

Abstract
Purpose
This study evaluates the Argus™ II Retinal Prosthesis System in blind subjects with severe outer retinal degeneration.
Design
The study design is a single arm, prospective, multicenter clinical trial.
Participants
Thirty subjects were enrolled in the United States and Europe between 6 June 2007 and 11 August 2009. All subjects were followed for a minimum of six months and up to 2.7 years.
Methods
The electronic stimulator and antenna of the implant was sutured onto the sclera using an encircling silicone band. Next, a pars plana vitrectomy was performed and the electrode array and cable were introduced into the eye via a pars plana sclerotomy. The microelectrode array was then tacked to the epiretinal surface.
Main Outcome Measures
The primary safety endpoint for the trial was the number, severity, and relation of adverse events. Principal performance endpoints were assessments of visual function as well as performance on orientation and mobility tasks.
Results
Subjects performed statistically better with system ON vs. OFF in the following tasks: object localization (96% of subjects); motion discrimination (57%); and discrimination of oriented gratings (23%). The best recorded visual acuity to date is 20/1260. Subjects’ mean performance on Orientation and Mobility tasks was significantly better when the System was ON vs. OFF.
Seventy percent of the patients did not have any serious adverse events (SAEs). The most common SAE reported was either conjunctival erosion or dehiscence over the extraocular implant and was successfully treated in all subjects except in one which required explantation of the device without further complications.
Conclusions
The long-term safety results of Second Sight’s retinal prosthesis system are acceptable and the majority of subjects with profound visual loss perform better on visual tasks with system than without.
Introduction
Several different treatment avenues using biological and bioelectronic approaches have been proposed to restore sight to the blind.[1–4]
Some of the major challenges for bioelectronic implants include long-term stable performance of the implanted electronics as well as a safe surgical implantation procedure. Previous studies have shown that electrical stimulation of the retinal ganglion cell side (epiretinal stimulation) can produce discrete phosphenes and that spatial resolution and partial restoration of vision is possible.[5–13] Herein, from an ongoing international clinical trial evaluating the Argus II Retinal Prosthesis System (Second Sight Medical Products, Inc., Sylmar, CA), we report our experience from 45.6 cumulative subject-years in 30 subjects implanted at 10 clinical centers.
Methods
Statement of compliance
This multi-center feasibility study for Second Sight’s retinal prosthesis system is being conducted in accordance with the Declaration of Helsinki and the national regulations for medical device clinical trials in the countries in which the study is being conducted. The study has been approved by the national ministries of health in these countries and the Ethics Committees or Institutional Review Boards of participating institutions. All subjects signed informed consent to participate. The clinical trial is posted on www.clinicaltrials.gov, trial registration number NCT00407602.
Purpose of Study and Description of Subjects
The study is a single-arm, prospective, unmasked study to evaluate the safety and utility of the prosthesis in providing functional vision to blind subjects with end-stage outer retinal degenerations.
A total of 32 subjects have been implanted with the prosthesis. The first two subjects were part of a pilot study in Mexico (the first country to grant regulatory approval for clinical use); because these subjects received a significantly different device (the electrode array was placed outside of the macula), this report will focus on the 30 subjects who had an electrode array that could be placed at least partly in the macular region. These 30 subjects were 50 years or older (18 or older at some clinical sites) with a diagnosis of retinitis pigmentosa (or other outer retinal degeneration at some sites; one participant had Leber Congenital Amaurosis and one had choroideremia) with remaining vision of bare or no light perception (visual acuity worse than 2.9 log MAR in both eyes). All subjects had a history of useful form vision. Exclusion criteria addressed any inability to implant the device physically, concurrent complicating ocular pathology, and any inability to commit to the expectations and duration of the study. Refer to www.clinicaltrials.gov for full subject selection criteria.
Subjects had a median age of 57.5 ± 9.9 (range 27 – 77) at the time of implantation, and all but one subject were at least 45 years old. Thirty percent (30%) of subjects were female and 70% were male. Some subjects (33%) had undergone previous cataract removal surgery in the implanted eye and one subject had had several previous ocular surgeries in the implanted eye (prior pars plana vitrectomy to clear vitreous debris and sub-conjunctival placental tissue injections). The last 15 subjects were implanted with a modified device that was slightly re-engineered to have a more flexible cable and an electrode array that allowed closer apposition of the electrodes to the underlying retina. All subjects were followed for a minimum of six months and up to 2.7 years.
Description of Device
The Argus II Retinal Prosthesis System consists of an active implantable device surgically implanted on and in the eye, and an external unit worn by the user. The external unit consists of a small camera and transmitter mounted on glasses and a video processor and battery worn on the belt or a shoulder strap (Figure 1A).
Figure 1.
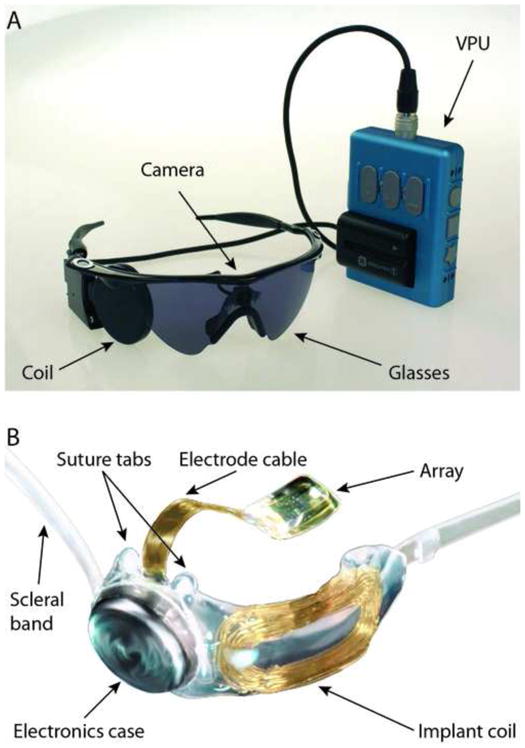
(A) A photograph of the external portion of the Argus II prosthesis system including glasses-mounted video camera, radio frequency (RF) coil, and Video Processing Unit (VPU) with rechargeable battery. (B) A photograph of the implanted portion of Argus II prosthesis system including the 6×10 electrode array, electronics case, and implant RF coil.
The implanted portion (Figure 1B) consists of a receiving and transmitting coil and electronics case (which are fixed to the sclera outside of the eye), and an electrode array (60 electrodes) that is surgically positioned onto the surface of the retina by a retinal tack. The electrode array is connected to the electronics case by a metallized polymer ribbon cable that penetrates the sclera in the pars plana. The camera captures video and sends the information to the processor, which converts the image to electronic signals that are then sent to the transmitter coil on the glasses. The episcleral implanted receiver coil/antenna wirelessly receives these data and sends the signals via the ribbon cable to the electrode array, where electrical stimulation pulses are emitted. The spatially controlled microelectrode electrical stimulation of the retinal cells induces cellular responses in the retina that travel through the optic nerve to the central visual system, resulting in visual percepts.
Although magnification or zoom is possible in the system, for this study, the field of view of the camera was cropped to match the predicted visual field of the array on the retina (assuming 1° visual angle = 300 μm on the retina)[14]. The cropped image was then down-sampled to 10×6 pixels. The pixels were mapped 1:1 onto the electrodes of the implanted array, so the chosen field of view of the camera matched the field of view of the array – approximately 20° on the diagonal.
Surgery
At the start of the implant procedure, 8 mg of dexamethasone (to reduce inflammation) and 1 g cefazolin (or equivalents) were administered by intravenous injection. In phakic eyes, the lens was removed via clear cornea phacoemulsification (with the exception of pars plana lensectomy in one subject). Next a 360-degree limbal conjunctival peritomy was performed followed by isolating the rectus muscles using 2-0 black silk.
The coil was placed temporally on the globe and centered under the lateral rectus muscle. The electronics package was centered in the superior temporal quadrant. The inferior part of the scleral band was passed under the inferior and the medial rectus muscles, and the superior portion of the band under the superior rectus muscle. The implant was fixed to the eye via sutures passed through suture tabs on the implant in both temporal quadrants and with the use of mattress sutures around the encircling band in the nasal quadrants with the Watzke® sleeve (Labtician Ophthalmics, Inc., Oakville, Ontario, Canada) positioned in the superonasal quadrant.
Core and peripheral vitrectomies were performed and were followed by dissection of any epiretinal membrane in the area where the surgeon intended to tack the array. The microelectrode array was then inserted through a temporal sclerotomy (approximately 5 mm in width) and was placed onto the retina in the macula and then tacked using a custom retinal tack (Second Sight Medical Products, Inc., Sylmar, CA). The extraocular portion of the cable was sutured to the sclera and all sclerotomies were sutured.
An allograft (Tutoplast®, IOP Inc., Costa Mesa, CA), or suitable alternative in countries where allografts were not permitted, was sutured and draped over the electronics package to reduce the likelihood of conjunctival irritation. Finally, the Tenon’s capsule and the conjunctiva were sutured.
At the end of the surgery, 2 mg of dexamethasone, 100 mg of cefazolin, and 2 mlof lidocaine (or equivalents) were injected under the conjunctiva. Midway through the trial, in order to reduce the likelihood of endophthalmitis, the surgical procedure was modified by the addition of prophylatic intravitreal injections of antibiotics (0.1cc intravitreal vancomycin [1 mg/0.1cc] and ceftazidime [2.25 mg/0.1 cc]) at the end of the implant procedure.
Post-operatively the following medications were administered per protocol: 500 mg Ciprofloxacin twice a day for 7–14 days, 1 drop Gatifloxacin four times a day for 7–14 days, 60mg daily Prednisolone (orally) for 2 weeks, immediately followed by a Methylprednisolone (Medrol) taper pack (8mg), until the pack was completed (or equivalent taper of Prednisolone), 1 drop Pred Forte 1% four times a day for 2 weeks, and 1 drop daily Atropine 1% for 2 weeks.
Clinical Evaluation
Subjects were evaluated on Day 1, Weeks 1, 2 and 4, and Months 3, 6, 9, 12, 18, 24, 30 and 36. At each of these follow-up time points, complete eye exams (including measurement of intraocular pressure), retinal fundus photography, fluorescein angiography, and optical coherence tomography (OCT) were performed.
Serious device- or surgery- related events were reported to the relevant competent authorities and ethics committees in accordance with the local reporting requirements. During the trial, all adverse events were subject to detailed review by an independent medical safety monitor (IMSM), both as individual events and collated data.
Subjects were allowed to use the system outside outpatient clinical setting in their daily lives once it had been individually programmed and they had completed training (usually after the first month post implantation).
Full-field stimulus light threshold
Subjects’ residual native light perception (i.e., without the use of the prosthesis) was measured before and after implantation using the following protocol: The subjects’ eyes were dilated and dark-adapted for 30 minutes. Monocular thresholds were obtained by patching the other eye during testing. Dark-adapted light thresholds of implanted and fellow eyes to full-field white light stimuli were measured using the Espion D-FST test within the commercially available E2 clinical electrophysiology software package (version 5.0; Diagnosys LLC, Littleton, Massachusetts). In one center, custom-written software was used to obtain FST thresholds.[15, 16] Further details are provided in Appendix 2 (available online at http://aaojournal.org). Subjects at sites without an Espion system or with no measureable threshold below the maximum luminance provided by the system were tested for having residual bare light perception (BLP) using a photographic camera flash (Uniblitz 82ABSZ; Rochester, NY). This method used a forced-choice paradigm with 20 blocks, with four presentations per block. A 95% significance criterion was used to determine if a subject was BLP (i.e.,≥9/20 correct blocks. According to the binomial distribution, given a chance rate of 0.25, the probability (p) of scoring 9 or more correct out of 20 by chance is less than 0.05).
Outside Outpatient Clinic Use
An important objective in the months following implantation was to have the subjects start using their system outside outpatient clinical setting. As soon as possible after implantation, subjects were trained to independently set up and use the system, and to respond to audible alarm states (e.g., low battery alarm). Subjects were also trained to use the system to perform activities of daily living (video clips 1 and 2 showing subjects using their systems to perform tasks both indoors and outdoors are available at http://aaojournal.org).
Electrode Reliability
Although all microelectrode arrays comprised 60 electrodes, the number of enabled electrodes (i.e., electrodes available for stimulation) in the delivered, finished device in this clinical trial varied from 46 to 60 due to a conservative policy of shutting off electrodes that didn’t meet stringent electrical stimulation criteria. The median number of enabled electrodes at the time of implant was 55. Following implantation, impedance measurements on each electrode were used to determine if additional electrodes should be disabled or if previously disabled electrodes should be re-enabled.
Tests of Visual Acuity and Real-world Utility
Three types of visual acuity tests were performed using computer monitors. In “Square Localization,” subjects were asked to localize a white square on a black background, in “Direction of Motion,” subjects were asked to indicate the path of a white line swept across a black background, and in “Grating Visual Acuity,” subjects were asked to differentiate the orientation of black and white bars of a range of widths. Two types of real-world utility tests were performed. In the “Door Test,” subjects were asked to find a door across a room, and in the “Line Test,” subjects were asked to follow a white line on the floor. For further details on these testing methods, please see Appendix 2 (available online at http://aaojournal.org).
Results
Surgery
If the subjects’ eyes did not have equal visual acuity at baseline, implantation was performed in their worse-seeing eye. If the subjects’ eyes had equal visual acuity, the right eye was selected for implantation. Twenty-six subjects were implanted in their right eye, and four were implanted in their left eye.
During the implantation procedure, 67% of subjects had their natural lens removed via clear cornea phacoemulsification (with the exception of pars plana lensectomy in two cases) and they were left aphakic (no lens). Patients with lens implants (i.e., pseudophakic) did not undergo lens explantation with the exception of one case in whom the intraocular lens was subluxed prior to surgery necessitating its removal. The width of the sclerotomy where the cable was inserted averaged 5.0 ± 0.5 mm (range 4.5 – 6.0 mm). Fifty-seven percent (57%) of subjects had either a well-adhered posterior hyaloid and/or an epiretinal membrane which required peeling. Most subjects had an allograft (either Tutoplast® (IOP Inc.; Costa Mesa, CA) sclera [57%] or Tutoplast® (IOP Inc.; Costa Mesa, CA) pericardium [30%]) placed over the extraocular portion of the device (under the conjunctiva) at the end of the surgery. France does not permit the use of these allografts, so the subjects in France received either a polytetrafluoroethylene (PTFE or Teflon®) patch (one subject) or an autologous aponeurosis graft (three subjects).
The median implant surgery time was 4 hours, 4 minutes (range 1:53 to 8:32 hours). The longest implant procedure was prolonged by the fact that the subject had had several previous surgeries on his implanted eye (prior pars plana vitrectomy to clear vitreous debris and sub-conjunctival placental injections) which had resulted in extensive conjunctival scarring. In addition, this subject’s lateral rectus muscle was fibrosed and disinserted in these prior surgeries and required re-insertion. Figure 2A shows a fundus photograph of an implanted array, and Figure 2B shows an Optical Coherence Tomagraphy image of an implanted array.
Figure 2.
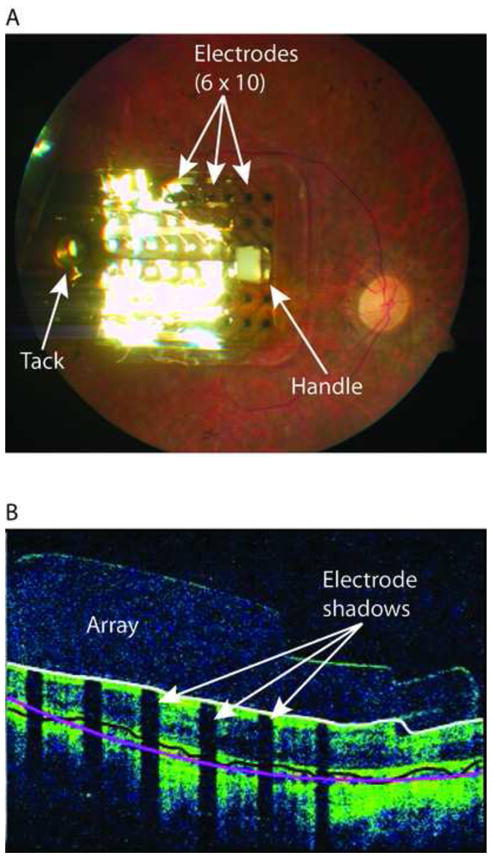
(A) Fundus photograph of implanted Argus II array in the macular region. The electrode array is secured to the retina with a retinal tack; the white square visible on the distal side of the array is an opaque section of tubing (the “handle”) used by the surgeon to position the array. (B) An optical coherence tomography (OCT) image of an implanted Argus II array. Shadows cast on the retinal image (white arrows) are due to occlusion of the scanning light source by the metal electrodes.
Clinical Safety
Serious Adverse Events
Serious adverse events (SAEs) were defined according to ISO 14155 as medical occurrences that either: caused death; were life threatening; caused permanent impairment of a body function or permanent damage to body structure, or necessitated medical or surgical intervention to preclude such impairment or damage; required hospitalization or prolonged hospitalization; or caused fetal death or abnormality (though pregnancy was an exclusion criterion).There were 17 serious adverse events that were determined to be device or surgery related as of 1 March 2010. Table 1 presents a summary of these events. SAEs were often clustered (i.e., more than one event occurred in the same subject) and 70% of subjects did not experience any SAEs.
Table 1.
Serious Adverse Events (Device- or Surgery-Related)
| Serious Adverse Events | All Subjects (n = 30) | Last 15 subjects enrolled in study (n = 15) | |
|---|---|---|---|
| # Subjects with Event | 95% Confidence Interval | # Subjects with Event | |
| Conjunctival dehiscence | 3 | 2.1 – 26.5% | 1 |
| Conjunctival erosion | 2 | 0.8 – 22.1% | 0 |
| Presumed Endophthalmitis | 3 | 2.1 – 26.5% | 0 |
| Hypotony | 3 | 2.1 – 26.5% | 1 |
| Re-tack | 2 | 0.8 – 22.1% | 1 |
| Retinal detachment - rhegmatogenous | 1 | 0.1 – 17.2% | 1 |
| Retinal Detachment - tractional | 1 | 0.1 – 17.2% | 0 |
| Retinal Tear | 1 | 0.1 – 17.2% | 0 |
| Uveitis – inflammatory | 1 | 0.1 – 17.2% | 0 |
The vast majority of SAEs occurred within the first 6 months post-implant. Eighty-two percent (82% or 14/17) of SAEs occurred within the first 6 months post-implant, and 70% (12/17) occurred within the first 3 months post-implant. Subjects enrolled later in the study experienced fewer SAEs (n = 4 SAEs in 2 subjects) than those enrolled earlier in the study (n = 13 SAEs in 7 subjects). This improvement was attributed to improvements in the surgical technique and minor design improvements made midway through the study.
Conjunctival erosion and dehiscence over the extraocular implant, when combined, were the most common occurrences and were treated in all but one subject with additional sutures and/or placement of additional tissue (conjunctiva or sclera). In one subject, during the repair the suture tab on the device was damaged, precluding the ability to restore the device; after recurrent erosions, this device was explanted without any further complications.
Culture-negative presumed endophthalmitis occurred and resolved in three subjects in the first group of 15 subjects. None of the cases were associated with observed pre-existing conjunctival erosion or hypotony. All cases were treated with intravitreal (0.1cc vancomycin [1 mg/0.1cc] and ceftazidime [2.25 mg/0.1 cc]) subconjunctival, topical, and systemic antibiotics.
The first endophthalmitis case developed in the very immediate post-operative period in a subject from a U.S. site. The subject was treated with intravitreal vancomycin and ceftazidime as well as oral tablets of moxifloxacin. At week 1, antibiotics (moxifloxacin tablets and topical gatifloxacin) were tapered and Pred Forte (prednisolone acetate) was given over the next week. The second and third endophthalmitis cases developed approximately five and eight weeks post-operatively. These subjects were implanted at the same center in the U.K. on the same day. The first of these subjects was treated with intravitreal injections of amikacin and vancomycin, topical application of chloramphenicol and dexamethasone, and oral tablets of moxifloxacin. The final subject with endophthalmitis was treated with intra-vitreal vancomycin, ceftazidime, and amphotericin B. It should be noted that although the second and third subjects were implanted at the same center on the same day, the intravitreal antibiotics chosen for the second subject were different from the third. The principal investigator consulted with the infectious disease specialist who recommended the use of ceftazidime instead of amikacin and the addition of an antifungal. The subject also continued oral tablets of moxifloxacin and prednisolone, as well as topical chloramphenicol and dexamethasone. Four days later, the subject received a second round of intra-vitreal vancomycin and ceftazidime.
None of the presumed endophthalmitis cases required explantation of the device, and none occurred in subjects implanted later in the trial (the last 15 subjects) after a protocol change was implemented that included prophylactic intravitreal antibiotics at the end of the case.
In the trial, hypotony was defined as intraocular pressure (IOP) <5 mmHg that persisted for greater than two weeks or shorter duration if the low IOP was associated with appositional choroidals or with a flat anterior chamber. Three subjects had hypotony that required surgical intervention. Of these three cases, two occurred within the first six months (at 1 and 4 months) of implantation and the third at one year in the patient whose suture to secure the implant had broken and whose device had migrated anteriorly. As described above, this third subject’s device was eventually explanted, which led to normalization of the IOP. Of the other two subjects, one was treated with intraocular silicone oil tamponade, which normalized the IOP. The second had an associated rhegmatogenous retinal detachment requiring repair; the subject was later treated with silicone oil tamponade, which resulted in stabilization of the IOP between 6–7 mmHg.
Two cases of retinal detachment, which eventually required surgical intervention to treat, occurred at the 5–6 month post-implant period. The first had a rhegmatogenous detachment, associated with 360 circumferential bands and choroidal effusion; this is the same subject described above. At approximately 5 months post-operatively, a second subject incurred blunt trauma to the implanted eye resulting in proliferative vitreoretinopathy which progressed to a tractional retinal detachment. The RD was successfully repaired with vitrectomy, partial retinectomy, and silicone oil.
Two subjects required the array to be re-tacked to the retina shortly after the implant surgery. In both cases it became apparent in the first few days post-operatively that the tack was not securely implanted at the time of the initial surgery. In both cases the tack was successfully re-attached near the same retinal site.
Non-serious Adverse Events
Non-serious adverse events (non-SAEs) were those events related to the device or surgery that did not require surgical intervention (they resolved after treatment with topical or oral medications, or did not require any treatment). Conjunctival edema that was considered to be more extensive or lasting longer than what is typically seen post operatively occurred in 10 subjects and was considered a non-serious adverse event. The following non-SAEs occurred in 5–7 subjects: intraocular inflammation, hypotony without significant choroidal detachments, suture irritation, and ocular pain (mostly foreign body sensation). The following non-SAEs occurred in 2–3 people: inflammatory conjunctivitis, corneal filaments, epiretinal membrane, high intraocular pressure controlled by topical anti-glaucoma medications, epiphora, mild hyphema, inflammatory uveitis with few keratic precipitates, and mild vitreous hemorrhage. The following non-SAEs had only a single occurrence and resolved: limited conjunctival dehiscence, corneal abrasion, mild peripheral corneal vascularization, cystoid macular edema (CME), decrease in light perception, dry eye, transient headache, iris vessel engorgement that receded secondary to surgery to resuture sclerotomy (to treat hypotony), a stable tractional retinal detachment, transient nausea, transient increased nystagmus, scleritis, and transient vertigo.
Full-field stimulus light threshold
Across all subjects measured there was no significant difference between threshold obtained before and after surgery for both implanted and fellow eyes (Student’s two-tailed, paired t-test, p > 0.05).
All but one subject had bare light perception (BLP) in both eyes prior to implant. The one subject who had no light perception (NLP) recorded in one eye before implantation was subsequently was categorized as BLP in that eye after implantation, when the photographic flash test was available. Ninety-three percent (93%) of subjects still had BLP in both eyes as of the latest follow-up time point (as of November 30 2010). Of the two whose vision declined from BLP to no light perception, one showed the decline in both eyes, and one declined only in the non-implanted eye. For subjects with quantifiable light thresholds in both eyes, implanted and fellow eye thresholds were correlated (R2=0.41, p<0.01), thus providing validation of method.
Outside Outpatient Clinical Use
Subjects took the system home at an average of 2.3 ± 0.7 months post-implant (range 1.4 – 3.7 months; median 2.1 months). As of March 1, 2010, 29 out of 30 subjects were using the system at home (one subject’s device was explanted as described above), and subjects had been using their systems at home for an average of 15.8 ± 9.7 months (range 4.2 – 28.8 months; median 14.3 months).
Electrode Reliability
The implants are designed with an electrode array that contains 60 electrodes arranged in a rectangular grid of 6 × 10. 94.4% of the electrodes that were enabled at the time of implant remained enabled and functional throughout the study (as of March 1, 2010).
Perception Thresholds for Electrical Stimulation
All subjects (100%) were able to perceive light when their Systems were stimulated (thresholds were measurable on at least one electrode). An average of 55.5% (standard deviation = 32.0) of all enabled electrodes across subjects had measurable thresholds below a charge-density of 1.0 mC/cm2.
Square Localization
Figure 3 shows the mean distance from the center of the target (accuracy) for System ON and OFF for each subject at the latest follow-up time point (the smaller the mean distance, the closer the subject’s response was to the target). These data show that, as of the most recent follow-up time point, 27 out of 28 subjects (96%) perform this test better with System ON versus OFF, and no subjects performed significantly better with the System OFF.
Figure 3.
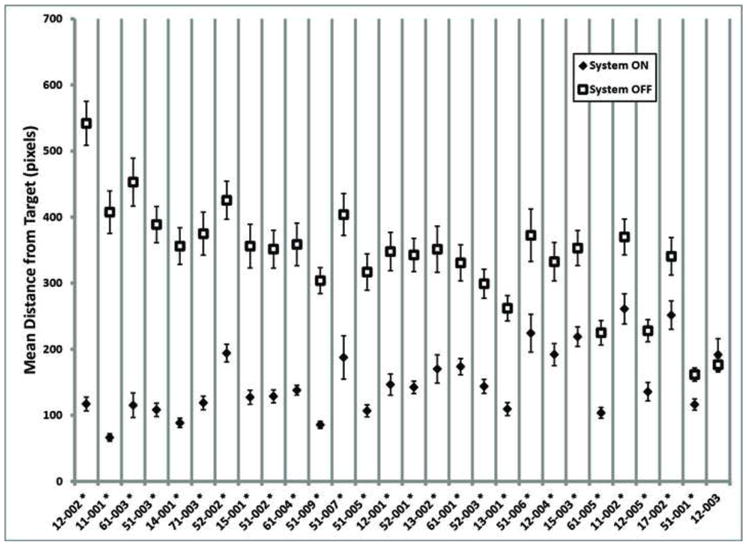
Mean accuracy on the Square Localization task for each subject, System ON (filled diamonds) and OFF (open squares). Subjects are ordered along the x-axis from greatest difference between ON and OFF performance to least difference. Error bars indicate standard error. Asterisks indicate subjects for whom the mean System ON performance was significantly different from the mean System OFF performance (t-test assuming unequal variances, two-tailed, p<0.05). Data are the latest available for each subject as of 1 March 2010.
Direction of Motion
Figure 4 shows the mean response error (stimulus angle minus the response angle) with the System ON and OFF for each subject at the latest follow-up time point (the smaller the mean response error, the closer the subject’s response was to the stimulus direction). These data show that 16 out of 28 subjects (57%) perform this test better with System ON versus OFF.
Figure 4.
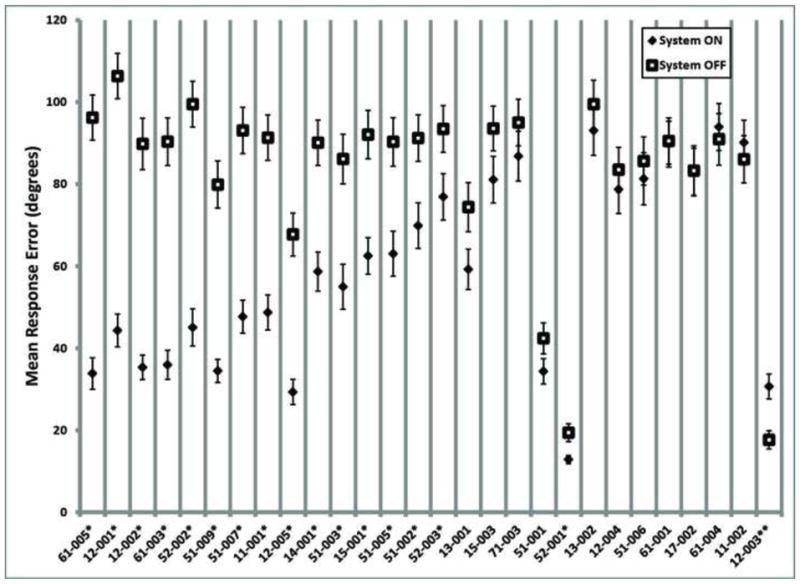
Mean response error for each subject on the Direction of Motion task, System ON (filled diamonds) and system OFF (empty squares). Subjects are ordered along the x-axis from greatest difference between ON and OFF performance to least difference. Single asterisks indicate subjects for whom the System ON mean was significantly different (smaller) than the System OFF mean, and the double asterisk indicates a subject for whom System OFF was significantly different (smaller) than System ON (t-test assuming unequal variances, two-tailed, p<0.05). Data are the latest available for each subject as of 1 March 2010.
Grating Visual Acuity
Per the protocol inclusion criteria, all subjects’ acuity was measured at worse than 2.9 log MAR – off the acuity scale used for this test – in both eyes before implantation (at month 0). To date, none of the subjects have been able to reliably score on the visualacuity scale in either eye with the System OFF. Seven subjects have been able to reliably score on the scale (with visual acuity between 2.9 and 1.6 log MAR with the System ON. The best result to date is 1.8 log MAR (equivalent to Snellen 20/1262). Note, these tests were performed without magnification or zoom.
Orientation and Mobility
The observed results at each time point for the ON vs. OFF conditions are shown in Figure 5 and Figure 6 for the Door and Line Tasks, respectively. A repeated measures analysis of variance (RM ANOVA) model was used to compute and compare the difference between the mean success rates (over all subjects) for the Door task with System ON and OFF at each follow-up time point. These results are provided in Table 2 and Table 3.
Figure 5.
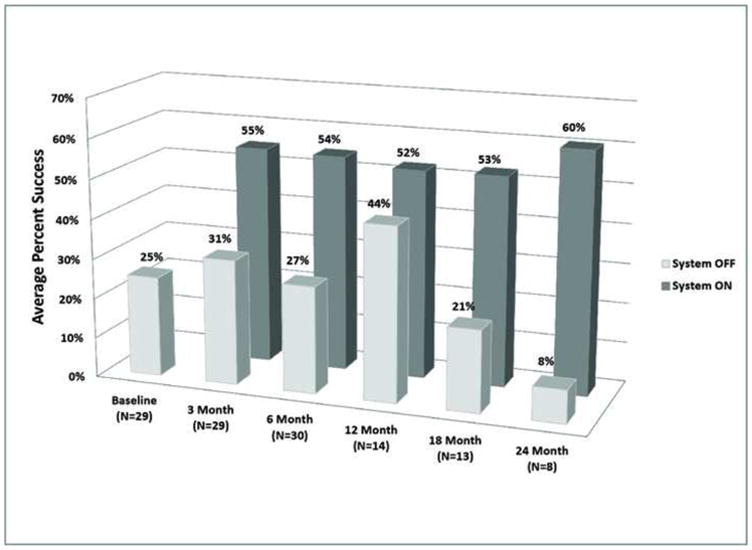
Average percent success at each clinical visit for the “find the door” orientation and mobility task.
Figure 6.
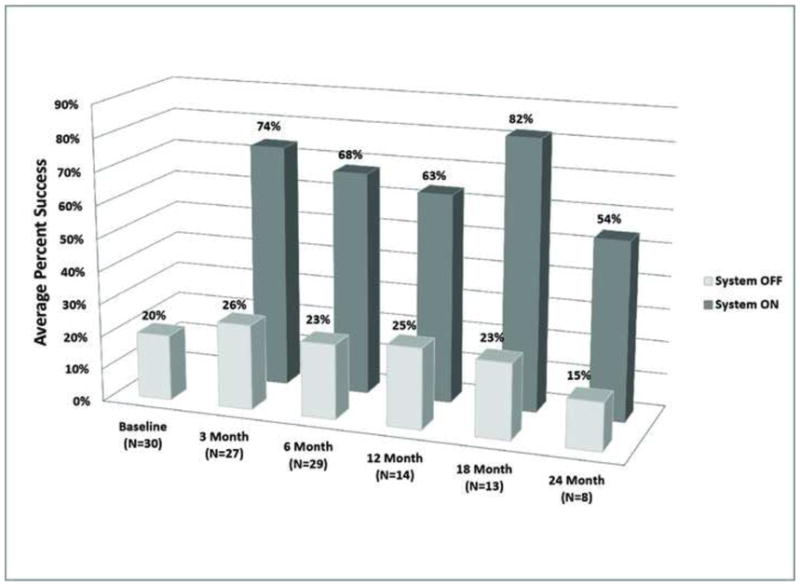
Average percent success at each clinical visit for the “follow the line” orientation and mobility task.
Table 2.
Repeated Measure Analysis of Variance (RM ANOVA) - Door Task
| Door Task | Baseline | 3 Month* | 6 Month* | 12 Month | 18 Month* | 24 Month* |
|---|---|---|---|---|---|---|
| RM ANOVA Difference (On-Off) in Mean % Success |
Not applicable | 24% | 27% | 10% | 32% | 48% |
| P-value | Not applicable | 0.001 | 0.0001 | N/S | 0.002 | 0.0004 |
statistical analysis of the door task results. Mean percent-rates success with the system ON minus the mean percent-success rates with the System OFF (over all subjects) at each time point. Means were computed using a repeated measure analysis of variance model as described in Appendix B. The mean percent differences were tested against zero for each time point (i.e., differences significantly greater than zero indicate that the outcome was significantly better with the system ON compared to OFF).Means were significantly different for all time points except at 12 months post-implant (asterisks). Due to different enrollment times and a method change partway through the study (to make the task more difficult to perform by chance), the 12-month clinical visit consisted of a larger proportion of subjects using the new (more difficult) method.
Table 3.
Repeated Measure Analysis of Variance (RM ANOVA) - Line Task
| Line Task | Baseline | 3 Month* | 6 Month* | 12 Month* | 18 Month* | 24 Month* |
|---|---|---|---|---|---|---|
| RM ANOVA Difference (On-Off) in Mean % Success |
Not applicable | 48% | 45% | 44% | 65% | 42% |
| P-value | Not applicable | <0.0001 | <0.0001 | 0.0002 | <0.0001 | 0.005 |
statistical analysis of the line task results. Mean percent-rates success with the system ON minus the mean percent-success rates with the System OFF (over all subjects) at each time point. Means were computed using a repeated measure analysis of variance model as described in Appendix B. The mean percent differences were tested against zero for each time point (i.e., differences significantly greater than zero indicate that the outcome was significantly better with the system ON compared to OFF. Means were significantly different at all time points (asterisks).
This analysis demonstrated that with the exception of the 12-month time point, subjects’ performance on the Door test was significantly better with the System ON compared with System OFF (p < 0.05; indicated with asterisks).
This analysis demonstrated that subjects’ performance on the Line task with the System ON was significantly better (p<0.05; indicated with asterisks) than it was with the System OFF at all follow-up time points.
Discussion
This study represents 45.6 cumulative subject-years in 30 human subjects implanted with the Argus II Retinal Prosthesis System.
There are no other suitable retinal prostheses with which the safety or efficacy of this system can be compared. While other retinal prostheses are currently being developed by both commercial and academic entities, none of these devices are commercially approved, none are approved for longer than one month implantation, few are even in clinical trials, and none have published long-term clinical results or multi-center data. However, there are other commercially available ophthalmic devices that have some similar characteristics (e.g., extra-ocular or intra-ocular components, requirement for vitrectomy to install, among others) as this system. But even in this comparison, the adverse event rates quoted are reflective of rates for established and practiced therapies; the rates of adverse events for these same therapies at the time they were introduced to the market (similar to this device at this stage) would likely have been higher.
As an example, to help evaluate the incidence of conjunctival erosion in prosthesis subjects, glaucoma drainage devices (GDD, shunts ) are a potentially useful comparator device since, like this system, they have an intra- and extraocular portion and have a portion of similar volume implanted under the conjunctiva. Studies conducted by Gedde et al. and Lankaranian et al., report the rate of conjunctival erosion of GDDs as 5–16%, respectively.[17, 18] Gedde reported that wound dehiscence also occurred at a rate of 11%. In this trial, there were two cases of conjunctival erosion (6.7%) and three cases of conjunctival dehiscence (10.0%) which are within the range of those seen with GDDs.
The incidence of presumed endophthalmitis was 10% (3 of 30)and occurred within 2 months post-implant. In all three subjects, clinical symptoms were reported and signs of endophthalmitis were observed, although no positive cultures were identified and all three resolved. Although a comprehensive investigation was conducted for each incidence of presumed endophthalmitis no conclusive source of infection could be determined. Potential contributing factors included slightly longer surgical times than average in these cases, and a greater-than-usual number of personnel moving in and out of the operating room, some of whom did not wear face masks.
Typically, the incidence of post-surgical endophthalmitis in ophthalmic procedures is low due to sterile surgical technique. The literature reveals an endophthalmitis rate of 1–5% of subjects with glaucoma drainage devices.[18, 19]
With the addition of a temporary sleeve to cover the array region before it is introduced intraocularly, stricter sterile techniques during implant procedures especially in the handling of the implant, reduction of the number of observers present, and the routine use of prophylactic intraoperative broad spectrum antibiotics, the risk of endophthalmitis has been reduced. In fact, no cases of endophthalmitis were observed after these procedural changes (N = 15 cases). It is also important to note that none of these subjects’ implants were explanted and all remained functional after the presumed endophthalmitis events were treated with antibiotics. The presumed endophthalmitis cases occurred early (within two months post-implantation) and were not associated with conjunctival erosions so it is not likely that the conjunctival erosions led to the infections as has been seen in glaucoma filtering procedures. Moreover, there was no pocket of localized infection seen either atthe scleral entry site of the cable or around the extraocular device.
In one of the two subjects (2/30, or 6.7%) with retinal detachments, the subject had experienced a blunt trauma to the eye that most likely caused the retinal detachment. The second subject was a young subject (27 years old) with very adherent hyaloid which led to incomplete vitreous removal from posterior retina and subsequent tractional retinal detachment.
Again, it is difficult to find a large clinical trial in which the surgical procedure is similar to the one required for implantation of a retinal prosthesis. But in an effort to draw some comparisons, the following are the results from more complex procedures with implants: The rate of retinal detachment for sclerally fixated IOLs is reported between 8.5 – 9.5%.[20, 21] The rate of retinal detachment with Retisert implantation is between 1.5 – 2.2%,[22, 23] and with Vitrasert implantation has been reported as high as 13.8%[24]. Of course, one must consider that the target subject population for the Vitrasert (AIDS-related CMV) carried with it a higher risk of retinal detachment.
Finally, it is worth noting that there were two dislodged retinal tacks in this trial (6.7%). This is comparable to the percentage of dislodged retinal tacks reported in the literature (5.3%).[25]
As discussed above, the safety profile of the prosthesis, an active implantable device, is encouraging. The serious adverse event rates are comparable to similar implantable devices, particularly when considering that the comparator devices are mature, established therapies. Furthermore, in later enrollees (the second group of 15 subjects) there is a lower rate of adverse events, suggesting an improving safety profile even over the course of this study.
The stability of dark-adapted light perception of implanted and fellow eyes speaks to the stable mechanical and electrical interface between the electrode array and the underlying retina. Three of the four subjects who went from bare light perception (BLP) to no light perception (NLP) as determined by the photographic flash test did so in both implanted and fellow eyes, suggesting the loss in sensitivity may be due to the natural time course of disease progression. Note that in all four subjects the system has remained functional despite loss of light sensitivity.
Performance data are encouraging: threshold testing demonstrated that all subjects were able to perceive percepts when their implant was activated, and that they were able to do this throughout their entire follow-up duration to date.
Reliability of the prosthesis was also high. Over the follow up time period of this study, all but one device remain implanted and the vast majority of electrodes (94.4%) remain functional. All subjects who received an implant use their systems outside of outpatient clinical setting. In addition, 96% of subjects can localize high-contrast objects on a computer screen significantly better with the System ON than OFF; 57% can detect the direction of motion of a high-contrast bar significantly better with the System ON than OFF; 23% were able to score on a Grating Visual Acuity test; and subjects performed two orientation and mobility tasks significantly better with the System ON than OFF in all but one time point (12 months post-implant for the Door task). The exception was likely due to the change in method. Partway through the trial, the task was made harder to perform by chance (as described in Appendix 2, available online at http://aaojournal.org). Because subjects were implanted over the course of two years, each time point represents a slightly different population of subjects. Due to the time at which the method was changed, nearly all of the 14 subjects represented in the 12 month post-implant time point were tested with the old method. The corresponding higher chance rate (reflected in the higher average success rate with the system OFF seen in Figure 5) resulted in no significant difference between ON and OFF performance at this one time point.
In conclusion, the prosthesis system is reliable over the long-term (45.6 subject years so far in this study) and provided benefit to implanted subjects over this period. The data in this report suggest that on average, prosthesis subjects have improved visual acuity from light perception to at least hand motion, with some improving to at least counting fingers.[26, 27]
These visual acuity data combined with the safety and other performance results to date (e.g., da Cruz et al., Invest Ophthalmol Vis Sci 2010; ARVO E-Abstract) demonstrate the ability of this retinal implant to provide meaningful visual perception and utility to subjects blind due to end-stage outer retinal degenerations.
Supplementary Material
Acknowledgments
The authors would like to thank Suber Huang, M.D., the Independent Medical Safety Monitor for this study.
Financial Support
NIH grant: 5R01EY012893-10 Greenberg (PI) 07/2000 – 02/2011 NEI, Research/Development of Artificial Retinas for the Blind.
Sponsor: Second Sight Medical Products, Inc. The sponsor participated in the design of the study, conducting the study, data collection, data management, data analysis, interpretation of the data, preparation, and review of the manuscript.
Footnotes
Conflict of Interest: Authors with financial interests or relationships to disclose are listed after the references.
Conflict of Interest related to the study sponsor, Second Sight Medical Products (SSMP):
Mark S. Humayun has financial interests in SSMP and is employed by an institution that receives funds from SSMP
Jessy D. Dorn and Robert J. Greenberg are employed by and have stock options in SSMP
Aries Arditi has received a consulting fee from SSMP and was employed by an institution that receives funds from SSMP
Artur V. Cideciyan, Lyndon da Cruz, Gislin Dagnelie, Dean Eliott, Eugene Filley, Avinoam B. Safran, and José-Alain Sahel are or were employed by institutions that receive funds from SSMP
Jacque L.Duncan, Lucian V. Del Priore, Allen C. Ho, Arturo Santos, and Paulo E. Stanga have no conflict of interests to disclose
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.MacLaren RE, Pearson RA. Stem cell therapy and the retina. Eye (Lond) 2007;21:1352–9. doi: 10.1038/sj.eye.6702842. [DOI] [PubMed] [Google Scholar]
- 2.Radtke ND, Aramant RB, Petry HM, et al. Vision improvement in retinal degeneration patients by implantation of retina together with retinal pigment epithelium. Am J Ophthalmol. 2008;146:172–82. doi: 10.1016/j.ajo.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 3.Dobelle WH, Mladejovsky MG, Girvin JP. Artifical vision for the blind: electrical stimulation of visual cortex offers hope for a functional prosthesis. Science. 1974;183:440–4. doi: 10.1126/science.183.4123.440. [DOI] [PubMed] [Google Scholar]
- 4.Veraart C, Raftopoulos C, Mortimer JT, et al. Visual sensations produced by optic nerve stimulation using an implanted self-sizing spiral cuff electrode. Brain Res. 1998;813:181–6. doi: 10.1016/s0006-8993(98)00977-9. [DOI] [PubMed] [Google Scholar]
- 5.Humayun MS, de Juan E, Jr, Dagnelie G, et al. Visual perception elicited by electrical stimulation of retina in blind humans. Arch Ophthalmol. 1996;114:40–6. doi: 10.1001/archopht.1996.01100130038006. [DOI] [PubMed] [Google Scholar]
- 6.Humayun MS, de Juan E, Jr, Weiland JD, et al. Pattern electrical stimulation of the human retina. Vision Res. 1999;39:2569–76. doi: 10.1016/s0042-6989(99)00052-8. [DOI] [PubMed] [Google Scholar]
- 7.Rizzo JF, III, Wyatt J, Loewenstein J, et al. Methods and perceptual thresholds for short-term electrical stimulation of human retina with microelectrode arrays. Invest Ophthalmol Vis Sci. 2003;44:5355–61. doi: 10.1167/iovs.02-0819. [DOI] [PubMed] [Google Scholar]
- 8.Humayun MS, Weiland JD, Fujii GY, et al. Visual perception in a blind subject with a chronic microelectronic retinal prosthesis. Vision Res. 2003;43:2573–81. doi: 10.1016/s0042-6989(03)00457-7. [DOI] [PubMed] [Google Scholar]
- 9.de Balthasar C, Patel S, Roy A, et al. Factors affecting perceptual thresholds in epiretinal prostheses. Invest Ophthalmol Vis Sci. 2008;49:2303–14. doi: 10.1167/iovs.07-0696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yanai D, Weiland JD, Mahadevappa M, et al. Visual performance using a retinal prosthesis in three subjects with retinitis pigmentosa. Am J Ophthalmol. 2007;143:820–7. doi: 10.1016/j.ajo.2007.01.027. [DOI] [PubMed] [Google Scholar]
- 11.Caspi A, Dorn JD, McClure KH, et al. Feasibility study of a retinal prosthesis: spatial vision with a 16-electrode implant. Arch Ophthalmol. 2009;127:398–401. doi: 10.1001/archophthalmol.2009.20. [DOI] [PubMed] [Google Scholar]
- 12.Greenwald SH, Horsager A, Humayun MS, et al. Brightness as a function of current amplitude in human retinal electrical stimulation. Invest Ophthalmol Vis Sci. 2009;50:5017–25. doi: 10.1167/iovs.08-2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Horsager A, Greenwald SH, Weiland JD, et al. Predicting visual sensitivity in retinal prosthesis patients. Invest Ophthalmol Vis Sci. 2009;50:1483–91. doi: 10.1167/iovs.08-2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oyster CW. The Human Eye: Structure and Function. Sunderland, MA: Sinauer Associates; 1999. p. 660. [Google Scholar]
- 15.Roman AJ, Schwartz SB, Aleman TS, et al. Quantifying rod photoreceptor-mediated vision in retinal degenerations: dark-adapted thresholds as outcome measures. Exp Eye Res. 2005;80:259–72. doi: 10.1016/j.exer.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 16.Roman AJ, Cideciyan AV, Aleman TS, Jacobson SG. Full-field stimulus testing (FST) to quantify visual perception in severely blind candidates for treatment trials. Physiol Meas. 2007;28:N51–6. doi: 10.1088/0967-3334/28/8/N02. [DOI] [PubMed] [Google Scholar]
- 17.Lankaranian D, Reis R, Henderer JD, et al. Comparison of single thickness and double thickness processed pericardium patch graft in glaucoma drainage device surgery: a single surgeon comparison of outcome. J Glaucoma. 2008;17:48–51. doi: 10.1097/IJG.0b013e318133fc49. [DOI] [PubMed] [Google Scholar]
- 18.Gedde SJ, Schiffman JC, Feuer WJ, et al. Tube Versus Trabeculectomy Study Group. Three-year follow-up of the Tube Versus Trabeculectomy Study. Am J Ophthalmol. 2009;148:670–84. doi: 10.1016/j.ajo.2009.06.018. [DOI] [PubMed] [Google Scholar]
- 19.Ang GS, Varga Z, Shaarawy T. Postoperative infection in penetrating versus non-penetrating glaucoma surgery. Br J Ophthalmol. 2010;94:1571–6. doi: 10.1136/bjo.2009.163923. [DOI] [PubMed] [Google Scholar]
- 20.Bading G, Hillenkamp J, Sachs HG, et al. Long-term safety and functional outcome of combined pars plana vitrectomy and scleral-fixated sutured posterior chamber lens implantation. Am J Ophthalmol. 2007;144:371–7. doi: 10.1016/j.ajo.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 21.Kim SW, Kim MJ, Yang KS, et al. Risk factors for pseudophakic retinal detachment after intraocular lens scleral fixation with or without pars plana vitrectomy. Retina. 2009;29:1479–85. doi: 10.1097/IAE.0b013e3181aade61. [DOI] [PubMed] [Google Scholar]
- 22.Jaffe GJ, Martin D, Callanan D, et al. Fluocinolone Acetonide Uveitis Study Group. Fluocinolone acetonide implant (Retisert) for noninfectious posterior uveitis: thirty-four-week results of a multicenter randomized clinical study. Ophthalmology. 2006;113:1020–7. doi: 10.1016/j.ophtha.2006.02.021. [DOI] [PubMed] [Google Scholar]
- 23.Pavesio C, Zierhut M, Bairi K, et al. Fluocinolone Acetonide Uveitis Study Group. Evaluation of an intravitreal fluocinolone acetonide implant versus standard systemic therapy in noninfectious posterior uveitis. Ophthalmology. 2010;117:567–75. doi: 10.1016/j.ophtha.2009.11.027. [DOI] [PubMed] [Google Scholar]
- 24.Kappel PJ, Charonis AC, Holland GN, et al. Southern California HIV/Eye Consortium. Outcomes associated with ganciclovir implants in patients with AIDS-related cytomegalovirus retinitis. Ophthalmology. 2006;113:673–83. doi: 10.1016/j.ophtha.2005.11.022. [DOI] [PubMed] [Google Scholar]
- 25.Abrams GW, Williams GA, Neuwirth J, McDonald HR. Clinical results of titanium retinal tacks with pneumatic insertion. Am J Ophthalmol. 1986;102:13–9. doi: 10.1016/0002-9394(86)90202-3. [DOI] [PubMed] [Google Scholar]
- 26.Lange C, Feltgen N, Junker B, et al. Resolving the clinical acuity categories “hand motion” and “counting fingers” using the Freiburg Visual Acuity Test (FrACT) Graefes Arch Clin Exp Ophthalmol. 2009;247:137–42. doi: 10.1007/s00417-008-0926-0. [DOI] [PubMed] [Google Scholar]
- 27.Bach M, Wilke M, Wilhelm B, et al. Basic quantitative assessment of visual performance in patients with very low vision. Invest Ophthalmol Vis Sci. 2010;51:1255–60. doi: 10.1167/iovs.09-3512. [DOI] [PubMed] [Google Scholar]
- 28.Hauswirth WW, Aleman TS, Kaushal S, et al. Treatment of Leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: short-term results of a phase I trial. Hum Gene Ther. 2008;19:979–90. doi: 10.1089/hum.2008.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klein M, Birch DG. Psychophysical assessment of low visual function in patients with retinal degenerative diseases (RDDs) with the Diagnosys full-field stimulus threshold (D-FST) Doc Ophthalmol. 2009;119:217–24. doi: 10.1007/s10633-009-9204-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Little RJ, Rubin DB. Statistical Analysis with Missing Data. Hoboken, NJ: Wiley; 2002. pp. 117–23. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.