

Abstract
The gene ULK1 is an excellent candidate for Crohn's disease (CD) due to its role in autophagy. A recent study provided evidence for the involvement of ULK1 in the pathogenesis of CD (Henckaerts et al., 2011). We attempted to validate this association, using a candidate gene SNP study of ULK1 in CD. We identified tagging SNPs and genotyped these SNPs using the Sequenom platform in a Caucasian New Zealand dataset consisting of 406 CD patients and 638 controls. In this sample, we were able to demonstrate an association between CD and several different ULK1 SNPs and haplotypes. Phenotypic analysis showed an association with age of diagnosis 17–40 years and inflammatory behaviour. The findings of this study provide evidence to suggest that genetic variation in ULK1 may play a role in interindividual differences in CD susceptibility and clinical outcome.
1. Introduction
Crohn's disease (CD) is a form of inflammatory bowel disease (IBD) characterized by chronic, relapsing gastrointestinal inflammation. It results from multiple genetic and multiple environmental risk factors, operating additively and interactively. In recent years, the search for genetic determinants of CD has changed dramatically with the introduction of the Genome Wide Association Study (GWAS) technology from which results have been excellent. As well as helping to identify multiple susceptibility loci involved in the genetic susceptibility to CD, GWAS has also provided evidence for the involvement of biological pathways such as autophagy. Autophagy is a well-conserved regulatory process by which protein and organelle turnover occurs in cells by autodigestion through lysosomal degradation. The pathway can interact with other vital processes such as programmed cell death, inflammation, and immune mechanisms. Autophagy has several roles in innate and adaptive immunity including pattern recognition receptor signalling, regulation of cell death, elimination of bacteria and viruses, and immune cell homeostasis [1–3]. Thus, it is thought that CD may result from a defective autophagy pathway causing an impaired antibacterial response and so an ineffective control of bacterial infection, dysbiosis of the intestinal microbiota, and chronic inflammation.
There are many genes in the autophagy pathway that have been previously associated with CD. They code for proteins involved in the detection of autophagic triggers (IRGM [4–6], NOD2 [7–9], VDR [10], and DAP1 [11, 12]), orchestrating autophagosome formation (ATG16L1 [13, 14]), or autophagosomal maturation (LRRK2 [15]). Mutations in these autophagy-related genes may lead to loss of autophagic function and the subsequent development of Crohn's disease.
A recent study investigating a number of autophagy genes for their involvement in CD has described a novel association between Unc-51-like kinase-1 (ULK1) and CD [16]. ULK1 is a serine/threonine protein kinase that plays a critical role in the initial stages of autophagy, although the exact molecular mechanism is unknown.
Here, we attempted to validate the association of ULK1 with CD in a well-characterised case-control New Zealand dataset. We considered not only allele and genotype frequencies, but also the question as to whether genotype could predict phenotype since this is an essential tool in understanding disease behaviour and future treatment requirements [17].
2. Methods
2.1. Samples
A total of 1044 subjects from New Zealand were included in the study: 406 CD patients and 638 controls. All participants self-reported European ancestry.
Clinical records were analysed to confirm diagnosis, and IBD status was defined using standard diagnostic criteria [18]. Cases were phenotyped according to the Montreal Classification systems. Clinical characteristics of the CD patients are shown in Table 1.
Table 1.
Summary of clinical data of CD patients.
| CD | ||
|---|---|---|
| Gender | F | 265 (65.6) |
| M | 139 (34.4) | |
| Age at diagnosis | <17 | 46 (12.6) |
| 17 to 40 | 257 (70.2) | |
| 40< | 63 (17.2) | |
| CD behaviour | Inflammatory | 201 (55.1) |
| Stricturing | 118 (32.3) | |
| Penetrating | 46 (12.6) | |
| CD location | Ileal | 136 (37.2) |
| Colonic | 119 (32.5) | |
| Ileocolonic | 111 (30.3) | |
| Bowel resection | N | 270 (66.7) |
| Y | 135 (33.3) | |
| Other IBD family | N | 330 (89.7) |
| Y | 38 (10.3) | |
| Perianal disease | N | 329 (85.7) |
| Y | 55 (14.3) |
Participants consented to collection of peripheral blood or a buccal swab for DNA extraction and genotyping, and DNA was extracted from the blood/buccal samples using Qiagen DNA extraction kit and following the manufacturer's instructions.
The study was conducted under ethical protocol MEC/04/12/011, authorised through the New Zealand Multi-Region Human Ethics Committee. All study subjects gave informed consent.
2.2. SNP Selection
Tag SNPs in ULK1 were selected using HapMap release 28 and the tagger functionality within Haploview with pairwise tagging to identify SNPs using an r 2 > 0.8 and a minor allele frequency >5%. As a result, nine tag SNPs were selected for genotyping: rs10902469, rs7133672, rs7953348, rs7488085, rs11616018, rs12303764, rs4964879, rs3088051, and rs3923716.
2.3. Genotyping
Genotyping was performed with the MassARRAY and iPlex systems of the Sequenom genotyping platform (Sequenom, San Diego, CA), which uses the MALDI-TOF primer extension assay [19, 20], according to manufacturers' recommendations.
Assays were optimized in 24 samples consisting of 20 reference Centre d'Etude du Polymorphisme Humain (CEPH) samples and 4 blanks.
All sample plates contained cases, controls, blanks, CEPH, and duplicate samples. Quality control measures included independent double genotyping and, where available, comparison of our CEPH genotypes to those in the Hapmap database (http://www.hapmap.org/).
2.4. Statistical Analysis
SNPs were tested for deviation from HWE in both cases and controls using a chi-square goodness-of-fit test. To determine if there were differences between cases and controls, allele frequencies for each SNP were analyzed using 2 × 2 chi-square tables.
Genotype and phenotype associations were assessed by comparing allele frequencies between controls and patient subgroups defined using the clinical characteristics. These analyses were carried out using R (R: a language and environment for statistical computing, R Foundation for Statistical Computing, Vienna, Austria. ISBN 3–900051-07-0, URL http://www.R-project.org/) and SAS (V9.1 SAS Institute., Cary, NC, USA).
To determine linkage disequilibrium (LD) between SNPs and to define haplotype blocks, we uploaded our data into Haploview [21]. Haplotype blocks were defined using the default algorithm, which uses confidence intervals [22]. Haplotype analysis was carried using HAPLO.SCORE in R to test for association of these haplotypes with CD.
For all analyses we considered a P value less than 0.05 to indicate statistical significance.
The false discovery rate (FDR) was used to correct for multiple testing [23, 24].
3. Results
Two SNPs, rs7133672 and rs4964879, failed in the genotyping assay. The remaining seven SNPs were all genotyped successfully and were in Hardy-Weinberg equilibrium in both cases and controls.
3.1. Association Analysis
From the seven genotyped SNPs, we saw association with CD for two SNPs. The G allele of SNP rs10902469 was more frequent (95.4%) in the cases compared to controls (92.5%), OR = 1.69, P = 0.0084. The T allele of SNP rs7488085 was more frequent (93.7%) in the cases compared to controls (91.1%), OR = 1.46, P = 0.030. These SNPs remain statistically significant if we correct for multiple testing using the false discovery rate. Genotype and allele counts/frequencies and P values for all genotyped SNPs are shown in Table 2.
Table 2.
Genotype and allele counts (and frequencies) in CD patients and in controls.
| SNP | Case | Control | Case | Control | OR (955 CI) | P | ||
|---|---|---|---|---|---|---|---|---|
| rs10902469 | G/G | 367 (90.8) | 544 (0.86) | G | 771 (95.4) | 1177 (92.5) | 1.69 (1.14–2.50) | 0.0084* |
| C/G | 37 (9.2) | 89 (0.14) | C | 37 (4.6) | 95 (7.5) | |||
| C/C | 0 (0.00) | 3 (0.00) | ||||||
| rs7953348 | T/T | 276 (69.0) | 406 (65.8) | T | 663 (82.9) | 993 (80.5) | 1.17 (0.93–1.46) | 0.17 |
| C/T | 111 (27.8) | 181 (29.3) | C | 137 (17.1) | 241 (19.5) | |||
| C/C | 13 (3.3) | 30 (4.9) | ||||||
| rs7488085 | T/T | 355 (87.7) | 529 (82.8) | T | 759 (93.7) | 1164 (91.1) | 1.46 (1.03–2.06) | 0.030* |
| C/T | 49 (12.1) | 106 (16.6) | C | 51 (6.3) | 114 (8.9) | |||
| C/C | 1 (0.2) | 4 (0.6) | ||||||
| rs11616018 | T/T | 281 (70.6) | 419 (66.3) | T | 668 (83.9) | 1022 (80.9) | 1.23 (0.98–1.55) | 0.078 |
| C/T | 106 (26.6) | 184 (29.1) | C | 128 (16.1) | 242 (19.1) | |||
| C/C | 11 (2.8) | 29 (4.6) | ||||||
| rs12303764 | T/T | 165 (40.8) | 254 (40.3) | T | 515 (63.7) | 788 (62.5) | 1.06 (0.88–1.26) | 0.58 |
| G/T | 185 (45.8) | 280 (44.4) | G | 293 (36.3) | 472 (37.5) | |||
| G/G | 54 (13.4) | 96 (15.2) | ||||||
| rs3088051 | T/T | 193 (47.7) | 331 (52.2) | T | 560 (69.1) | 921 (72.6) | 1.20 (0.99–1.46) | 0.086 |
| C/T | 174 (43.0) | 259 (40.9) | C | 250 (30.9) | 347 (27.4) | |||
| C/C | 38 (9.4) | 44 (6.9) | ||||||
| rs3923716 | C/C | 333 (82.4) | 506 (79.9) | C | 735 (91.0) | 1134 (89.6) | 1.17 (0.86–1.59) | 0.30 |
| A/C | 69 (17.1) | 122 (19.3) | A | 173 (9.0) | 132 (10.4) | |||
| A/A | 2 (0.5) | 5 (0.8) |
*Remain statistically significant after applying a multiple testing correction using FDR.
3.2. Phenotypic Analysis
The two SNPs that were associated with CD (rs10902469 and rs7488085) were both associated with age of diagnosis 17 to 40 years (OR = 1.90, P = 0.010 and OR = 1.53, P = 0.044) and inflammatory disease (OR = 2.63, P = 0.002 and OR = 1.79, P = 0.018). SNP rs10902469 was also associated with colonic disease (OR = 2.33, P = 0.025). SNP rs3088051 was associated with stricturing (OR = 1.45, P = 0.015) and ileal (OR = 1.34, P = 0.042) disease and bowel resection (OR = 1.58, P = 0.002). The other 4 SNPs did not demonstrate any associations with any subphenotypes. Full phenotype results are shown in Table 3. All of the significant findings remained significant after multiple testing correction, with the exception of the association of rs7488085 with age of diagnosis 17–40.
Table 3.
Phenotypic analysis results.
| rs10902469: G | rs7953348: T | rs7488085: T | rs11616018: T | rs12303764: T | rs3088051: C | rs3923716: C | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OR (95% CI) | P | OR (95% CI) | P | OR (95% CI) | P | OR (95% CI) | P | OR (95% CI) | P | OR (95% CI) | P | OR (95% CI) | P | ||
| Age at diagnosis | <17 | 0.98 (0.44–2.18) | 0.953 | 0.97 (0.58–1.64) | 0.912 | 0.90 (0.44–1.85) | 0.770 | 1.15 (0.66–2.01) | 0.634 | 0.94 (0.61–1.43) | 0.761 | 1.05 (0.65–1.70) | 0.836 | 0.93 (0.46–1.86) | 0.827 |
| 17 to 40 | 1.90 (1.17–3.10) | 0.010* | 1.11 (0.86–1.44) | 0.436 | 1.53 (1.01–2.32) | 0.044 | 1.15 (0.88–1.50) | 0.298 | 1.17 (0.95–1.45) | 0.142 | 1.18 (0.94–1.48) | 0.165 | 1.03 (0.73–1.46) | 0.852 | |
| 40< | 1.37 (0.62–3.04) | 0.435 | 1.35 (0.82–2.22) | 0.241 | 1.14 (0.57–2.25) | 0.715 | 1.35 (0.81–2.26) | 0.245 | 1.11 (0.77–1.61) | 0.580 | 1.33 (0.90–1.97) | 0.151 | 1.53 (0.75–3.11) | 0.243 | |
| CD behaviour |
Inflammatory | 2.63 (1.42–4.86) | 0.002* | 1.25 (0.93–1.68) | 0.139 | 1.79 (1.10–2.91) | 0.018* | 1.25 (0.93–1.69) | 0.143 | 1.07 (0.85–1.35) | 0.554 | 1.06 (0.82–1.36) | 0.667 | 1.19 (0.80–1.76) | 0.396 |
| Stricturing | 1.19 (0.68–2.10) | 0.547 | 1.00 (0.71–1.41) | 0.994 | 1.19 (0.70–2.01) | 0.516 | 1.17 (0.82–1.68) | 0.392 | 1.20 (0.89–1.60) | 0.229 | 1.45 (1.08–1.95) | 0.015* | 1.02 (0.64–1.63) | 0.943 | |
| Penetrating | 0.84 (0.39–1.81) | 0.658 | 0.97 (0.58–1.63) | 0.912 | 0.72 (0.37–1.39) | 0.323 | 0.95 (0.57–1.60) | 0.850 | 1.29 (0.83–2.01) | 0.265 | 1.17 (0.73–1.86) | 0.509 | 0.85 (0.44–1.65) | 0.633 | |
| CD location |
Ileal | 1.62 (0.89–2.94) | 0.116 | 1.20 (0.85–1.69) | 0.295 | 1.57 (0.91–2.70) | 0.104 | 1.22 (0.87–1.73) | 0.254 | 1.11 (0.85–1.45) | 0.430 | 1.34 (1.01–1.78) | 0.042* | 1.15 (0.73–1.82) | 0.541 |
| Colonic | 2.33 (1.11–4.89) | 0.025* | 1.34 (0.92–1.95) | 0.124 | 1.58 (0.88–2.81) | 0.123 | 1.33 (0.91–1.95) | 0.137 | 1.08 (0.82–1.44) | 0.586 | 1.00 (0.73–1.37) | 0.992 | 1.27 (0.77–2.09) | 0.345 | |
| Ileocolonic | 1.19 (0.66–2.13) | 0.565 | 0.89 (0.63–1.25) | 0.499 | 0.99 (0.59–1.64) | 0.954 | 1.01 (0.71–1.45) | 0.951 | 1.19 (0.89–1.60) | 0.245 | 1.21 (0.88–1.66) | 0.237 | 0.86 (0.54–1.36) | 0.517 | |
| Other IBD family | Y | 6.10 (0.84–44.4) | 0.075 | 1.01 (0.73–1.39) | 0.968 | 3.67 (0.88–15.2) | 0.073 | 1.40 (0.72–2.73) | 0.327 | 1.08 (0.68–1.74) | 0.737 | 1.24 (0.75–2.04) | 0.412 | 2.07 (0.74–5.81) | 0.169 |
| Bowel resection | Y | 1.12 (0.66–1.90) | 0.674 | 0.99 (0.56–1.74) | 0.963 | 1.09 (0.68–1.77) | 0.717 | 1.22 (0.86–1.73) | 0.270 | 1.18 (0.90–1.56) | 0.225 | 1.58 (1.19–2.10) | 0.002* | 0.95 (0.61–1.46) | 0.802 |
| Perianal disease | Y | 1.70 (0.67–4.29) | 0.262 | 0.74 (0.48–1.16) | 0.186 | 0.79 (0.42–1.50) | 0.473 | 0.82 (0.52–1.30) | 0.398 | 1.46 (0.96–2.23) | 0.079 | 1.10 (0.71–1.71) | 0.676 | 0.62 (0.36–1.09) | 0.099 |
*Remain statistically significant after applying a multiple testing correction using FDR.
3.3. Haplotypic Analysis
Figure 1 shows the LD plot for the ULK1 SNPs in our New Zealand dataset. Five SNPs are in the same haplotype block: rs10902469, rs7953348, rs7488085, rs11616018, and rs12303764. Table 4 summarises haplotype analysis results but in brief three haplotypes were found to be statistically significant in their association with CD. Haplotype CCCCT was protective in that it was more frequent in the controls (0.066) compared to cases (0.036), P = 0.005. Haplotype GCTTT was protective in that it was more frequent in the controls (0.019) compared to cases (0.006), P = 0.038. Haplotype GTTTT was more frequent in the cases (0.455) compared to controls (0.406), P = 0.027. However, after applying multiple testing correction these haplotypes were no longer statistically significant.
Figure 1.
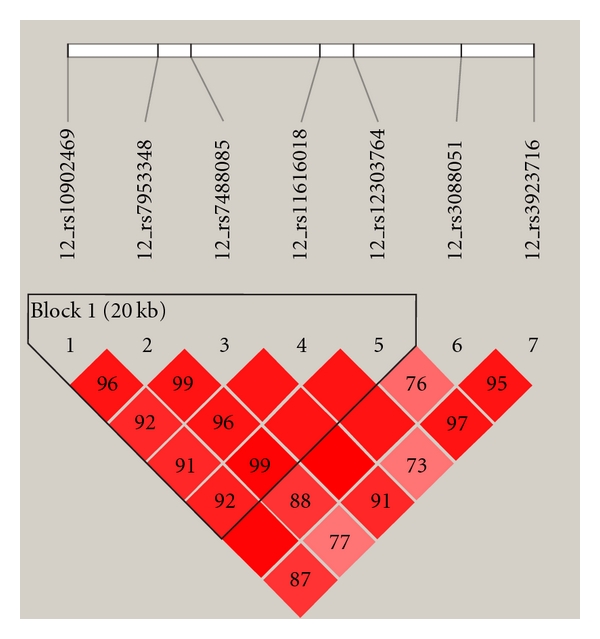
ULK1 LD plot.
Table 4.
ULK1 haplotype analysis.
| rs10902469 | rs7953348 | rs7488085 | rs11616018 | rs12303764 | Hap-score | P | Hap-frequency | |
|---|---|---|---|---|---|---|---|---|
| Control | Case | |||||||
| C | C | C | C | T | −2.828 | 0.005 | 0.066 | 0.036 |
| G | C | T | T | T | −2.071 | 0.038 | 0.019 | 0.006 |
| G | T | T | T | G | 0.011 | 0.991 | 0.361 | 0.363 |
| G | C | C | C | T | 0.151 | 0.880 | 0.019 | 0.020 |
| G | C | T | C | T | 0.333 | 0.739 | 0.094 | 0.098 |
| G | T | T | T | T | 2.212 | 0.027 | 0.406 | 0.455 |
4. Discussion
ULK1 is an autophagy gene that has recently been reported for the first time to be associated with CD [16]. In order to confirm the role of ULK1 in CD susceptibility we performed an independent association study in a New Zealand case–control sample set. We were able to demonstrate evidence of association for two SNPs. However, the associations we observed were different from those reported by the previous study. Henckaerts et al. [16] had the strongest association with CD for rs12303764, but this SNP was not associated with CD in our samples. They also reported weaker associations for rs10902469, rs7953348, and rs3923716. From these we only saw association in our dataset for rs10902469. We also saw association with rs7488085, which was not genotyped in the previous study. To further determine whether ULK1 is a CD susceptibility gene, we examined the data (data not shown) from a recent CD genome-wide meta-analysis [25] for SNPs in this gene. Three SNPs in ULK1 were included in the analysis: rs11246867 that is LD (r 2 = 1) with rs10902469 that was associated with CD in both our study and the study by Henckaerts et al. showed no association in the GWAS meta-analysis, rs3923716 was associated with CD in the study of Henckaerts et al. but not in our study and likewise not in the GWAS meta-analysis, and rs3088051 was not associated with CD neither in the study of Henckaerts et al. nor our study (although there is a difference in cases and controls which is approaching statistical significance) but showed association in the GWAS meta-analysis (uncorrected nominal, P = 0.00068). It is by no means certain that support for ULK1 as a CD susceptibility gene requires the same pattern of association to be obtained. Genetic heterogeneity, variation in phenotypes, and lack of power may explain some of the discrepancies. Further studies are needed in other cohorts to determine the robustness of these observations in different populations and to be certain whether ULK1 can be described as CD susceptibility gene.
Phenotypic analysis demonstrated association for the 2 CD associated SNPs with a young adult age at first diagnosis (17–40 years) and not with disease diagnosed after 40 years nor with early-onset (paediatric) disease (before 17 years). The age of diagnosis of CD in adults is known to have a bimodal distribution: the first peak occurs between the ages of 15 and 30 years, and the second peak occurs between the ages of 60 and 80 years [26]. Younger age-at-diagnosis patients represent a separate and often more severe phenotype of CD [27]. The different phenotypes seen in the different age groups are likely to be as a result of each of these groups having a different genetic component to their disease. The study we report here concludes that ULK1 has a role in patients who are diagnosed as young adults and is unlikely to be important in patients who are diagnosed after 40 years. Likewise there is no evidence to suggest ULK1 has a role in paediatric CD, although this cannot be ruled out entirely as the numbers in this group are small and so the power to detect an association here is limited.
Phenotypic analysis also demonstrated association with inflammatory CD behaviour. In terms of disease behavior inflammatory disease is the milder and less complicated form and over time some patients may develop penetrating or stricturing complications. So a strong association with inflammatory disease is difficult to interpret. But the association of ULK1 with inflammation is not surprising. ULK1 plays a critical role in the initial stages of autophagy and it is possible that genetic variation in this gene may result in autophagy-mediated control of commensal bacteria being compromised, subsequently leading to an intestinal inflammatory response to bacteria.
The results from phenotype analysis also suggested that other subphenotypes may also be affected as SNP rs3088051 demonstrated association with stricturing behaviour, ileal location, and bowel resection. This SNP was not associated with CD in the main case-control analysis. However there was a difference in allele frequency between cases and controls that was approaching statistical significance.
In conclusion, the findings of this study provide some evidence to suggest that genetic variation in ULK1 may play a role in interindividual differences in CD susceptibility and clinical outcome. However it remains unclear which variants are most important. There could be other genetic variants such as rare variants and/or copy number variations that exist at this locus and are in LD with one or more of the SNPs we have investigated. It is known that the ULK1 gene is located in a region of copy number variation [28, 29]. Future efforts should aim to identify the causative variants in this region by sequencing and functional experiments.
Acknowledgments
The authors would like to thank the many patients with IBD who participated in this study and also the control subjects. Nutrigenomics New Zealand is collaboration between AgResearch Ltd., Plant and Food Research and The University of Auckland with funding through the Ministry of Science and Innovation (MSI).
References
- 1.Levine B, Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nature Reviews Immunology. 2007;7(10):767–777. doi: 10.1038/nri2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Deretic V. Multiple regulatory and effector roles of autophagy in immunity. Current Opinion in Immunology. 2009;21(1):53–62. doi: 10.1016/j.coi.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469(7330):323–335. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Massey DCO, Parkes M. Genome-wide association scanning highlights two autophagy genes, ATG16L1 and IRGM, as being significantly associated with Crohn’s disease. Autophagy. 2007;3(6):649–651. doi: 10.4161/auto.5075. [DOI] [PubMed] [Google Scholar]
- 5.Parkes M, Barrett JC, Prescott NJ, et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn’s disease susceptibility. Nature Genetics. 2007;39(7):830–832. doi: 10.1038/ng2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roberts RL, Hollis-Moffatt JE, Gearry RB, Kennedy MA, Barclay ML, Merriman TR. Confirmation of association of IRGM and NCF4 with ileal Crohn’s disease in a population-based cohort. Genes and Immunity. 2008;9(6):561–565. doi: 10.1038/gene.2008.49. [DOI] [PubMed] [Google Scholar]
- 7.Cooney R, Baker J, Brain O, et al. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nature Medicine. 2010;16(1):90–97. doi: 10.1038/nm.2069. [DOI] [PubMed] [Google Scholar]
- 8.Homer CR, Richmond AL, Rebert NA, Achkar J, McDonald C. ATG16L1 and NOD2 interact in an autophagy-dependent antibacterial pathway implicated in crohn’s disease pathogenesis. Gastroenterology. 2010;139(5):1630–1641. doi: 10.1053/j.gastro.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Travassos LH, Carneiro LAM, Ramjeet M, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nature Immunology. 2010;11(1):55–62. doi: 10.1038/ni.1823. [DOI] [PubMed] [Google Scholar]
- 10.Simmons JD, Mullighan C, Welsh KI, Jewell DP. Vitamin D receptor gene polymorphism: association with Crohn’s disease susceptibility. Gut. 2000;47(2):211–214. doi: 10.1136/gut.47.2.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koren I, Reem E, Kimchi A. DAP1, a novel substrate of mTOR, negatively regulates autophagy. Current Biology. 2010;20(12):1093–1098. doi: 10.1016/j.cub.2010.04.041. [DOI] [PubMed] [Google Scholar]
- 12.Kuester D, Guenther T, Biesold S, et al. Aberrant methylation of DAPK in long-standing ulcerative colitis and ulcerative colitis-associated carcinoma. Pathology Research and Practice. 2010;206(9):616–624. doi: 10.1016/j.prp.2010.05.004. [DOI] [PubMed] [Google Scholar]
- 13.Cummings JRF, Cooney R, Pathan S, et al. Confirmation of the role of ATG16L1 as a Crohn’s disease susceptibility gene. Inflammatory Bowel Diseases. 2007;13(8):941–946. doi: 10.1002/ibd.20162. [DOI] [PubMed] [Google Scholar]
- 14.Rioux JD, Xavier RJ, Taylor KD, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nature Genetics. 2007;39(5):596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barrett JC, Hansoul S, Nicolae DL, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nature Genetics. 2008;40(8):955–962. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Henckaerts L, Cleynen I, Brinar M, et al. Genetic variation in the autophagy gene ULK1 and risk of Crohn's disease. Inflammatory Bowel Diseases. 2011;17(6):1392–1397. doi: 10.1002/ibd.21486. [DOI] [PubMed] [Google Scholar]
- 17.Tarrant KM, Barclay ML, Frampton CMA, Gearry RB. Perianal disease predicts changes in Crohn’s disease phenotype—results of a population-based study of inflammatory bowel disease phenotype. American Journal of Gastroenterology. 2008;103(12):3082–3093. doi: 10.1111/j.1572-0241.2008.02212.x. [DOI] [PubMed] [Google Scholar]
- 18.Lennard-Jones JE. Classification of inflammatory bowel disease. Scandinavian Journal of Gastroenterology. 1989;24(170, supplement):2–6. doi: 10.3109/00365528909091339. [DOI] [PubMed] [Google Scholar]
- 19.Jurinke C, van den Boom D, Cantor CR, Köster H. The use of MassARRAY technology for high throughput genotyping. Advances in Biochemical Engineering/Biotechnology. 2002;77:57–74. doi: 10.1007/3-540-45713-5_4. [DOI] [PubMed] [Google Scholar]
- 20.Storm N, Darnhofer-Patel B, van den Boom D, Rodi CP. MALDI-TOF mass spectrometry-based SNP genotyping. Methods in Molecular Biology. 2003;212:241–262. doi: 10.1385/1-59259-327-5:241. [DOI] [PubMed] [Google Scholar]
- 21.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21(2):263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 22.Gabriel SB, Schaffner SF, Nguyen H, et al. The structure of haplotype blocks in the human genome. Science. 2002;296(5576):2225–2229. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- 23.Bender R, Lange S. Adjusting for multiple testing–when and how? Journal of Clinical Epidemiology. 2001;54(4):343–349. doi: 10.1016/s0895-4356(00)00314-0. [DOI] [PubMed] [Google Scholar]
- 24.Jones HE, Ohlssen DI, Spiegelhalter DJ. Use of the false discovery rate when comparing multiple health care providers. Journal of Clinical Epidemiology. 2008;61(3):232–240. doi: 10.1016/j.jclinepi.2007.04.017. [DOI] [PubMed] [Google Scholar]
- 25.Franke A, McGovern DPB, Barrett JC, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nature Genetics. 2010;42(12):1118–1125. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rose JDR, Roberts GM, Williams G, Mayberry JF, Rhodes J. Cardiff Crohn’s disease jubilee: the incidence over 50 years. Gut. 1988;29(3):346–351. doi: 10.1136/gut.29.3.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Polito JM, Childs B, Mellits ED, Tokayer AZ, Harris ML, Bayless TM. Crohn’s disease: influence of age at diagnosis on site and clinical type of disease. Gastroenterology. 1996;111(3):580–586. doi: 10.1053/gast.1996.v111.pm8780560. [DOI] [PubMed] [Google Scholar]
- 28.Pinto D, Marshall C, Feuk L, Scherer SW. Copy-number variation in control population cohorts. Human molecular genetics. 2007;16:R168–R173. doi: 10.1093/hmg/ddm241. [DOI] [PubMed] [Google Scholar]
- 29.Wong KK, DeLeeuw RJ, Dosanjh NS, et al. A comprehensive analysis of common copy-number variations in the human genome. American Journal of Human Genetics. 2007;80(1):91–104. doi: 10.1086/510560. [DOI] [PMC free article] [PubMed] [Google Scholar]