complexes.
Like humans, but unlike mice, rat NK cells can express CD8αα homodimers (48). On CD8+ T cells, CD8 is expressed as an αα homodimer or αβ heterodimer, and acts as a co-receptor, simultaneously binding MHC-I with the T cell receptor to enhance responsiveness (49). It is unclear what function CD8 plays on rat NK cells, however it is presumably capable of binding MHC-I, and at a site below the peptide-binding groove involving at least the MHC-I α3 domain (50). Since this is the same area on MHC-I that Ly49 binds, it implies that binding of individual MHC-I molecules by Ly49 and CD8 on rat NK cells is mutually exclusive. Our results indicate that Ly49i2 is peptide selective in MHC-I binding, such that a subset of RT1-A1c molecules, perhaps only those with a Pro or Val in the P2 position of bound peptide are recognized by Ly49i2. In contrast, mouse CD8 is not peptide selective in its interaction with MHC-I ligands (50, 51), as is also likely for rat CD8. Thus, there would likely be some RT1-A1c molecules on the surface of a potential target cell that are ligands for either Ly49i2 or CD8, and others that are ligands only for CD8. This adds complexity to receptor/ligand interactions between NK cells and target cells, perhaps influencing the balance of signals and ultimately whether a target is killed or spared. This would be the case for normal rat NK cells and possibly also with RNK-16 and RNK-16Wi2 used in our studies, as RNK-16 also naturally expresses CD8αα (52, 53).
Finally, since differences in peptides bound to MHC-I can alter Ly49 recognition, as we have shown here with rat Ly49i2 and has been shown with mouse Ly49C and I, this may have implications for immune surveillance. It has been noted that viral infection can alter the repertoire of peptides presented by MHC-I (54, 55). Such changes may alter the type of self peptides bound by MHC molecules as well as provide substituted or added presentation of virus protein derived peptides, which may affect the identity of anchor residues of MHC-bound peptides. Should certain viral infections or tumor transformation skew the peptide profile bound to MHC-I in this way, Ly49 recognition could potentially be affected, possibly enhancing or diminishing interactions with inhibitory or activating Ly49 receptors, and correspondingly, NK cell effector functions.
Acknowledgments
We thank Dr. Kerry Lavender for helpful discussions and critical reading of the manuscript. We also thank Dong-Er Gong for her excellent technical assistance.
Footnotes
This work was supported by an operating grant from the Canadian Institutes for Health Research (to K.P.K). B.J.M. was supported by Canadian Institutes for Health Research and Alberta Heritage Foundation for Medical Research studentships.
The abbreviations used are: KIR, killer cell immunoglobulin-like receptor; MFI, mean fluorescence intensity; MHC-I, MHC class I; β2m, β2-microglobulin
References
- 1.Trinchieri G. Biology of natural killer cells. Adv Immunol. 1989;47:187–376. doi: 10.1016/S0065-2776(08)60664-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Biron CA, Nguyen KB, Pien GC, Cousens LP, Salazar-Mather TP. Natural killer cells in antiviral defense: function and regulation by innate cytokines. Annu Rev Immunol. 1999;17:189–220. doi: 10.1146/annurev.immunol.17.1.189. [DOI] [PubMed] [Google Scholar]
- 3.Lanier LL. NK cell receptors. Annu Rev Immunol. 1998;16:359–393. doi: 10.1146/annurev.immunol.16.1.359. [DOI] [PubMed] [Google Scholar]
- 4.Pegram HJ, Andrews DM, Smyth MJ, Darcy PK, Kershaw MH. Activating and inhibitory receptors of natural killer cells. Immunol Cell Biol. 2010 doi: 10.1038/icb.2010.78. [DOI] [PubMed] [Google Scholar]
- 5.Lanier LL. NK cell recognition. Annu Rev Immunol. 2005;23:225–274. doi: 10.1146/annurev.immunol.23.021704.115526. [DOI] [PubMed] [Google Scholar]
- 6.Tormo J, Natarajan K, Margulies DH, Mariuzza RA. Crystal structure of a lectin-like natural killer cell receptor bound to its MHC class I ligand. Nature. 1999;402:623–631. doi: 10.1038/45170. [DOI] [PubMed] [Google Scholar]
- 7.Matsumoto N, Mitsuki M, Tajima K, Yokoyama WM, Yamamoto K. The functional binding site for the C-type lectin-like natural killer cell receptor Ly49A spans three domains of its major histocompatibility complex class I ligand. J Exp Med. 2001;193:147–158. doi: 10.1084/jem.193.2.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dam J, Guan R, Natarajan K, Dimasi N, Chlewicki LK, Kranz DM, Schuck P, Margulies DH, Mariuzza RA. Variable MHC class I engagement by Ly49 natural killer cell receptors demonstrated by the crystal structure of Ly49C bound to H-2Kb. Nat Immunol. 2003;4:1213–1222. doi: 10.1038/ni1006. [DOI] [PubMed] [Google Scholar]
- 9.Sette A, Sidney J. Nine major HLA class I supertypes account for the vast preponderance of HLA-A and -B polymorphism. Immunogenetics. 1999;50:201–212. doi: 10.1007/s002510050594. [DOI] [PubMed] [Google Scholar]
- 10.Sidney J, Peters B, Frahm N, Brander C, Sette A. HLA class I supertypes: a revised and updated classification. BMC Immunol. 2008;9:1. doi: 10.1186/1471-2172-9-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Naper C, Hayashi S, Joly E, Butcher GW, Rolstad B, Vaage JT, Ryan JC. Ly49i2 is an inhibitory rat natural killer cell receptor for an MHC class Ia molecule (RT1-A1c) Eur J Immunol. 2002;32:2031–2036. doi: 10.1002/1521-4141(200207)32:7<2031::AID-IMMU2031>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 12.Stevens J, Jones RC, Bordoli RS, Trowsdale J, Gaskell SJ, Butcher GW, Joly E. Peptide specificity of RT1-A1c, an inhibitory rat major histocompatibility complex class I natural killer cell ligand. J Biol Chem. 2000;275:29217–29224. doi: 10.1074/jbc.M002565200. [DOI] [PubMed] [Google Scholar]
- 13.Lavender KJ, Kane KP. Cross-species dependence of Ly49 recognition on the supertype defining B-pocket of a class I MHC molecule. J Immunol. 2006;177:8578–8586. doi: 10.4049/jimmunol.177.12.8578. [DOI] [PubMed] [Google Scholar]
- 14.Reynolds CW, Bere EW, Jr, Ward JM. Natural killer activity in the rat. III. Characterization of transplantable large granular lymphocyte (LGL) leukemias in the F344 rat. J Immunol. 1984;132:534–540. [PubMed] [Google Scholar]
- 15.Ljunggren HG, Stam NJ, Öhlen C, Neefjes JJ, Höglund P, Heemels MT, Bastin J, Schumacher TN, Townsend A, Kärre K, et al. Empty MHC class I molecules come out in the cold. Nature. 1990;346:476–480. doi: 10.1038/346476a0. [DOI] [PubMed] [Google Scholar]
- 16.Butcher GW. A list of monoclonal antibodies specific for alloantigens of the rat. J Immunogenet. 1987;14:163–176. doi: 10.1111/j.1744-313x.1987.tb00377.x. [DOI] [PubMed] [Google Scholar]
- 17.Beckett D, Kovaleva E, Schatz PJ. A minimal peptide substrate in biotin holoenzyme synthetase-catalyzed biotinylation. Protein Sci. 1999;8:921–929. doi: 10.1110/ps.8.4.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 19.Schagger H. Tricine-SDS-PAGE. Nat Protoc. 2006;1:16–22. doi: 10.1038/nprot.2006.4. [DOI] [PubMed] [Google Scholar]
- 20.Schagger H, von Jagow G. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal Biochem. 1987;166:368–379. doi: 10.1016/0003-2697(87)90587-2. [DOI] [PubMed] [Google Scholar]
- 21.Ma BJ, Silver ET, Hazes B, Kane KP. Reciprocal transfer of class I MHC allele specificity between activating Ly-49P and Ly-49W receptors by exchange of β4-β5 loop residues. J Immunol. 2003;171:5337–5344. doi: 10.4049/jimmunol.171.10.5337. [DOI] [PubMed] [Google Scholar]
- 22.Lavender KJ, Ma BJ, Silver ET, Kane KP. The rat RT1-A1c MHC molecule is a xenogeneic ligand recognized by the mouse activating Ly-49W and inhibitory Ly-49G receptors. J Immunol. 2004;172:3518–3526. doi: 10.4049/jimmunol.172.6.3518. [DOI] [PubMed] [Google Scholar]
- 23.Rammensee H, Bachmann J, Emmerich NP, Bachor OA, Stevanovic S. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics. 1999;50:213–219. doi: 10.1007/s002510050595. [DOI] [PubMed] [Google Scholar]
- 24.Stevens J, Wiesmuller KH, Barker PJ, Walden P, Butcher GW, Joly E. Efficient generation of major histocompatibility complex class I-peptide complexes using synthetic peptide libraries. J Biol Chem. 1998;273:2874–2884. doi: 10.1074/jbc.273.5.2874. [DOI] [PubMed] [Google Scholar]
- 25.Garboczi DN, Hung DT, Wiley DC. HLA-A2-peptide complexes: refolding and crystallization of molecules expressed in Escherichia coli and complexed with single antigenic peptides. Proc Natl Acad Sci USA. 1992;89:3429–3433. doi: 10.1073/pnas.89.8.3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Altman JD, Moss PA, Goulder PJ, Barouch DH, McHeyzer-Williams MG, Bell JI, McMichael AJ, Davis MM. Phenotypic analysis of antigen-specific T lymphocytes. Science. 1996;274:94–96. doi: 10.1126/science.274.5284.94. [DOI] [PubMed] [Google Scholar]
- 27.Correa I, Raulet DH. Binding of diverse peptides to MHC class I molecules inhibits target cell lysis by activated natural killer cells. Immunity. 1995;2:61–71. doi: 10.1016/1074-7613(95)90079-9. [DOI] [PubMed] [Google Scholar]
- 28.Orihuela M, Margulies DH, Yokoyama WM. The natural killer cell receptor Ly-49A recognizes a peptide-induced conformational determinant on its major histocompatibility complex class I ligand. Proc Natl Acad Sci USA. 1996;93:11792–11797. doi: 10.1073/pnas.93.21.11792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Silver ET, Lavender KJ, Gong DE, Hazes B, Kane KP. Allelic variation in the ectodomain of the inhibitory Ly-49G2 receptor alters its specificity for allogeneic and xenogeneic ligands. J Immunol. 2002;169:4752–4760. doi: 10.4049/jimmunol.169.9.4752. [DOI] [PubMed] [Google Scholar]
- 30.Lavender KJ, Chau HH, Kane KP. Distinctive interactions at multiple site 2 subsites by allele-specific rat and mouse Ly49 determine functional binding and class I MHC specificity. J Immunol. 2007;179:6856–6866. doi: 10.4049/jimmunol.179.10.6856. [DOI] [PubMed] [Google Scholar]
- 31.Natarajan K, Boyd LF, Schuck P, Yokoyama WM, Eliat D, Margulies DH. Interaction of the NK cell inhibitory receptor Ly49A with H-2Dd: identification of a site distinct from the TCR site. Immunity. 1999;11:591–601. doi: 10.1016/s1074-7613(00)80134-x. [DOI] [PubMed] [Google Scholar]
- 32.Franksson L, Sundbäck J, Achour A, Bernlind J, Glas R, Kärre K. Peptide dependency and selectivity of the NK cell inhibitory receptor Ly-49C. Eur J Immunol. 1999;29:2748–2758. doi: 10.1002/(SICI)1521-4141(199909)29:09<2748::AID-IMMU2748>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 33.Scarpellino L, Oeschger F, Guillaume P, Coudert JD, Lévy F, Leclercq G, Held W. Interactions of Ly49 family receptors with MHC class I ligands in trans and cis. J Immunol. 2007;178:1277–1284. doi: 10.4049/jimmunol.178.3.1277. [DOI] [PubMed] [Google Scholar]
- 34.Hanke T, Takizawa H, McMahon CW, Busch DH, Pamer EG, Miller JD, Altman JD, Liu Y, Cado D, Lemonnier FA, Bjorkman PJ, Raulet DH. Direct assessment of MHC class I binding by seven Ly49 inhibitory NK cell receptors. Immunity. 1999;11:67–77. doi: 10.1016/s1074-7613(00)80082-5. [DOI] [PubMed] [Google Scholar]
- 35.Falk K, Rotzschke O, Stevanovic S, Jung G, Rammensee HG. Allele-specific motifs revealed by sequencing of self-peptides eluted from MHC molecules. Nature. 1991;351:290–296. doi: 10.1038/351290a0. [DOI] [PubMed] [Google Scholar]
- 36.Molano A, Erdjument-Bromage H, Fremont DH, Messaoudi I, Tempst P, Nikolic-Zugic J. Peptide selection by an MHC H-2Kb class I molecule devoid of the central anchor (“C”) pocket. J Immunol. 1998;160:2815–2823. [PubMed] [Google Scholar]
- 37.Rudolph MG, Stevens J, Speir JA, Trowsdale J, Butcher GW, Joly E, Wilson IA. Crystal structures of two rat MHC class Ia (RT1-A) molecules that are associated differentially with peptide transporter alleles TAP-A and TAP-B. J Mol Biol. 2002;324:975–990. doi: 10.1016/s0022-2836(02)01095-1. [DOI] [PubMed] [Google Scholar]
- 38.Shields MJ, Hodgson W, Ribaudo RK. Differential association of β2-microglobulin mutants with MHC class I heavy chains and structural analysis demonstrate allele-specific interactions. Mol Immunol. 1999;36:561–573. doi: 10.1016/s0161-5890(99)00077-2. [DOI] [PubMed] [Google Scholar]
- 39.Li H, Natarajan K, Malchiodi EL, Margulies DH, Mariuzza RA. Three-dimensional structure of H-2Dd complexed with an immunodominant peptide from human immunodeficiency virus envelope glycoprotein 120. J Mol Biol. 1998;283:179–191. doi: 10.1006/jmbi.1998.2091. [DOI] [PubMed] [Google Scholar]
- 40.Ribaudo RK, Margulies DH. Polymorphism at position nine of the MHC class I heavy chain affects the stability of association with beta 2-microglobulin and presentation of a viral peptide. J Immunol. 1995;155:3481–3493. [PubMed] [Google Scholar]
- 41.Deng L, Cho S, Malchiodi EL, Kerzic MC, Dam J, Mariuzza RA. Molecular architecture of the MHC-binding site of Ly49 natural killer cell receptors. J Biol Chem. 2008;283:16840–16849. doi: 10.1074/jbc.M801526200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Logdberg L, Bjorck L, Cigen R. Heterologous interaction between beta 2-microglobulin and Ag-B antigens. Transplantation. 1980;30:233–235. doi: 10.1097/00007890-198009000-00017. [DOI] [PubMed] [Google Scholar]
- 43.Mitsuki M, Matsumoto N, Yamamoto K. A species-specific determinant on β2-microglobulin required for Ly49A recognition of its MHC class I ligand. Int Immunol. 2004;16:197–204. doi: 10.1093/intimm/dxh017. [DOI] [PubMed] [Google Scholar]
- 44.Logdberg L, Bjorck L. On the heterologous interaction between beta 2-microglobulin and the heavy chain of rat major histocompatibility complex class 1 antigens. Scand J Immunol. 1984;20:61–68. doi: 10.1111/j.1365-3083.1984.tb00978.x. [DOI] [PubMed] [Google Scholar]
- 45.Boyington JC, Motyka SA, Schuck P, Brooks AG, Sun PD. Crystal structure of an NK cell immunoglobulin-like receptor in complex with its class I MHC ligand. Nature. 2000;405:537–543. doi: 10.1038/35014520. [DOI] [PubMed] [Google Scholar]
- 46.Fan QR, Long EO, Wiley DC. Crystal structure of the human natural killer cell inhibitory receptor KIR2DL1-HLA-Cw4 complex. Nat Immunol. 2001;2:452–460. doi: 10.1038/87766. [DOI] [PubMed] [Google Scholar]
- 47.Fadda L, Borhis G, Ahmed P, Cheent K, Pageon SV, Cazaly A, Stathopoulos S, Middleton D, Mulder A, Claas FH, Elliott T, Davis DM, Purbhoo MA, Khakoo SI. Peptide antagonism as a mechanism for NK cell activation. Proc Natl Acad Sci USA. 2010;107:10160–10165. doi: 10.1073/pnas.0913745107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Torres-Nagel N, Kraus E, Brown MH, Tiefenthaler G, Mitnacht R, Williams AF, Hunig T. Differential thymus dependence of rat CD8 isoform expression. Eur J Immunol. 1992;22:2841–2848. doi: 10.1002/eji.1830221113. [DOI] [PubMed] [Google Scholar]
- 49.Laugel B, van den Berg HA, Gostick E, Cole DK, Wooldridge L, Boulter J, Milicic A, Price DA, Sewell AK. Different T cell receptor affinity thresholds and CD8 coreceptor dependence govern cytotoxic T lymphocyte activation and tetramer binding properties. J Biol Chem. 2007;282:23799–23810. doi: 10.1074/jbc.M700976200. [DOI] [PubMed] [Google Scholar]
- 50.Wang R, Natarajan K, Margulies DH. Structural basis of the CD8 αβ/MHC class I interaction: focused recognition orients CD8β to a T cell proximal position. J Immunol. 2009;183:2554–2564. doi: 10.4049/jimmunol.0901276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang J, Edwards LJ, Evavold BD, Zhu C. Kinetics of MHC-CD8 interaction at the T cell membrane. J Immunol. 2007;179:7653–7662. doi: 10.4049/jimmunol.179.11.7653. [DOI] [PubMed] [Google Scholar]
- 52.Reynolds CW, Sharrow SO, Ortaldo JR, Herberman RB. Natural killer activity in the rat. II. Analysis of surface antigens on LGL by flow cytometry. J Immunol. 1981;127:2204–2208. [PubMed] [Google Scholar]
- 53.Silver ET, Gong DE, Chang CS, Amrani A, Santamaria P, Kane KP. Ly-49P activates NK-mediated lysis by recognizing H-2Dd. J Immunol. 2000;165:1771–1781. doi: 10.4049/jimmunol.165.4.1771. [DOI] [PubMed] [Google Scholar]
- 54.Van Bleek GM, Nathenson SG. Isolation of an endogenously processed immunodominant viral peptide from the class I H-2Kb molecule. Nature. 1990;348:213–216. doi: 10.1038/348213a0. [DOI] [PubMed] [Google Scholar]
- 55.Meiring HD, Soethout EC, Poelen MC, Mooibroek D, Hoogerbrugge R, Timmermans H, Boog CJ, Heck AJ, de Jong AP, van Els CA. Stable isotope tagging of epitopes: a highly selective strategy for the identification of major histocompatibility complex class I-associated peptides induced upon viral infection. Mol Cell Proteomics. 2006;5:902–913. doi: 10.1074/mcp.T500014-MCP200. [DOI] [PubMed] [Google Scholar]