

Abstract
A concise stereoselective total synthesis of (+)-saxitoxin is described. A silver-(I)-initiated hydroamination cascade constructs the bicyclic guanidinium ion core from a propargyl-bis-guanidine. This sequence creates 2 C-N bonds, 1 C-O bond, and three rings as a single stereoisomer in a single synthetic transformation. This process enabled us to complete the synthesis of (+)-saxitoxin in 14 steps from N-Boc-L-serine methyl ester.
Keywords: Saxitoxin, hydroamination, alkaloids, toxins, guanidine
Unraveling the molecular basis for the toxicity of paralytic shellfish poisoning agents has proven indispensible to understand the complex biology of voltage gated sodium channels.1 Paralytic shellfish toxins (PSTs) represent a family of at least 35 different chemical entities, and are generally characterized by the presence of one or more cyclic guanidinium ion(s).2 The flag bearer for the PSTs is undoubtedly (+)-saxitoxin (STX), its significant toxicity has not only led to its use as an ion channel probe but also to its use as a chemical warfare agent.3 STX has received a great deal of attention from the synthetic community concerning the significant challenges associated with the chemical synthesis of such a polar, heteroatom rich and densely functionalized natural product. Advances in synthetic technology to access this natural product architecture holds promise to further develop isoform specific small molecule inhibitors of Nav channels with both improved potency and selectivity toward therapeutically relevant Nav isoforms.4 With these goals in mind we engaged the synthesis of STX.
To date, STX has succumbed to synthetic campaigns by Kishi, Jacobi, Du Bois and Nagasawa.5–8 Numerous other accounts have detailed approaches to related natural product including a recent report by Nishikawa which exploits a halonium induced propargylguanidine cyclization, conceptually related to that detailed below.9 Our group has been interested in the metal-catalyzed hydroaminations of propargylguanidines, in particular from the vantage of controlling the selectivity in these cyclizations between 5-exo and 6-endo manifolds.10,11 Armed with the ability to control these processes, STX became an intriguing skeletal system in which to further probe hydroamination selectivity. As shown in Figure 1, we envisaged that the pyrrolidine ring of STX could arise from the alkylation of a pendant electrophile at C10, as in A. In turn the N3-C4 or N9-C4 bonds could be formed by the oxidative cyclization of a neighboring guanidine on the ene-guanidines B or C. These intermediates were ultimately slated to arise from the competitive 5-exo vs. 6-exo hydroamination of the propargyl-bis-guanidine D. From our previous studies, we were confident that we could control 5-exo vs. 6-endo selectivity using Ag(I), to give B but were concerned that the 6-exo-dig process would be kinetically competitive to give C. It is likely that the 7-endo pathway would be much slower relative to the other three modes of cyclization. In the event that the 5-exo and 6-exo pathways are indeed competitive, both B and C should be viable intermediates for the oxidative cyclization to give A. Although this might deliver a mixture of epimers at C12, it would be inconsequential as the oxidation state of C12 would later be adjusted. Thus both intermediates should provide the correct stereochemistry at C4 presuming the oxidative cyclization is thermodynamically controlled by the stereochemistry at C5. With this synthetic redundancy in mind, we embraced this strategy to access STX.
Figure 1.

A strategy for (+)-saxitoxin.
As exemplified by Du Bois in his second generation synthesis of STX, Merino’s stereoselective addition of acetylides to aldonitrones provides a straight forward and readily scaleable access to the requisite propargyldiamine and thus became a natural entry point for our campaign also (Scheme 1).7b,12 Addition of the magnesiated homopropargyl benzyl ether to L-serine aldonitrone (1) gave the desired N-hydroxydiamine 2 in good chemical yield and diastereoselectivity for the anti-isomer (86%, 9 : 1 d.r.). Reductive cleavage of the N-O bond and acidic removal of both the silyl and carbamate protecting groups gave the diamine 3 as its bis-hydrochloride salt.13 Fearing yet another competitive cyclization pathway from an unprotected primary alcohol in our hydroamination strategy, at this point we installed the primary carbamate. This was accomplished with potassium cyanate in concentrated MsOH to give 4 as its free base after workup.14 Mercuric oxide assisted guanylation with di-Boc-pseudothiourea cleanly delivered the propargyl-bis-guanidine 5.
Scheme 1.

Preparation of the propargyl-bis-guanidine.
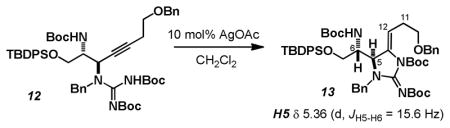
Exposure of 5 to our previously reported Ag(I) catalyzed cyclization conditions afforded a single regio- and stereoisomer of a cyclic ene-guanidine product (Scheme 2).10b,15 Given our experience with these compounds, chemical shift and multiplicity analysis persuaded us to assign the structure of this product as 6, arising from 5-exo-dig cyclization. More careful analysis, however, revealed that the protons in the 6-exo-dig product 7 would occupy identical spin systems and that this connectivity was likely indistinguishable through routine 1D or 2D-NMR techniques. Particularly troubling was the large 3J coupling between the methine protons H5 and H6 (3J =15.6 Hz) which is more indicative of a trans-diaxial orientation as expected in 7. However, a model compound which lacks the guanidine capable of forming the 6-exo-product, displays analogous 3J coupling, 1H chemical shift and multiplicities for H5, the vinylic proton (H12) and the two allylic methylene protons (H11) suggesting that the 5-exo-dig product 6 is indeed favored.16
Scheme 2.

Stepwise access to the key tricyclic carbamate.
Attempts to oxidatively trigger 6 to form the bicyclic guanidine, via epoxidation with DMDO or mCPBA, proved unsuccessful. This led us to consider the possibility of using an electrophilic halogen source to trigger the formation of the C4 aminal. While this would place a halogen at C12 instead of an oxygen atom, we were hopeful that the resulting halide might be displaced by an adjacent Boc group, reminiscent of the Woodward dihydroxylation.17 This strategic change would permit differentiation of the two guanidines needed to successfully execute the final annulation sequence. To our delight, treatment of 6 with iodine promoted cyclization thus providing the bicyclic aminal 8 as a single diastereomer. The stereochemistry of the secondary iodide at C12 has not been confirmed but is likely that arising from iodonium ion formation on the β-face of the alkene. Treatment of this secondary alkyliodide with Ag(I) and AcOH promoted the clean formation of the oxazolidinone 9, in which only a single Boc group had participated.17b,18
We recognized that the tricyclic carbamate 9 arose from alternating Ag(I)-catalyzed processes (5→6→8→9). This was conveniently streamlined to a one pot operation providing 9 in 57–67% isolated yield (Scheme 3). This successfully forged 2 C-N bonds, 1 C-O bond and three rings from an acyclic precursor in a single synthetic manipulation.
Scheme 3.

One pot synthesis of the key tricyclic carbamate.
To ready this intermediate for the final annulation sequence, the O- and N- benzyl groups were cleaved by hydrogenolysis and the resulting free alcohol mesylated to give 10 (Scheme 4). We anticipated that the oxazolidinone could be preferentially hydrolyzed because of both steric accessibility and its increased electrophilicity. Treatment of this intermediate with Cs2CO3 in EtOH cleanly and selectively cleaved the cyclic carbamate without hydrolysis of the t-butyl or primary carbamates.19 The release of N1 resulted in the final 5-membered ring annulation to give 11.
Scheme 4.

Completing the synthesis of (+)-saxitoxin.
Deprotection of the remaining Boc groups in 11 with 1:1 TFA:CH2Cl2 cleanly gave β-saxitoxinol. Alternatively, oxidation of the secondary alcohol could be accomplished with Dess-Martin periodinane to give the corresponding ketone. This intermediate proved to be relatively unstable but could be immediately deprotected to give (+)-saxitoxin in 81% yield over the final two steps.20
This strategy delivered (+)-saxitoxin in 14 steps from the commercially available N-Boc-L-serine methyl ester, comparable to Du Bois’ second generation synthesis.7b The approach is highlighted by a regio- and stereoselective one-pot Ag(I)-catalyzed cyclization cascade which generates 2 C-N bonds, 1 C-O bond and three rings in a single synthetic manipulation. This work is poised to complement that of others in delivering new small molecule modulators of ion channel function.
Supplementary Material
Acknowledgments
We are thankful to Dr. Jim Muller for help with mass spectral analysis and to Prof. Jon Rainier and Prof. Matt Sigman for insightful discussions.
* This work is dedicated to the memory of Prof. David Y. Gin.
FUNDING SOURCES
We are grateful to the National Institutes of Health, General Medical Sciences (R01 GM 090082) for financial support of this research.
Footnotes
Supporting Information. Experimental procedures, analytical data for all new compounds including 1H and 13C NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.a) Novakovic SD, Eglen RM, Hunter JC. Trends Neurosci. 2001;24:473. doi: 10.1016/s0166-2236(00)01884-1. [DOI] [PubMed] [Google Scholar]; b) Lai HC, Jan LY. Nat ReV Neurosci. 2006;7:548. doi: 10.1038/nrn1938. [DOI] [PubMed] [Google Scholar]; c) Rush AM, Cummins TR, Waxman SG. J Physiol. 2007;579:1. doi: 10.1113/jphysiol.2006.121483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Shimizu Y, Hsu CP, Fallon WE, Oshima Y, Miura Y, Nakanishi K. J Am Chem Soc. 1978;100:67. [Google Scholar]; Llewellyn LE. Nat Prod Rep. 2006;23:200. doi: 10.1039/b501296c. [DOI] [PubMed] [Google Scholar]
- 3.(a) Schantz EJ. Ann NY Acad Sci. 1986;479:15. doi: 10.1111/j.1749-6632.1986.tb15557.x. [DOI] [PubMed] [Google Scholar]; (b) Tucker JB. International Security. 2002;27:107. [Google Scholar]
- 4.For derivatives and analogues of saxitoxin see Koehn FE, Ghazarossian VE, Schantz EJ, Schnoes HK, Strong FM. Bioorg Chem. 1981;10:412.Andresen BM, Du Bois J. J Am Chem Soc. 2009;131:12524. doi: 10.1021/ja904179f.Mao H, Fieber LA, Gawley RE. ACS Med Chem Lett. 2010;1:135. doi: 10.1021/ml100035t.Shinohara R, Akimoto T, Iwamoto O, Hirokawa T, Yotsu-Yamashita M, Yamaoka K, Nagasawa K. Chem Eur J. 2011;17:12144. doi: 10.1002/chem.201101058.Mulcahy JV, Du Bois J. J Am Chem Soc. 2008;130:12630. doi: 10.1021/ja805651g.Iwamoto O, Nagasawa K. Org Lett. 2010;12:2150. doi: 10.1021/ol1006696.Sawayama Y, Nishikawa T. Angew Chem In Ed Engl. 2011;50:7176. doi: 10.1002/anie.201102494.
- 5.Tanino H, Nakata T, Kaneko T, Kishi Y. J Am Chem Soc. 1977;99:2818. doi: 10.1021/ja00450a079. [DOI] [PubMed] [Google Scholar]
- 6.Jacobi PA, Martinelli MJ, Polanc S. J Am Chem Soc. 1984;106:5594. [Google Scholar]
- 7.(a) Fleming JJ, Du Bois J. J Am Chem Soc. 2006;128:3926. doi: 10.1021/ja0608545. [DOI] [PubMed] [Google Scholar]; (b) Fleming JJ, McReynolds MD, Du Bois J. J Am Chem Soc. 2007;129:9964. doi: 10.1021/ja071501o. [DOI] [PubMed] [Google Scholar]
- 8.Iwamoto O, Shinohara R, Nagasawa K. Chem Asian J. 2009;4:277. doi: 10.1002/asia.200800382. [DOI] [PubMed] [Google Scholar]
- 9.(a) Nishikawa T, Sawayama Y. Angew Chem In Ed Engl. 2011;50:7176. doi: 10.1002/anie.201102494. [DOI] [PubMed] [Google Scholar]; (b) Sawayama Y, Nishikawa T. Synlett. 2011;5:651. [Google Scholar]
- 10.(a) Giles RL, Sullivan JD, Steiner AM, Looper RE. Angew Chem Int Ed. 2009;48:3162. doi: 10.1002/anie.200900160. [DOI] [PubMed] [Google Scholar]; (b) Gainer MJ, Bennet NR, Takahashi Y, Looper RE. Angew Chem Int Ed. 2011;50:684. doi: 10.1002/anie.201006087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.For other methods for preparing cyclic guanidines see Overman LE, Rabinowitz MH. J Org Chem. 1993;58:3235.Cohen F, Overman LE, Sakata SKL. Org Lett. 1999;1:2169. doi: 10.1021/ol991269u.Kim M, Vulcahy JV, Espino CG, Du Bois J. Org Lett. 2006;8:1073. doi: 10.1021/ol052920y.Snider BB, Xie CY. Tetrahedron Lett. 1998;39:7021.Arnold MA, Day KA, Durón SG, Gin DY. J Am Chem Soc. 2006;128:13255. doi: 10.1021/ja063860+.
- 12.(a) Merino P, Lanaspa A, Merchan FL, Tejero T. Tetrahedron Asymmetry. 1998;9:629–646. [Google Scholar]; (b) Merino P, Fransco S, Merchan FL, Tejero T. J Org Chem. 1998;63:5627. [Google Scholar]
- 13.Dhavale DD, Genntilucci L, Piazza MG, Trombini C, Liebigs Ann Chem. 1992:1289. [Google Scholar]
- 14.Choi Yong-Moon, Kim Min Woo. 20050080268. U S patent. 2005:A1.
- 15.Ermolatev D, Bariwal J, Steenackers H, De Keersmaecker S, Van der Eycken E. Angew Chem In Ed Engl. 2010;49:9465. doi: 10.1002/anie.201004256. [DOI] [PubMed] [Google Scholar]
-
16.Model compound 13 was prepared by the AgOAc catalyzed cyclization of 12. See supporting information for details and comparisons of 1H-NMR spectra.
- 17.(a) Woodward RB, Brutcher FV., Jr J Am Chem Soc. 1958;80:209–211. [Google Scholar]; (b) Kang SH, Ryu DH. Tetrahedron Lett. 1997;38:607–610. [Google Scholar]; (c) Kobayashi S, Isobe T, Ohno M. Tetrahedron Lett. 1984;25:5079. [Google Scholar]
- 18.The stereochemistry of the C-O bond at C12 has not been unambiguously assigned, but is inferred by the ultimate conversion of this intermediate to β-saxitoxinol.
- 19.Kunieda T, Ishizuka T. Tetrahedron Lett. 1987;28:4185. [Google Scholar]
- 20.Both synthetically prepared saxitoxin and β-saxitoxinol had physical properties in agreement with those reported for the natural products; see refs. 4a and 7b.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.