

Abstract
The aryl hydrocarbon receptor nuclear translocator (ARNT) heterodimerizes with hypoxia inducible factor-1α (HIF-1α), followed by upregulation of genes that are essential for carcinogenesis. We utilized a novel peptide (Ainp1) to address whether the HIF-1α signaling could be suppressed by an ARNT-mediated mechanism. Ainp1 suppresses the HIF-1α-dependent luciferase expression in Hep3B cells and this suppression can be reversed by ARNT. Ainp1 reduces the interaction between ARNT and HIF-1α, suppresses the formation of the HIF-1 gel shift complex, and suppresses the ARNT recruitment to the vegf promoter. These effects are partly mediated by redistribution of the nuclear ARNT contents to the cytoplasm.
Keywords: ARNT-interacting peptide, HIF-1α, ARNT, AhR, anticancer, Hep3B
1. Introduction
ARNT (aka HIF-1β) belongs to the basic-helix-loop-helix Per-ARNT-Sim (bHLH-PAS) protein family. It is a nuclear protein which takes part in various cellular functions. For example, it is essential for the signaling pathways of AhR and HIF-1α by interacting with them through its PAS domain [1,2,3]. ARNT also participates in other cellular functions as follows: (1) ARNT interacts with ERα and ERβ through its C-terminal part and acts as a potent coactivator of the ER signaling [4]; (2) ARNT interacts with RelB to form an inhibitory complex at the NFκB promoter and in turn reduces the RelA/p50 transcriptional activity [5] and (3) ARNT interacts with the TNF receptor CD30 which activates the NFkB signaling that drives the growth of Hodgkin’s lymphoma [5]. ARNT knockout is embryonically lethal due to failure in angiogenesis [6,7]. Recently, ARNT has been found to be reduced in islets [8] and livers [9] of diabetic individuals. Knockdown of ARNT expression in β-cells compromises the metabolic event affecting insulin secretion [10]. Liver-specific ARNT knockout mice showed features that are consistent with type 2 diabetes, suggesting that ARNT plays a role in the development of diabetes [9]. Taken together, it would be important to understand how the ARNT levels can be altered in a cell.
HIF-alpha is overexpressed in most solid tumors (e.g. bladder, brain, breast, colon, ovarian, pancreatic, prostate, and kidney [11]) and is responsible for angiogenesis and upregulation of transporters (e.g. GLUT1) and enzymes (e.g. LDH, aldolase, PFK, endolase, and aldolase) for efficient glucose metabolism via glycolysis. More than 100 HIF-1α target proteins have been identified which are related to tumorigenesis [12]. HIF-1α is rapidly degraded by the pVHL-dependent ubiquitin-proteasome pathway under normoxic condition; this mechanism is mediated through the oxygen-dependent degradation domain (ODDD) of HIF-1α. During hypoxia, the HIF-1α protein escapes degradation and heterodimerizes with ARNT in the nucleus to form an active transcription factor HIF-1. HIF-1 binds to the HRE enhancer, causing recruitment of coactivators (such as CBP/p300 [13], TIF2 [14], SRC-1 [14], and TRIP230 [15]) and activation of gene transcription. In addition to protein degradation to modulate the amount of HIF-1α protein for function, transcription of the HIF-1α message is also regulated via the PI3K/Akt pathway [16]. Phosphorylation of HIF-1α by MAPKs p42/p44 [17] and ERK [18] appears to affect its transcriptional activity. Collectively, various mechanisms can be possible for cancer cells to increase HIF-1α expression under normoxia.
We are interested in suppressing the HIF-1α function in tumor cells via an ARNT-mediated mechanism. We hypothesize that ARNT-interacting peptides may be viable anticancer therapeutics by suppressing an ARNT-dependent function which is essential for tumor growth. Previously we used the phase display method to identify Ainp1 as one of the ARNT-interacting peptides [19]. Our objective was to test whether Ainp1 suppresses the HIF-1 function via an ARNT-dependent mechanism. Here, we provided evidence supporting that Ainp1 reduces the formation of the HIF-1α::ARNT heterodimer and in turn suppresses the recruitment of ARNT to the vegf promoter. Ainp1 suppresses the nuclear ARNT levels by redistributing ARNT from nucleus to cytoplasm. Ainp1 also appears to cause the ARNT-dependent, but HIF-1α-independent, cell death.
2. Material and methods
2.1 Reagents
Human hepatoma Hep3B cells were grown at 37 °C and 5% CO2 in Advanced MEM (Gibco, Carlsbad, CA) supplemented with 5% FBS, 2mM L-glutamate, 100 units/ml of penicillin, and 0.1 mg/ml of streptomycin. Cobalt chloride (CoCl2) was used to chemically mimic hypoxia in Hep3B cells. Anti-ARNT rabbit polyclonal IgG (H-172, sc-5580), anti-ARNT goat polyclonal IgG (C-19, sc-8076), and anti-GFP rabbit polyclonal IgG (sc-8334) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-HIF-1α rabbit monoclonal IgG (2015-1) was purchased from Epitomics (Burlingame, CA). Anti-caspase 3 mouse monoclonal IgG (3G2) was purchased from Cell Signaling Technology (Danvers, MA). Baculovirus HIF-1α was generated according to our previously published protocols for the generation of baculovirus ARNT [20]. This full-length human HIF-1α contained a C-terminal 6His tag and was affinity purified from the infected Sf9 cells using the TALON resin (Clontech, Mountain View, CA). The pSport-ARNT plasmid containing the full-length human ARNT cDNA and the pSport-HIF-1α plasmid containing the full-length human HIF-1α cDNA were gifts from Dr. Chris Bradfield (University of Wisconsin, Madison) and used to generate ARNT and HIF-1α using the rabbit reticulocyte lysate (RRL) system (Promega, Madison, WI) according to the manufacturer’s protocol. Chimeric bacteria expression plasmid containing the Ainp1 cDNA was generated by cloning the cDNA into BamHI and HindIII sites of pQE80-Ainp1 plasmid (Qiagen, Valencia, CA). Thioredoxin was expressed using the parent pThioHisC plasmid (Invitrogen, Carlsbad, CA).
Both recombinant proteins were affinity purified from the IPTG-treated JM109 culture using the TALON resin according to our previously published protocol [19]. The Ainp1 cDNA was cloned into BglII and XhoI sites of the pGFP2-C1 plasmid (Perkin Elmer, Waltham, MA) to generate the pGFP2-Ainp1 plasmid. Double-stranded oligonucleotides containing nuclear localization sequence in tandem (3XNLS, underlined) were purchased from Invitrogen with XhoI and ApaI half sites at the termini (sense, OL287, 5’-TCGAGGATCCAAAAAAGAAGAGAAAGGTA GATCCAAAAAAGAAGAGAAAGGTAGATCCAAAAAAGAAGAGAAAGGTAGGGCC-3’; antisense, OL288, 5’-CTACCTTTCTCTTCTTTTTTGGATCTACCTTTCTCTTCTTTTTTGGATCTACCTTTCTCT TTTTTGGATCC-3’). The annealed 3XNLS oligonucleotides were cloned into XhoI and ApaI sites of pGFP2-C1 to generate the pGFP2-3XNLS plasmid. The Ainp1 cDNA was then cloned into the pGFP2-3XNLS plasmid to generate the pGFP2-Ainp1-3XNLS plasmid. Same strategy was used to generate the pGFP2-NES and pGFP2-Ainp1-NES plasmids containing one copy of NES. The NES sequence (LQLPPLERLTLDC) was obtained from HIV-1 Rev protein [21]. The corresponding oligonucleotides were 5’-TCGAGCTTCAGCTACCACCGCTTGAGAGACTTACTCTTGATTGTGGGCC-3’ (OL564) and 5’-CACAATCAAGAGTAAGTCTCTCAAGCGGTGGTAGCTGAAGC-3’ (OL565). The Ainp1 cDNA was cloned into BamHI and XhoI sites of the pCMV-Tag4A plasmid (Stratagene, La Jolla, CA) to generate the pCMV-Tag4A-Ainp1 plasmid for transfection experiments. The pGL3-Epo reporter luciferase and pCMV-C 553 plasmids were generated as described previously [22]. The β-galactosidase expressing plasmid pCH110 was purchased from Amersham-Pharmacia Biotech (Piscataway, NJ). IRdye700 conjugated HRE and 10X orange loading dye were purchased from LI-COR (Lincoln, NE). The wild type HRE was generated by annealing two primers purchased from Invitrogen (sense, OL568, 5’-AGCTTGCCCTACGTGCTGTCTCAGA-3’; antisense, OL569, 5’-TCTGAGACAGCACGTAGGGCAAGCT-3’), the mutated HRE was also generated by annealing two primers purchased from Invitrogen (sense, OL570, 5’-AGCTTGCCCTAGCTGCTGTCTCAGA-3’, antisense, OL571, 5’-TCTGAGACAGCAGCTAGGGCAAGCT-3’).
2.2 Transient transfection studies
Hep3B cells were grown in a 24-well plate to about 90% confluence prior to transfection. Before transient transfection, media was exchanged to Advanced MEM containing 3% FBS and 2 mM L-glutamate. In each well, cells were transfected with 50 μl of Opti-MEM (Invitrogen, Carlsbad, CA) containing 1.6 μl of Fugene HD (Roche, Indianapolis, IN), and 0.8 μg of total plasmid DNA. Cells were incubated in this transfection mix for 24 h at 37 °C, 5% CO2. Afterwards, media was exchanged to fresh complete media before treatment with CoCl2. Cells were treated with CoCl2 (100 μM) or water for 18 h at 37° C, 5% CO2 before harvested using the Dual-Light luciferase kit (Applied Biosystems, Carlsbad, CA).
2.3 Co-immunoprecipitation studies
For ARNT::Ainp1 interaction study, bacterially expressed Ainp1 (40 μg) or bacterially expressed thioredoxin (40 μg) was incubated with 15 μl of RRL expressed ARNT or uncharged RRL as the negative control in HEDG buffer (25 mM HEPES, pH 7.4, 1 mM EDTA, 1 mM DTT, and 10% glycerol) containing 0.1 M KCl for 30 min at 30 °C. For HIF-1α::Ainp1 interaction study, bacterially expressed Ainp1 (40 μg) or bacterially expressed thioredoxin (40 μg) was incubated with baculovirus expressed HIF-1α or the Sf9 lysate (50 μg) in HEDG buffer containing 0.1 M KCl for 30 min at 30 °C. For HIF-1α::ARNT interaction study in the presence or absence of Ainp1, RRL expressed ARNT and HIF-1α were used in the presence or absence of bacterially expressed 6His-Ainp1. RRL expressed ARNT (15 μl) and HIF-1α (15 μl) were incubated with bacterially expressed Ainp1 (60 μg), Ainp1 (30 μg)/ thioredoxin (30 μg), or thioredoxin (60 μg) in 400 μl of HEDG buffer containing 0.1 M KCl for 30 min at 30° C. In addition, five 100-mm plates of near confluent Hep3B cells were treated with 200 μM CoCl2 for 20 h. Cells were harvested into 1.5 ml of HEDG buffer containing 1 mM PMSF and 2 μg/ml of leupeptin. After three cycles of freeze-thaw, cells were centrifuged at 16,000g for 10 min at 4 °C. The pellet was resuspended into 1.5 ml of the same buffer except it contained 0.4 M KCl. After rotating for 1 h at 4 °C, the suspension was centrifuged at 16,000g for 20 min at 4 °C. The resulting supernatant was immediately diluted with HEDG buffer to 0.1 M final concentration of KCl and was used as the sample for co-immunoprecipitation. Sample (150 μg) was incubated with 6His-Ainp1 (60 μg), 6His-Ainp1/Sf9 lysate (30 μg of each) or Sf9 lysate alone (60 μg) in HEDG buffer containing 0.1 M KCl for 30 min at 30 °C. Co-immunoprecipitation began with protein G Dynabeads preclearance (2 μl, Invitrogen, Carlsbad, CA) by rotating for 30 min at 4 °C. Corresponding immunoprecipitation antibody was then added to each precleared sample. After incubation for 2 h at 4 °C, protein G Dynabeads (5 μl) was added and the samples were incubated for another 1 h at 4 °C. Beads were washed with HEDG buffer containing 0.4% Tween-20, 10mM β-mercaptoethanol (600 μl) for four times using DynaMag-2 magnet (Invitrogen). Proteins were retrieved from boiling the beads in 1X electrophoresis sample buffer (20 μl) for 5 min and then analyzed by Western analysis.
2.4 Generation of anti-Ainp1 mouse polyclonal antibody
Bacterially expressed purified 6His-Ainp1 (50 μg) was dissolved in 0.125 ml of PBS and 0.125 ml of the appropriate adjuvant: Ainp1 was combined with complete Freund’s adjuvant (F5881, Sigma, St. Louis, MO) for the first antigen injection while incomplete Freund’s adjuvant (F5506, Sigma) for the remaining injections. The protein/adjuvant was mixed thoroughly and administered via an intraperitoneal injection into a six-week-old female BALB/c mouse every two weeks for four times. Three days after the fourth injection, blood was collected from the heart with a sterile syringe. The plasma was decanted and clarified by centrifugation at 2,500g for 15 min to obtain the serum containing anti-Ainp1 polyclonal antibodies.
2.5 Western blot analysis
Tricine-SDS-PAGE was performed according to the published literature [23] except that samples were stacked at 30 V and resolved at 90 V using a Bio-Rad mini-Protean 3 system. Wet transfer was performed using the Bio-Rad transfer kit at 200 mA for 70 min. For normal SDS-PAGE, samples were run at 160 V and transferred at 300 mA for 100 min. The transferred nitrocellulose membrane was blocked in PBS containing 5% BSA and 0.1% Tween-20 for 1 h. Primary antibody incubation (1:2,000) was performed in the blocking buffer overnight at 4 °C. The membrane was then washed with PBS containing 0.1% Tween-20 for four times. Secondary antibody incubation (LI-COR donkey anti-mouse IgG IRDye 680 or anti-rabbit IgG IRDye 800CW, 1: 10,000) was performed in the blocking buffer for 1 h at room temperature. After washing four times with PBS containing 0.1% Tween-20 and once with PBS alone, the membrane was dried and analyzed using a LI-COR Odyssey imaging system (Lincoln, NE).
2.6 Gel shift assay
In a final volume of 11 μl, baculovirus expressed ARNT and HIF-1α and the Sf9 lysate (3 μg) were incubated in HEDG buffer (25 mM HEPES, pH 7.4, 1 mM EDTA, 1 mM DTT, and 10% glycerol) in the presence or absence of bacterially expressed Ainp1 at 30 °C for 30 min. After that, 2 μl of HEDG buffer containing 0.8 M KCl, poly-dIdC (0.75 μg), and the mutated HRE (75 ng) was added. In one case, the wild type HRE (75 ng) was used instead of the mutated HRE to show the specific HIF-1 gel shift complex formation. After 10 min at room temperature, IRDye700-conjugated HRE (75 fmol, 1 μl) was added for another 10 min incubation at room temperature. To trigger the supershifted complex formation, either specific or control IgG (1 μl, 0.1 μg) or HEDG buffer (1 μl) was added to a gel shift sample for additional 10 min at room temperature to prove the identity of the HIF-1 gel shift complex. After the addition of 10X orange loading dye, the samples were resolved on a 4 or 5% native polyacrylamide gel (1X TBE) at 4°C (185V) for 2.5 hours. After that, the gel was analyzed directly using a LI-COR Odyssey image system.
2.7 Chromatin co-immunoprecipitation (ChIP) assays
ChIP assay was performed as described previously [24] with the following modifications: Hep3B cells (80–90% confluence in a 60-mm plate) were treated with or without 300 μM CoCl2 in complete media for 4 h at 37°C. After that, cells were cross-linked with 1% formaldehyde in FBS-free DMEM for 10 min at room temperature. After glycine quenching, cells were washed three times with ice-cold PBS and the cell pellet was resuspended in lysis buffer (150 μl), followed by sonication. Diluted samples of 1.5 ml each were used for co-immunoprecipitation analysis. The washed ARNT IgG-precipitated samples were treated with RNaseA (0.75 μl of 10mg/ml) for 30 min at 37 °C, followed by the proteinase K treatment (0.225 μl of 50 mg/ml of proteinas K, 0.975 μl of 0.5M EDTA, and 1.95 μl of 1M Tris, pH6.5). PCR was performed using a primer set specific to the HRE region of the vegf promoter (forward primer, OL482: CAGGAACAAGGGCCTCTGTCT; reverse primer, OL483: TGTCCCTCTGACAATGTGCCATC).
2.8 Cell viability studies
Hep3B cells were seeded at 1.5x104 cells per well in a 96-well plate. After an overnight incubation at 37 °C and 5% CO2, transient transfection was performed using Fugene HD (0.4 μl/0.2 μg of pGFP2-C1 or pGFP2-C1-Ainp1/well). Cells were then treated with 100 μM CoCl2 or water right after transfection and cell viability was determined every 12 h using the CellTiter assay kit (Promega, Madison, WI).
2.9 Subcellular fractionation
Hep3B cells (4x106 in 0.4 ml) were transfected with 30 μg (in 30 μl of water) of pGFP2-C1, pGFP2-C1-Ainp1, pGFP2-C1-NLS, pGFP2-C1-Ainp1-NLS, pGFP2-C1-NES or pGFP2-C1-Ainp1-NES by electroporation (160 V, 70 ms, 1 pulse). The electroporated cells were seeded onto a 100-mm dish and grown in complete media for 36 h. After that, cells were washed with cold PBS twice before harvested into 200 μl of lysis buffer (25 mM HEPES, pH 7.9, 150 mM NaCl, 1% NP-40, 1 mM PMSF, and 2 μg/ml of leupeptin). After 30 min at 4 °C, the cell suspension was centrifuged at 3,000g for 3 min. The supernatant was defined as the cytoplasmic extract. The pellet was washed with cold PBS (500 μl) once and then resuspended into 100 μl of nuclear lysis buffer (25 mM HEPES, pH 7.9, 350 mM NaCl, 10% dextrose, 0.05% NP-40, 1 mM PMSF, and 2 μg/ml of leupeptin). After 4 °C for 1 h, the cell suspension was centrifuged at 16,000g for 10 min. The supernatant was defined as the nuclear extract. Protein contents were determined by BCA assays (Pierce, Rockford, IL).
2.10 Whole cell extract preparation
Hep3B cells (4x106 in 0.4 ml) were transfected with 30 μg (in 30 μl of water) of either pGFP2-C1 or pGFP2-C1-Ainp1 by electroporation (160 V, 70 ms, 1 pulse). The electroporated cells were seeded onto a 100-mm dish and grown in complete media for 36 h. Cells were then washed with cold PBS twice before resuspended into HEDG buffer (25 mM HEPES, pH 7.4, 1 mM EDTA, 1 mM DTT, and 10% glycerol) containing 0.4 M KCl, 1 mM PMSF, and 2 μg/ml of leupeptin. After three cycles of freeze-thaw, the cell suspension was incubated for 1 h at 4 °C before centrifuged at 16,000g for 10 min. The supernatant was defined as the whole cell extract (WCE).
2.11 Statistical analysis
We performed one-way ANOVA (Fig. 5 and S3) and two-way ANOVA (Fig. 3, 4D, and 6) followed by post hoc Bonferroni’s multiple comparison test to determine the statistical significance with 95% confidence intervals using the GraphPad Prism 5 software.
Fig. 5.
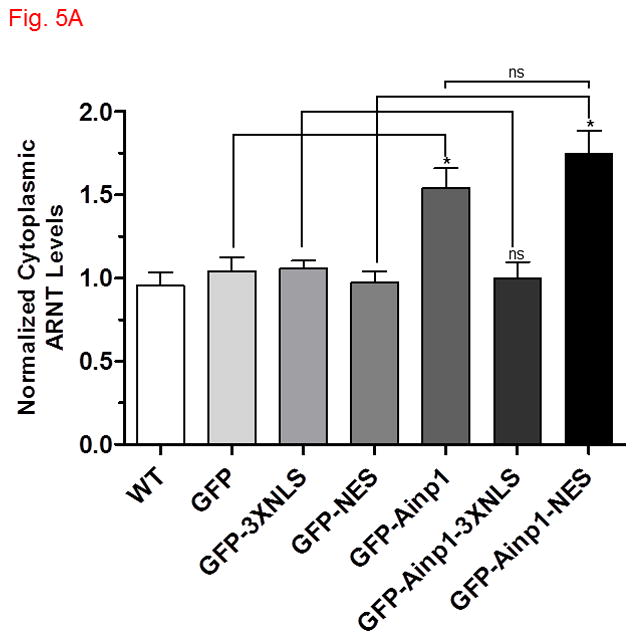
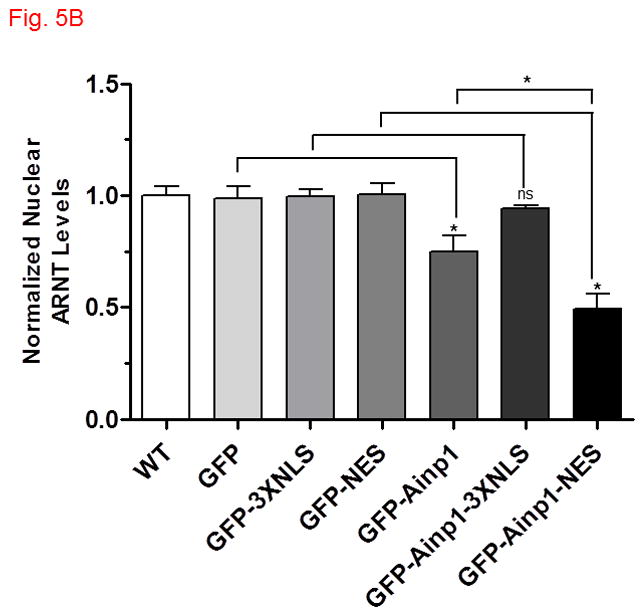
Ainp1 sequestered ARNT in the cytoplasm. The ARNT levels were determined in various cytoplasmic and nuclear fractions by Western using a LI-COR Odyssey imaging system. (For Western images, see Fig. S4A-E in supplementary content.) Hep3B cells were either untransfected (WT) or transfected with a plasmid expressing GFP, GFP-3XNLS, GFP-NES, GFP-Ainp1, GFP-Ainp1-3XNLS or GFP-Ainp1-NES. The cytoplasmic (A) and nuclear (B) ARNT levels were normalized by arbitrarily setting the corresponding average WT value as one. Statistics were calculated from four replicates (n = 4) of all conditions except for GFP-NES and GFP-Ainp1-NES from five replicates (n = 5). Error bars indicates mean ± SD. *p < 0.05; ns, not significant.
Fig. 3.
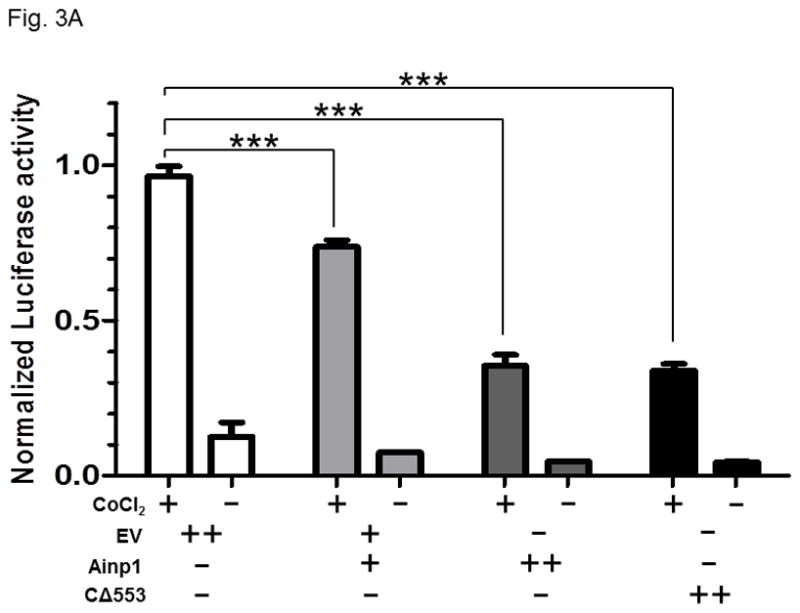
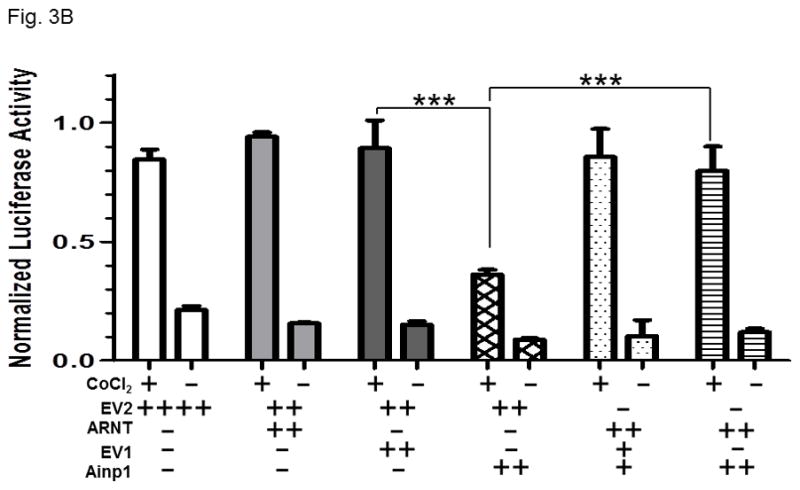
Ainp1 suppressed the CoCl2-activated, HRE-driven luciferase activity in Hep3B cells. A, cells (± 100 μM CoCl2) were transfected with the same amount of plasmid DNA (EV, pCMV- Tag4A; Ainp1, pCMV-Ainp1; C 553, pCMV-C 553). Amount of plasmid DNA used: +, 0.3 μg; ++, 0.6 μg; 150 ng of pGL3-Epo reporter luciferase plasmid; 50 ng of pCH110 plasmid. This experiment was repeated once with similar results. B, cells (± 100 μM CoCl2) were transfected with the same amount of plasmid DNA (EV1, pCMV-Tag4A; EV2, pSport; Ainp1, pCMV-Tag4-Ainp1; ARNT, pSport-ARNT). Amount of plasmid DNA used: +, 0.2 μg; ++, 0.4 μg; ++++, 0.8 μg; 75 ng of pGL3-Epo reporter luciferase plasmid; 25 ng of pCH110 plasmid. This experiment was repeated once with similar results. Luciferase activity was normalized by the β-galactosidase activity. Error bars indicate the variations of the means (mean ± SD, n = 3). ***p < 0.001.
Fig. 4.
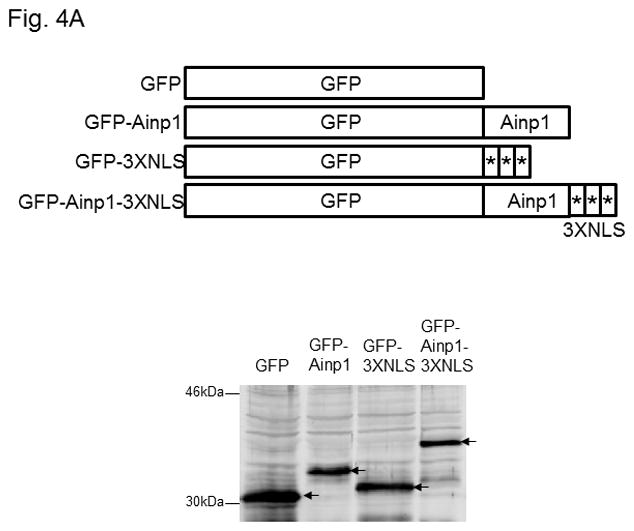
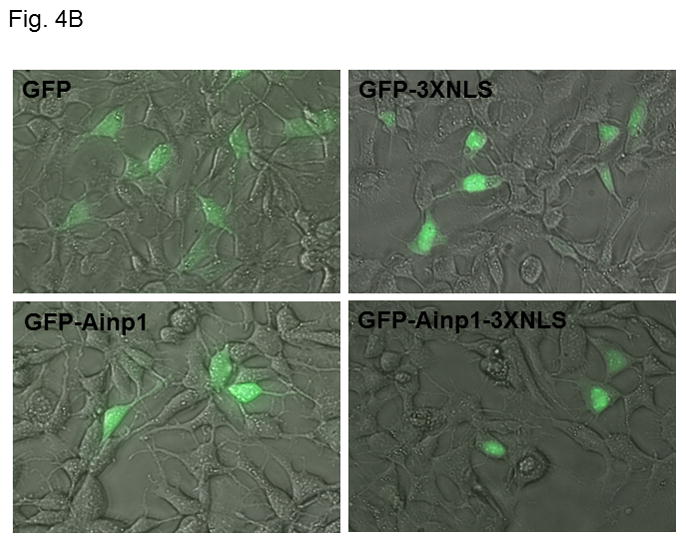
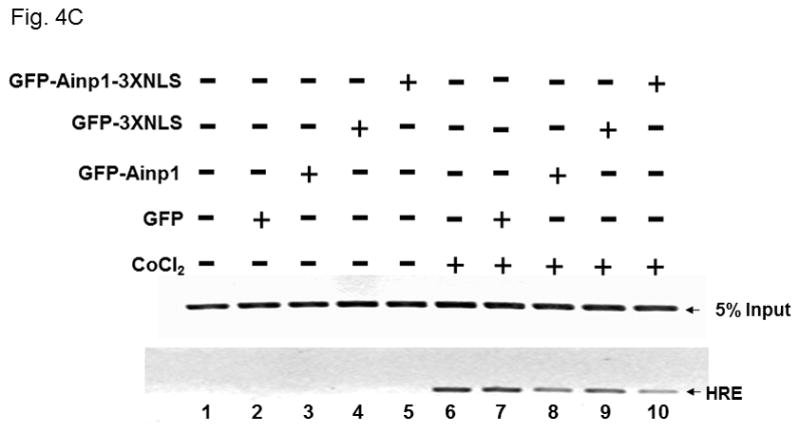
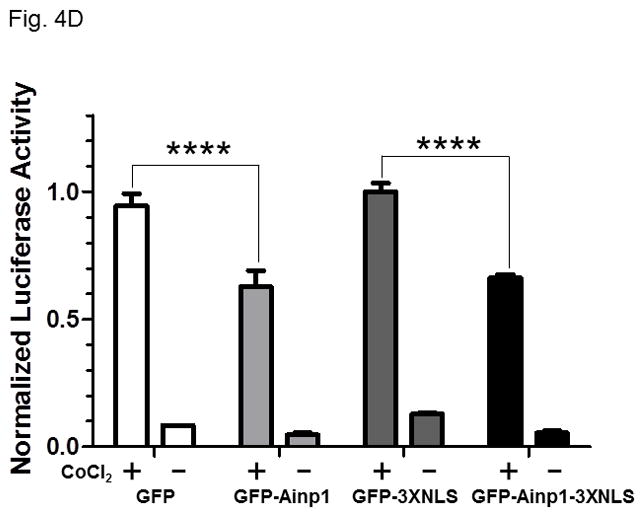
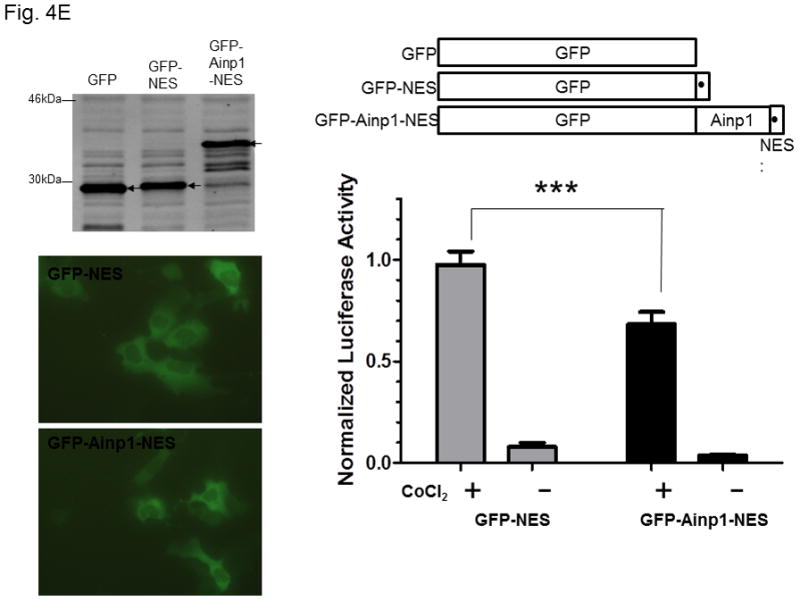
Ainp1 suppressed the HIF-1α function in Hep3B cells. Western analysis (A) and fluorescence imaging (B) were performed 24 hours after transfecting cells (1 × 105) with pGFP2-C1, pGFP2-Ainp1, pGFP2-3XNLS or pGFP2-Ainp1-3XNLS plasmid (1.6 μl FuGene HD/800 ng plasmid). Each Western lane contained 30 μg of WCE. Fluorescence images were superimposed with the phase contrast images to show the cell shape. C, ChIP data showing that Ainp1 suppressed recruitment of ARNT to the vegf promoter. Cells (4 × 106) ± 300 μM CoCl2 were transfected with GFP, GFP-Ainp1, GFP-3XNLS or GFP-Ainp1-3XNLS expressing plasmid (30 μg by electroporation at 160V, 70ms). Anti-ARNT IgG was used to precipitate the HRE-containing complex and the PCR products of the precipitated HRE fragment are indicated by the bottom arrow. 5% input refers to the band intensity of 5% of total ARNT content in the starting sample. D, GFP-Ainp1 and GFP-Ainp1-3XNLS suppressed the reporter luciferase activity to a similar extent in Hep3B cells. Cells (± 100 μM CoCl2) were transfected with the same amount of plasmid DNA (pGFP2-C1, pGFP2-Ainp1, pGFP2-3XNLS or pGFP2-Ainp1-3XNLS). Amount of plasmid DNA used: 0.6 μg of GFP, GFP-Ainp1, GFP-3XNLS or GFP-Ainp1-3XNLS, 150 ng of pGL3-Epo reporter luciferase plasmid, and 50 ng of pCH110 plasmid. GFP and GFP-3XNLS were set as 1 for comparison. This experiment was repeated once with similar results. Error bars indicate the variations of the means (mean ± SD, n = 3). ****p < 0.0001. E. Western analysis and fluorescence imaging were performed similarly as in A and B except that TurboFECT from Fermentas (1.6μl/800ng plasmid) was used in instead of Fugene HD. Arrows indicates the corresponding bands. Cytoplasmic localization of GFP-NES and GFP-Ainp1-NES was observed even without the superimposed phase contrast images. Luciferase experiment was performed using XtremeGENE 9 (Roche, Indianapolis, IN) (0.75 μl/ 0.25 μg plasmid). Amount of plasmid DNA used: 0.2 μg of GFP-NES or GFP-Ainp1-NES, 40 ng of pGL3-Epo reporter luciferase plasmid, and 10 ng of pCH110 plasmid. GFP-NES was set as 1 for comparison. This experiment was repeated once with similar results. Luciferase activity in 4D and E was normalized by the β-galactosidase activity. Error bars indicate the variations of the means (mean ± SD, n = 3). ***p < 0.001.
Fig. 6.
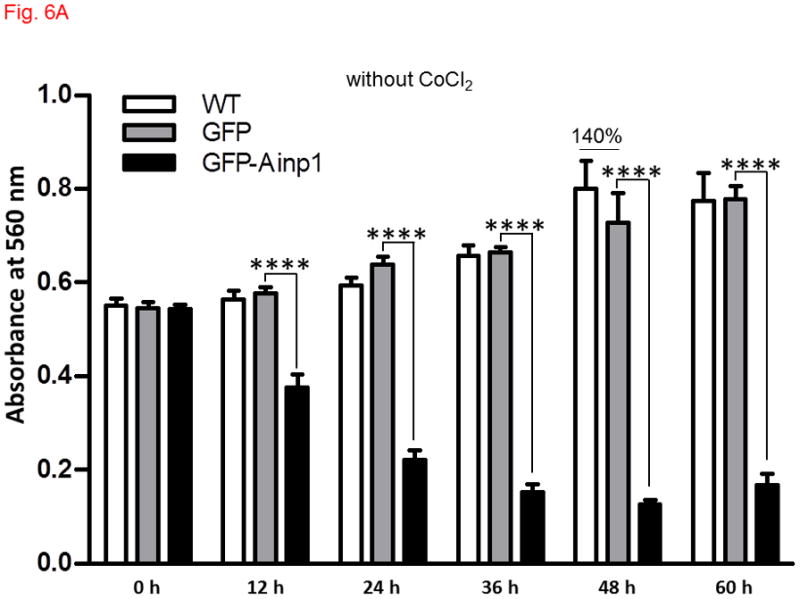
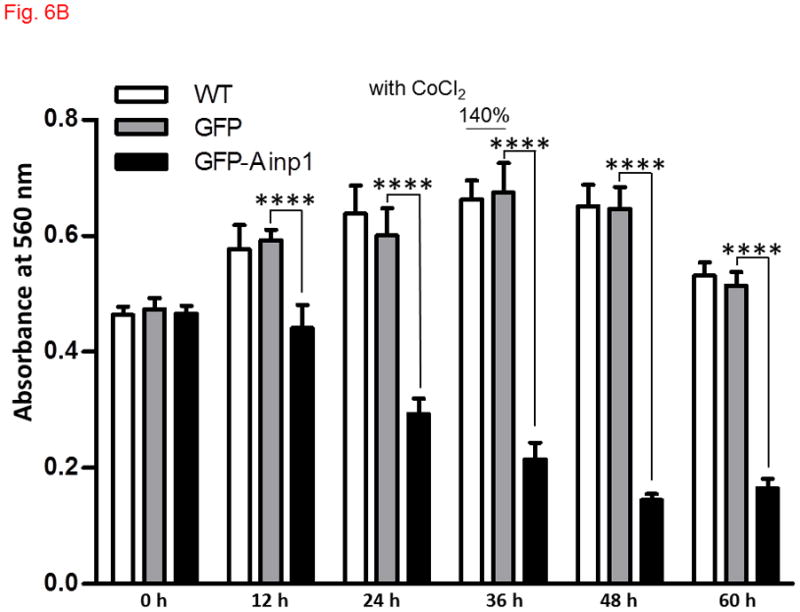
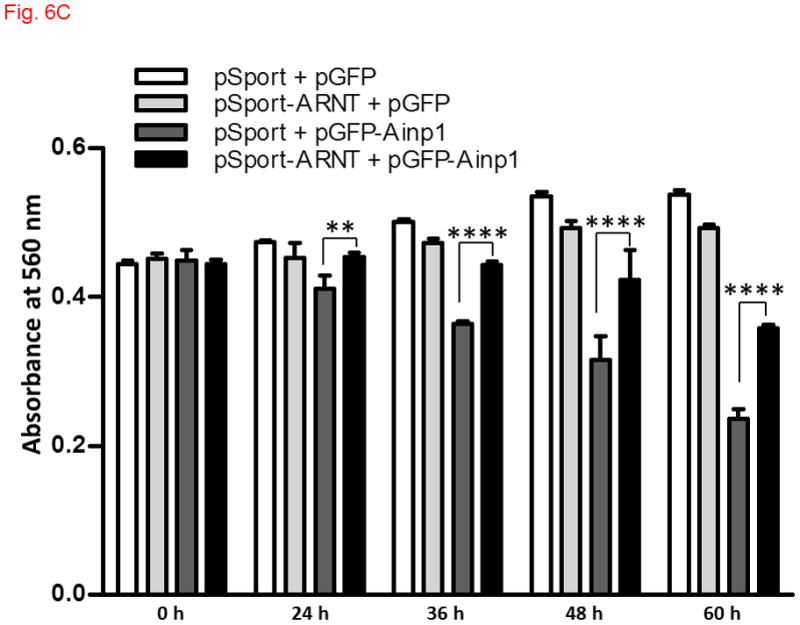
Ainp1 caused ARNT-dependent Hep3B cell death. Cells were transfected with either GFP or GFP-Ainp1 expressing plasmid, followed by treatment with water (A) or 100 μM CoCl2 (B). C, same as A except that pSport or pSport-ARNT plasmid was included in the transfection mix. Total cell count was determined by CellTiter assay at various time points (0-60 h). At zero time point, all conditions were started at a similar total cell count in all cases (A-C). The total amount of plasmid DNA (0.2 μg) in all conditions was the same in A-C so that a lesser amount of GFP or GFP-Ainp1 plasmid (0.1 μg) was used to accommodate pSport or pSport-ARNT plasmid (0.1 μg). Absorbance at 560 nm (y-axis) corresponds to the total cell number. This experiment was repeated once with similar results. Error bars indicate the variations of the means (mean ± SD, n = 3). **p < 0.01, ****p < 0.0001.
3. Results
3.1 Ainp1 interacts with ARNT and interferes with the formation of the HIF-1α::ARNT heterodimer and the HIF-1 gel shift complex
Ainp1 is a novel ARNT-interacting peptide of 59 amino acids in length (Fig. 1A). A search from the BLAST protein database did not provide its identity and there was no apparent consensus motif observed. Although we have not determined the full-length sequence of the primary transcript, Northern blot results revealed that Ainp1 is likely alternatively spliced and ubiquitously expressed in humans (Fig. 1B). However, it is unclear whether Hep3B cells express the full-length Ainp1 protein since our anti-Ainp1 antibody was not able to detect any particularly strong band in the Hep3B lysate (data not shown). It is conceivable that Ainp1 may be derived from a noncoding RNA and its endogenous role is remained to be determined. We abundantly expressed Ainp1 as a 6His fusion protein in the JM109 strain of E. coli and it was effectively purified using the TALON resin. The Coomassie blue staining gel revealed that this Ainp1 was roughly 90% homogeneity after affinity purification (Fig. S1, supplementary content). We used this bacterially expressed purified Ainp1 to examine whether it would interact with ARNT and HIF-1α in vitro. We observed that Ainp1 clearly interacted with RRL expressed ARNT when we used either anti-Ainp1 or anti-ARNT IgG, but not the control IgG, to immunoprecipitate the Ainp1::ARNT complex (Fig. 2A). We intentionally used uncharged reticulocyte lysate as the negative control for the co-immunoprecipitation of RRL expressed ARNT. Similarly, we used bacterially expressed thioredoxin as the negative control for bacterially expressed Ainp1since both Ainp1 and thioredoxin were purified similarly from the IPTG-treated JM109 (Fig. S1, supplementary content). On the contrary, we were not able to detect any appreciative interaction between Ainp1 and baculovirus expressed HIF-1α (Fig. S2, left panel, supplementary content). To make certain that the antibody was capable of immunoprecipitating the bait, we observed that anti-HIF-1α and anti-Ainp1 IgGs could specifically immunoprecipitate HIF-1α and Ainp1, respectively, whereas the same amount of the corresponding control IgG did not (Fig. S2, right panel, supplementary content). We noticed that our human HIF-1α preparations (from Sf9 cells, RRL, and Hep3B whole cell lysate), except the Hep3B nuclear extract, gave two bands on Western analysis, possibly due to post-translational modification; this phenomenon was also reported by the supplier of our anti-HIF-1α monoclonal (Epitomics) and other researchers [25,26]. Next, we addressed whether interaction between Ainp1 and ARNT would alter the HIF-1α::ARNT interaction. We observed that increased amounts of Ainp1 suppressed the ability of the anti-ARNT IgG to co-immunoprecipitate HIF-1α using two different preparations – either RRL expressed ARNT and HIF-1α (Fig. 2B) or CoCl2-treated Hep3B nuclear extract (Fig. S3, supplementary content). These results suggested that Ainp1 is capable of suppressing the HIF-1 complex formation. Next, we examined whether Ainp1 suppresses the formation of the HIF-1 gel shift complex. Baculovirus expressed ARNT and HIF-1α formed the HIF-1 gel shift complex (HIF-1α/ARNT/HRE); the authenticity of this gel shift complex was proven by the fact that wild type HRE, but not the mutated HRE which was an ingredient in all gel shift samples, abolished the complex and both anti-HIF-1α and anti-ARNT IgGs caused the formation of the supershifted complex while the control IgGs did not (Fig. 2C, lanes 1–8). We observed a dose-dependent suppression of the HIF-1 gel shift complex by Ainp1 (Fig. 2C, lanes 9–13), suggesting that Ainp1 could interfere with the HRE-driven downstream gene expression. Notice that the mobility of the HIF-1 gel shift complex was different between the two panels in Fig. 2C since the left gel shift panel was intentionally performed using a lower percentage of polyacrylamide gel (4 instead of 5%) to better visualize the supershifted complex.
Fig. 1.

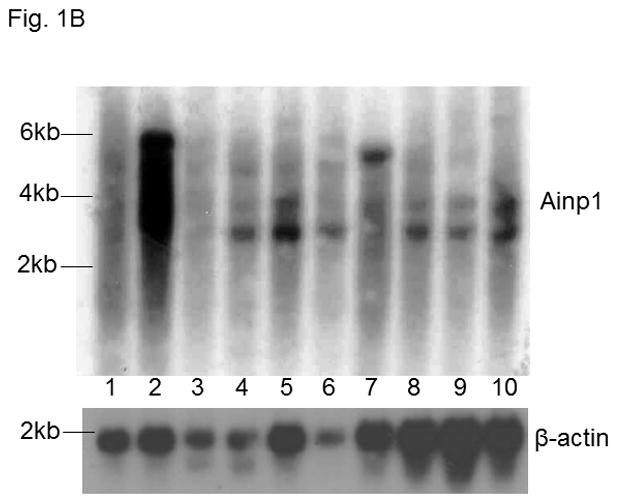
A. nucleotide (underlined) and amino acid sequence of Ainp1. B. Ainp1 mRNA is expressed in most human tissues. Northern blot analysis using Ambion FirstChoice human pre-made blot was performed as previously described [19]. 1, brain; 2, placenta; 3, skeletal muscle; 4, heart; 5, kidney; 6, pancreas; 7, liver; 8, lung; 9, spleen; 10, colon. A 500 bp antisense cDNA fragment of Ainp1 was used. β-actin was used as the standard for comparison.
Fig. 2.
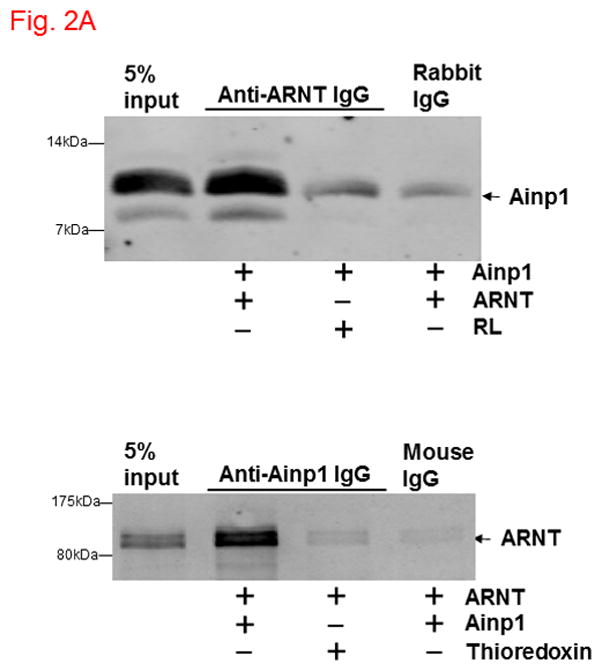
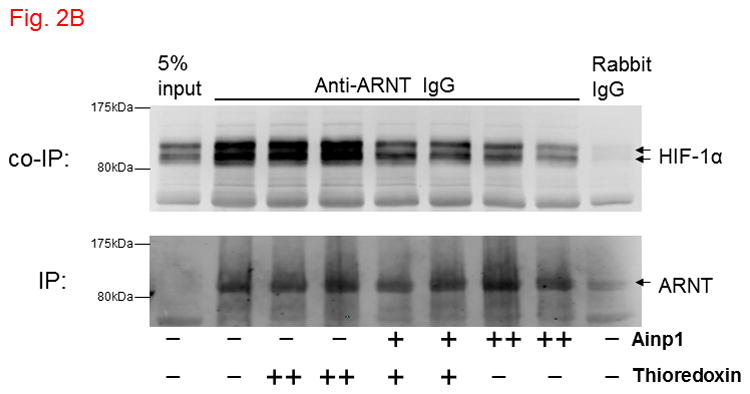
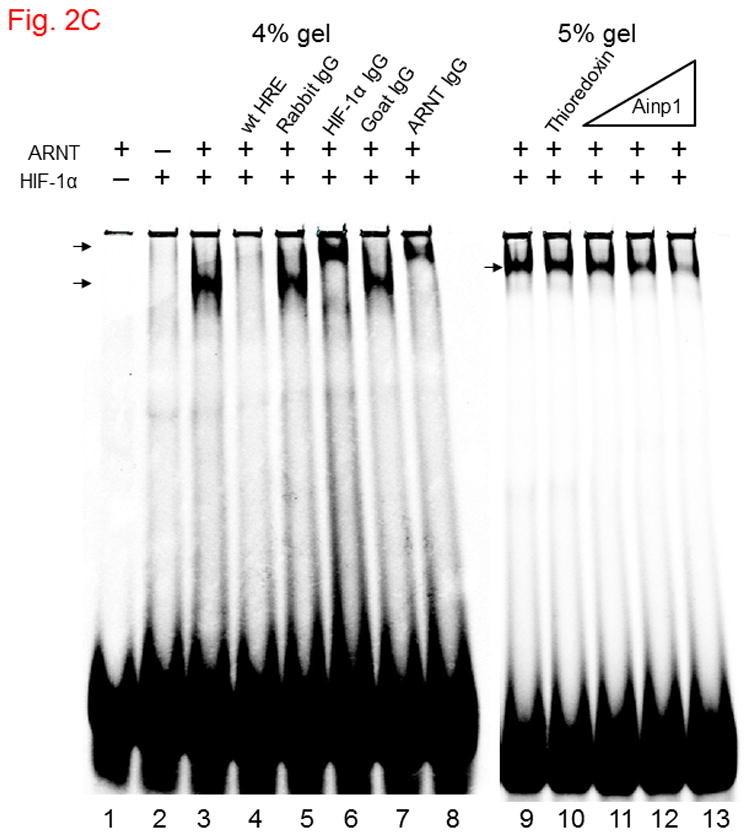
Ainp1 interacted with ARNT and suppressed the ARNT::HIF-1α interaction. A, upper panel, anti-ARNT IgG co-immunoprecipitated bacterially expressed Ainp1. RRL expressed ARNT (pSport-ARNT) was used as the bait; uncharged RRL (RL) and rabbit IgG as negative controls. Lower panel, anti-Ainp1 IgG co-immunoprecipitated RRL expressed ARNT. Bacterially expressed Ainp1 was used as the bait; thioredoxin and mouse IgG as negative controls. This experiment was repeated twice with similar results. B, anti-ARNT IgG co-immunoprecipitated RRL expressed HIF-1α (pSport-HIF-1 α) using RRL expressed ARNT as the bait with different amounts of bacterially expressed Ainp1 added (+, 30 μg; ++, 60 μg). Protein contents in some lanes were normalized by bacterially expressed thioredoxin. Rabbit IgG was the negative control. This experiment was repeated twice with similar results. co-IP shows co-immunoprecipitated HIF-1α whereas IP shows immunoprecipitated ARNT. 5% input (A and B) shows the band intensity of 5% of the target protein content in the starting sample. C. Gel shift data using baculovirus expressed ARNT and HIF-1α. All gel shift samples contained mutated HRE (75 ng) except lane 4 which contained wild type (wt) HRE (75 ng) instead. IgG was added to lane 5 (rabbit IgG), 6 (anti-HIF-1α rabbit IgG), 7 (goat IgG) and 8 (anti-ARNT goat IgG). Bacterially expressed thioredoxin (4 μg) was included in lane 10 whereas Ainp1 was included in lanes 11-13 (0.5, 1.5, and 4 μg). Lower arrow indicates the HIF-1 gel shift complex whereas upper arrow indicates the supershifted complex.
3.2 Ainp1 suppresses the CoCl2-driven, HRE-dependent luciferase expression
Next, we examined whether Ainp1 would inhibit the HIF-1 transcriptional activity in Hep3B cells. Results from our transfection studies showed that Ainp1 suppressed the HRE-driven reporter luciferase activity in a dose-dependent manner (Fig. 3A), supporting that Ainp1 suppressed the HIF-1α function. The extent of suppression by Ainp1 was similar to the suppression by the truncated human aryl hydrocarbon receptor construct C 553, which we had shown previously to suppress the xenograft tumor growth by blocking the HIF-1 function [27]. Next, we examined whether this Ainp1 suppressive effect is indeed ARNT-dependent. When we transiently transfected ARNT into Hep3B cells, we only observed a modest increase in the HRE-driven reporter luciferase activity (Fig. 3B). However, this transfected ARNT effectively reversed the Ainp1 effect in a statistically significant manner, suggesting that this Ainp1 suppression of the HIF-1 transcriptional activity is ARNT-mediated.
3.3 Ainp1 suppresses the CoCl2-driven recruitment of ARNT to the vegf promoter and this effect does not require Ainp1 to be nuclear
Next, we examined whether Ainp1 would interfere with the recruitment of ARNT to the HRE-containing promoter upon stimulus in Hep3B cells. We expressed the GFP fusion of Ainp1 so that we could use GFP as the negative control and ensure the expression of the GFP-Ainp1 protein (Fig. 4A, top panel). To examine whether the nuclear localization is necessary for the Ainp1 effect, we generated GFP-3XNLS and GFP-Ainp1-3XNLS by fusing three copies of the NLS sequence in tandem at the C-termini of these proteins. After transient transfection, expression of the GFP and its fusions was confirmed by Western analysis (Fig. 4A). We found that both GFP and GFP-Ainp1 were localized throughout the cells whereas GFP-3XNLS and GFP-Ainp1-3XNLS were primarily nuclear by fluorescence microscopy (Fig. 4B). Our ChIP results showed that ARNT was clearly recruited to the vegf promoter after the CoCl2 treatment (Fig. 4C, lanes 1 and 6) and the transfected GFP-Ainp1, but not GFP, suppressed this ARNT recruitment to the promoter (Fig. 4C, lanes 6–8). In addition, GFP-Ainp1-3XNLS did not seem to further suppress the CoCl2-driven recruitment of ARNT. The slight difference between GFP-Ainp1 and GFP-Ainp-3XNLS was likely due to the unexpected small suppression of the nuclear GFP (GFP-3XNLS) (Fig. 4C, lanes 7–10). To confirm that GFP-Ainp1 and GFP-Ainp1-3XNLS have a similar suppressive effect, we performed transfection studies to determine the CoCl2-activated, HRE-driven luciferase expression in the presence or absence of these proteins. We observed that 0.6 μg of the transfected GFP-Ainp1-3XNLS expressing plasmid suppressed more than 30% of the luciferase activity (Fig. 4D), which was similar to the extent of suppression by the same amount of the GFP-Ainp1 expressing plasmid. To further determine whether the presence of Ainp1 in the cytoplasm is sufficient to suppress the HIF-1α function, we utilized a NES sequence to localize GFP-Ainp1 in the cytoplasm. Both GFP-NES and GFP-Ainp1-NES were localized in the cytoplasm (Fig. 4E, left). GFP-Ainp1-NES suppressed the CoCl2-activated, HRE-driven luciferase activity to a similar extent as the GFP-Ainp1-3XNLS and GFP-Ainp1 (Fig. 4D and 4E, right). Collectively, we concluded that nuclear localization of the Ainp1 protein is not required to suppress the HIF-1α function.
3.4 Ainp1 decreases the nuclear ARNT levels by sequestering ARNT in the cytoplasm
Next, we explored whether the nuclear ARNT levels would be suppressed in the presence of Ainp1 in Hep3B cells. We observed that the nuclear ARNT levels were reduced in the presence of GFP-Ainp1, but not GFP, and the cytoplasmic ARNT levels were simultaneously increased when compared to the levels in untransfected Hep3B cells (Fig. 5, and S4A, supplementary content). The subcellular contents were validated by the nuclear and cytoplasmic marker proteins lamin A/C and GAPDH, recpectively (Fig. S4, supplementary content). The total ARNT content, however, was not altered in the presence of Ainp1 in Hep3B cells (Fig. S4B, supplementary content). In addition, both nuclear and cytoplasmic ARNT levels were not altered when Ainp1 was localized in the nucleus by fusing a nuclear localization signal (3XNLS) to Ainp1 (Fig. 5 and S4C, supplementary content), suggesting that ARNT may be retained by the cytoplasmic presence of Ainp1. To further explore this cytoplasmic retention of ARNT by Ainp1, we made the NES fusion of Ainp1 to examine whether ARNT could be retained more clearly in the cytoplasm when Ainp1 is cytoplasmic. We observed that the nuclear ARNT levels were reduced to a discernible extent when Ainp1 was mostly cytoplasmic while the cytoplasmic ARNT levels were increased (Fig. 4E, 5 and S4D, supplementary content). We were surprised to see substantial cytoplasmic ARNT levels in Hep3B cells since ARNT is considered primarily nuclear. In an effort to rule out any unforeseen artifact, we used the same extraction protocol to obtain the cytoplasmic and nuclear fractions of Hepa1c1c7 cells which are known to have nuclear ARNT. We observed that ARNT was primarily nuclear in Hepa1c1c7 cells (Fig. S4E, supplementary content), suggesting that the presence of cytoplasmic ARNT in Hep3B cells was likely cell line-specific rather than a protocol artifact. Taken together, our data suggested that Ainp1 sequesters ARNT in the cytoplasm which in turn decreases the nuclear ARNT levels.
3.5 Ainp1 causes ARNT-dependent cell death which is not CoCl2-dependent
Since Ainp1 suppressed the HIF-1 transcriptional activity, we addressed whether Ainp1 would suppress the HIF-1α-driven cell proliferation. We observed that CoCl2 caused a modest increase in the rate of Hep3B cell proliferation: Hep3B cells grew to 140% of the absorbance at 560 nm after 48 h whereas only 36 h was needed to reach the same percentage in the presence of CoCl2 (Fig. 6A and B, untreated (WT) cells). GFP did not seem to have any effect on cell proliferation in the presence or absence of CoCl2. But when cells were transfected with GFP-Ainp1, cell proliferation was suppressed with or without CoCl2, suggesting that Ainp1 suppresses cell growth regardless if HIF-1α is expressed or not. This suppression was partly reversed by ARNT (Fig. 6C), suggesting that the Ainp1 effect on cell viability may be ARNT-mediated.
4. Discussion
Inhibition of the HIF-1α function appears to be an effective means to treat solid tumors. Thus there is an immense interest in limiting the cellular HIF-1α levels via various mechanisms such as (1) suppression of its message levels using a HIF-1α-targeting antisense oligonucleotide [28]; (2) suppression of its protein synthesis by topoisomerase inhibitors [29]; (3) promotion of its protein degradation by Hsp90 inhibitors [30], and (4) suppression of the HIF-1 binding to enhancers by anthracyclines [31]. Although it has been shown that the endogenous ARNT levels are quite abundant [32], an adequate amount of an ARNT-interacting protein can be expressed in cells to interfere with the ARNT-mediated HIF-1function [22, 27]. Interestingly our data revealed that there might be a dynamic process of maintaining the nuclear ARNT levels in a cell. An ARNT-interacting peptide may affect the nuclear ARNT levels by its mere presence in the cytoplasm. Any mechanism that decreases the nuclear ARNT levels would undoubtedly inhibit the HIF-1α function. We propose that ARNT is potentially a good target for anticancer drug development and Ainp1 may serve as a prototype for protein therapeutics. For example, high affinity ARNT-interacting peptides which reside exclusively in the cytoplasm may downregulate the HIF-1α function by effectively reducing the nuclear ARNT levels via cytoplasmic retention. This approach of utilizing phage display peptides (such as Ainp1) for anticancer drug design has been employed to develop selective angiopoietin-2 inhibitors to block angiogenesis [33].
We are interested in utilizing Ainp1 to understand how the HIF-1α pathway can be suppressed. Our data revealed that Ainp1 is capable of interfering with the interaction between ARNT and HIF-1α by physically competing for ARNT binding in Hep3B cells. However, this is unlikely to be the sole mechanism for suppressing the HIF-1 function in Hep3B cells because the Ainp1 effect does not require it to be in the nucleus and the binding competition should only occur in the nucleus. We have previously observed a similar phenomenon: we discovered that human β4-tubulin interacts with ARNT; when we transfected β4-tubulin into Hep3B cells, the transfected protein resided primarily in the cytoplasm and caused suppression of the ARNT-dependent, DRE-driven luciferase expression [34]. Apparently, Ainp1 suppresses the HIF-1 function regardless of whether Ainp1 is in the cytoplasm or in the nucleus since all Ainp1 constructs – GFP-Ainp1, GFP-Ainp1-3XNLS, and GFP-Ainp1-NES – suppressed the reporter luciferase activity to a similar extent. Suppression of this luciferase activity is likely due to a summation of two effects: (1) Ainp1 reduces the nuclear ARNT levels by sequestering ARNT in the cytoplasm and (2) Ainp1 inhibits the HIF-1α::ARNT heterodimer formation in the nucleus. When we manipulated the Ainp1 peptide to be exclusively cytoplasmic (GFP-Ainp1 in Fig. 4B versus GFP-Ainp1-NES in Fig. 4E), we observed a further decrease of the nuclear ARNT levels and a concomitant increase of the cytoplasmic ARNT levels (Fig. 5). This shift reflects the Ainp1 ability of retaining ARNT in the cytoplasm. We did not observe a statistically significant increase of the cytoplasmic ARNT levels between GFP-Ainp1 and GFP-Ainp1-NES (0.05 < p < 0.1) probably owing to the challenge of quantifying a small amount of the cytoplasmic ARNT content compounded with the nonspecific fluorescence. In addition to the two Ainp1 effects we have described, one could argue that other mechanisms may be involved in the Ainp1 suppression of the HIF-1α function; for example, we cannot rule out the possibility that the cytoplasmic retention of ARNT would somehow hamper the activation of HIF-1α. Nevertheless, our data unambiguously showed that Ainp1 causes a shift of the cellular ARNT contents from nucleus to cytoplasm in Hep3B cells (Fig. 5). The precise mechanism that causes the redistribution of the cellular ARNT contents is unclear at present. We speculated that there are at least two mechanisms that would explain this cytoplasmic Ainp1 action: (1) trafficking of ARNT between the nuclear and cytoplasmic compartments may happen which allows the ARNT::Ainp1 interaction to occur in the cytoplasm and (2) Ainp1 binds to the newly synthesized ARNT in the cytoplasm and retards its nuclear entry.
HIF-1α appears to express only in the presence of CoCl2 or hypoxia in Hep3B cells. There is a modest but noticeable increase in the growth rate when cells were treated with 100 μM CoCl2, suggesting that HIF-1α increases the rate of cell proliferation to a limited extent. We were unclear about the role of ARNT in untreated cells and initially assumed that the ARNT function in Hep3B cells might only become evident when HIF-1α is expressed. Interestingly, our data supported that ARNT may promote growth in untreated Hep3B cells: Ainp1 causes cell death in a HIF-1α-independent manner and this Ainp1-triggered cell death can be partly reversed in the presence of the transfected ARNT. However, we cannot rule out the possibility that ARNT nullifies the Ainp1 effect by its association with Ainp1, and the Ainp1-mediated suppression of cell growth could be ARNT-independent. It has been reported that ARNT may play a role in the c-Jun/Sp1 function [35] and inhibition of the upstream MEF kinase, which activates c-Jun, causes apoptosis in HepG2 cells [36]. However, we did not observe any formation of the caspase 3 large fragment in the Ainp1-transfected Hep3B cells (Fig. S4B, supplementary content), suggesting that apoptosis is likely not involved in the Ainp1-mediated cell death. It is also possible that this Ainp1-induced cell death is somehow AhR-mediated since AhR is known to affect the cell cycle. Some investigators showed that AhR downregulates c-myc expression in human breast cancer cells without the addition of an exogenous ligand [37]. However, the same investigators were not able to observe any change in cell growth when the AhR function was inhibited by an ARNT-interacting protein (AhRR).
In summary, we concluded that Ainp1 is capable of inhibiting the HIF-1α function via an ARNT-dependent mechanism: Ainp1 suppresses the HIF-1α and ARNT heterodimer formation, suppresses the formation of the HIF-1/HRE gel shift complex, reduces the recruitment of ARNT at the HRE promoter, and suppresses the HIF-1-dependent activation of gene transcription. Interestingly, Ainp1 reduces the nuclear ARNT levels by sequestering ARNT in the cytoplasm. Thus, our data revealed that the nuclear ARNT levels can be reduced in the presence of an ARNT-interacting peptide in the cytoplasm.
Supplementary Material
Acknowledgments
This work is supported by the National Institutes of Health (R01 ES014050).
Abbreviations
- Ainp1
ARNT-interacting peptide-1
- ARNT
aryl hydrocarbon receptor nuclear translocator
- HIF-1α
hypoxia inducible factor-1 alpha
- AhR
aryl hydrocarbon receptor
- ERα
estrogen receptor alpha
- HRE
hypoxia response element
- DRE
dioxin response element
- ERE
estrogen receptor response element
- 3MC
3-methylchloranphrene
- E2
17 beta-estradiol
- HEDG
25 mM HEPES, pH 7.4, 1 mM EDTA, 1 mM DTT, and 10% glycerol
- EV
empty vector
- RRL
rabbit reticulocyte lysate
Footnotes
Conflict of Interest
All authors have no conflict of interest that would bias this work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Reisz-Porszasz S, Probst MR, Fukunaga BN, Hankinson O. Identification of functional domains of the aryl hydrocarbon receptor nuclear translocator protein (ARNT) Mol Cell Biol. 1994;14:6075–6086. doi: 10.1128/mcb.14.9.6075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic- helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wiesener MS, Turley H, Allen WE, Willam C, Eckardt KU, Talks KL, Wood SM, Gatter KC, Harris AL, Pugh CW, Ratcliffe PJ, Maxwell PH. Induction of endothelial PAS domain protein-1 by hypoxia: characterization and comparison with hypoxia- inducible factor-1alpha. Blood. 1998;92:2260–2268. [PubMed] [Google Scholar]
- 4.Brunnberg S, Pettersson K, Rydin E, Matthews J, Hanberg A, Pongratz I. The basic helix-loop-helix-PAS protein ARNT functions as a potent coactivator of estrogen receptor- dependent transcription. Proc Natl Acad Sci USA. 2003;100:6517–6522. doi: 10.1073/pnas.1136688100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wright CW, Duckett CS. The aryl hydrocarbon nuclear translocator alters CD30- mediated NF-kappaB-dependent transcription. Science. 2009;323:251–255. doi: 10.1126/science.1162818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maltepe E, Schmidt JV, Baunoch D, Bradfield CA, Simon MC. Abnormal angiogenesis and responses to glucose and oxygen deprivation in mice lacking the protein ARNT. Nature. 1997;386:403–407. doi: 10.1038/386403a0. [DOI] [PubMed] [Google Scholar]
- 7.Kozak KR, Abbott B, Hankinson O. ARNT-deficient mice and placental differentiation. Dev Biol. 1997;191:297–305. doi: 10.1006/dbio.1997.8758. [DOI] [PubMed] [Google Scholar]
- 8.Gunton JE, Kulkarni RN, Yim S, Okada T, Hawthorne WJ, Tseng YH, Roberson RS, Ricordi C, O'Connell PJ, Gonzalez FJ, Kahn CR. Loss of ARNT/HIF-1beta mediates altered gene expression and pancreatic-islet dysfunction in human type 2 diabetes. Cell. 2005;122:337–349. doi: 10.1016/j.cell.2005.05.027. [DOI] [PubMed] [Google Scholar]
- 9.Wang XL, Suzuki R, Lee K, Tran T, Gunton JE, Saha AK, Patti ME, Goldfine A, Ruderman NB, Gonzalez FJ, Kahn CR. Ablation of ARNT/HIF-1beta in liver alters gluconeogenesis, lipogenic gene expression, and serum ketones. Cell Metab. 2009;9:428–439. doi: 10.1016/j.cmet.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pillai R, Huypens P, Huang M, Schaefer S, Sheinin T, Wettig SD, Joseph JW. Aryl hydrocarbon receptor nuclear translocator/hypoxia-inducible factor-1β plays a critical role in maintaining glucose-stimulated anaplerosis and insulin release from pancreatic β-cells. J Biol Chem. 2011;286:1014–1024. doi: 10.1074/jbc.M110.149062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Talks KL, Turley H, Gatter KC, Maxwell PH, Pugh CW, Ratcliffe PJ, Harris AL. The expression and distribution of the hypoxia-inducible factors HIF-1alpha and HIF- 2alpha in normal human tissues, cancers, and tumor-associated macrophages. Am J Pathol. 2000;157:411–421. doi: 10.1016/s0002-9440(10)64554-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rankin EB, Giaccia AJ. The role of hypoxia-inducible factors in tumorigenesis. Cell Death Differ. 2008;15:678–685. doi: 10.1038/cdd.2008.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arany Z, Huang LE, Eckner R, Bhattacharya S, Jiang C, Goldberg MA, Bunn HF, Livingston DM. An essential role for p300/CBP in the cellular response to hypoxia. Proc Natl Acad Sci USA. 1996;93:12969–12973. doi: 10.1073/pnas.93.23.12969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carrero P, Okamoto K, Coumailleau P, O'Brien S, Tanaka H, Poellinger L. Redox- regulated recruitment of the transcriptional coactivators CREB-binding protein and SRC-1 to hypoxia-inducible factor 1alpha. Mol Cell Biol. 2000;20:402–415. doi: 10.1128/mcb.20.1.402-415.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beischlag TV, Taylor RT, Rose DW, Yoon D, Chen Y, Lee WH, Rosenfeld MG, Hankinson O. Recruitment of thyroid hormone receptor/retinoblastoma-interacting protein 230 by the aryl hydrocarbon receptor nuclear translocator is required for the transcriptional response to both dioxin and hypoxia. J Biol Chem. 2004;279:54620–54628. doi: 10.1074/jbc.M410456200. [DOI] [PubMed] [Google Scholar]
- 16.Laughner E, Taghavi P, Chiles K, Mahon PC, Semenza GL. HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1alpha (HIF-1alpha) synthesis: novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol Cell Biol. 2001;21:3995–4004. doi: 10.1128/MCB.21.12.3995-4004.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Richard DE, Berra E, Gothie E, Roux D, Pouyssegur J. p42/p44 mitogen-activated protein kinases phosphorylate hypoxia-inducible factor 1alpha (HIF-1alpha) and enhance the transcriptional activity of HIF-1. J Biol Chem. 1999;274:32631–32637. doi: 10.1074/jbc.274.46.32631. [DOI] [PubMed] [Google Scholar]
- 18.Minet E, Arnould T, Michel G, Roland I, Mottet D, Raes M, Remacle J, Michiels C. ERK activation upon hypoxia: involvement in HIF-1 activation. FEBS Lett. 2000;468:53–58. doi: 10.1016/s0014-5793(00)01181-9. [DOI] [PubMed] [Google Scholar]
- 19.Li Y, Luu TC, Chan WK. A novel ARNT-interacting protein Ainp2 enhances the aryl hydrocarbon receptor signaling. Arch Biochem Biophys. 2005;441:84–95. doi: 10.1016/j.abb.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 20.Chan WK, Chu R, Jain S, Reddy JK, Bradfield CA. Baculovirus expression of the Ah receptor and Ah receptor nuclear translocator. Evidence for additional dioxin responsive element-binding species and factors required for signaling. J Bio Chem. 1994;269:26464–26471. [PubMed] [Google Scholar]
- 21.Jin Q, Yan T, Ge X, Sun C, Shi X, Zhai Q. Cytoplasm-localized SIRT1 enhances apoptosis. J Cell Physiol. 2007;213:88–97. doi: 10.1002/jcp.21091. [DOI] [PubMed] [Google Scholar]
- 22.Jensen KA, Luu TC, Chan WK. A truncated Ah receptor blocks the hypoxia and estrogen receptor signaling pathways: a viable approach for breast cancer treatment. Mol Pharm. 2006;3:695–703. doi: 10.1021/mp0600438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schagger H. Tricine-SDS–PAGE. Nat Protoc. 2006;1:16–22. doi: 10.1038/nprot.2006.4. [DOI] [PubMed] [Google Scholar]
- 24.Li Y, Li Y, Zhang T, Chan WK. The aryl hydrocarbon receptor nuclear translocator- interacting protein 2 suppresses the estrogen receptor signaling via an ARNT-dependent mechanism. Arch Biochem Biophys. 2010;502:121–129. doi: 10.1016/j.abb.2010.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suzuki H, Tomida A, Tsuruo T. Dephosphorylated hypoxia-inducible factor 1alpha as a mediator of p53-dependent apoptosis during hypoxia. Oncogene. 2001;20:5779–5788. doi: 10.1038/sj.onc.1204742. [DOI] [PubMed] [Google Scholar]
- 26.Weir L, Robertson D, Leigh IM, Vass JK, Panteleyev AA. Hypoxia-mediated control of HIF/ARNT machinery in epidermal keratinocytes. Biochim Biophys Acta. 2011;1813:60–72. doi: 10.1016/j.bbamcr.2010.11.014. [DOI] [PubMed] [Google Scholar]
- 27.Wang D, Faridi JS, Li Y, Chan WK. A truncated human Ah receptor suppresses growth of human cervical tumor xenografts by interfering with hypoxia signaling. FEBS Lett. 2009;583:3039–3044. doi: 10.1016/j.febslet.2009.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Greenberger LM, Horak ID, Filpula D, Sapra P, Westergaard M, Frydenlund HF, Albaek C, Schroder H, Orum H. A RNA antagonist of hypoxia-inducible factor-1alpha, EZN-2968, inhibits tumor cell growth. Mol Cancer Ther. 2008;7:3598–3608. doi: 10.1158/1535-7163.MCT-08-0510. [DOI] [PubMed] [Google Scholar]
- 29.Rapisarda A, Uranchimeg B, Sordet O, Pommier Y, Shoemaker RH, Melillo G. Topoisomerase I-mediated inhibition of hypoxia-inducible factor 1: mechanism and therapeutic implications. Cancer Res. 2004;64:1475–1482. doi: 10.1158/0008-5472.can-03-3139. [DOI] [PubMed] [Google Scholar]
- 30.Isaacs JS, Jung YJ, Mimnaugh EG, Martinez A, Cuttitta F, Neckers LM. Hsp90 regulates a von Hippel Lindau-independent hypoxia–inducible factor-1 alpha-degradative pathway. J Biol Chem. 2002;277:29936–29944. doi: 10.1074/jbc.M204733200. [DOI] [PubMed] [Google Scholar]
- 31.Lee K, Qian DZ, Rey S, Wei H, Liu JO, Semenza GL. Anthracycline chemotherapy inhibits HIF-1 transcriptional activity and tumor-induced mobilization of circulating angiogenic cells. Proc Natl Acad Sci USA. 2009;106:2353–2358. doi: 10.1073/pnas.0812801106. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32.Pollenz RS, Davarinos NA, Shearer TP. Analysis of aryl hydrocarbon receptor- mediated signaling during physiological hypoxia reveals lack of competition for the aryl hydrocarbon nuclear translocator transcription factor. Mol Pharmacol. 56:1127–1137. doi: 10.1124/mol.56.6.1127. [DOI] [PubMed] [Google Scholar]
- 33.Huang H, Lai JY, Do J, Liu D, Li L, Del Rosario J, Doppalapudi VR, Pirie-Shepherd S, Levin N, Bradshaw C, Woodnutt G, Lappe R, Bhat A. Specifically targeting angiopoietin-2 inhibits angiogenesis, tie2-expressing monocyte infiltration, and tumor growth. Clin Cancer Res. 2011;17:1001–1011. doi: 10.1158/1078-0432.CCR-10-2317. [DOI] [PubMed] [Google Scholar]
- 34.Zhang T, Wang X, Shinn A, Jin J, Chan WK. Beta tubulin affects the aryl hydrocarbon receptor function via an ARNT-mediated mechanism. Biochem Pharmacol. 2010;79:1125–1133. doi: 10.1016/j.bcp.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang WC, Chen ST, Chang WC, Chang KY, Chen BK. Involvement of aryl hydrocarbon receptor nuclear translocator in EGF-induced c-Jun/Sp1-mediated gene expression. Cell Mol Life Sci. 2010;67:3523–3533. doi: 10.1007/s00018-010-0392-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huynh H, Nguyen TT, Chow KH, Tan PH, Soo KC, Tran E. Over-expression of the mitogen-activated protein kinase (MAPK) kinase (MEK)-MAPK in hepatocellular carcinoma: its role in tumor progression and apoptosis. BMC Gastroenterol. 2003;3:19. doi: 10.1186/1471-230X-3-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang X, Liu D, Murray TJ, Mitchell GC, Hesterman EV, Karchner SI, Merson RR, Hahn ME, Sherr DH. The aryl hydrocarbon receptor constitutively represses c-myc transcription in human mammary tumor cells. Oncogene. 2005;24:7869–7881. doi: 10.1038/sj.onc.1208938. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.