

Abstract
Clear cell carcinoma (CCC) of the ovary tends to show resistance to standard chemotherapy, which results in poor survival for patients with CCC. Developing a novel therapeutic strategy is imperative to improve patient prognosis. Epidermal growth factor receptor (EGFR) is frequently expressed in epithelial ovarian cancer. One of the major downstream targets of the EGFR signaling cascade is ERK. PEA-15, a 15-kDa phosphoprotein, can sequester ERK in the cytoplasm. MEK1/2 plays a central role in integrating mitogenic signals into the ERK pathway. We tested the hypothesis that inhibition of the EGFR-ERK pathway suppresses tumorigenicity in CCC, and we investigated the role of PEA-15 in ERK-targeted therapy in CCC. We screened a panel of four CCC cell lines (RMG-I, SMOV-2, OVTOKO, and KOC-7c) and observed that the EGFR tyrosine kinase inhibitor erlotinib inhibited cell proliferation of EGFR-overexpressing CCC cell lines through partial dependence on the MEK/ERK pathway. Further, erlotinib-sensitive cell lines were also sensitive to the MEK inhibitor selumetinib (AZD6244), which is under clinical development. Knockdown of PEA-15 expression resulted in reversal of selumetinib-sensitive cells to resistant cells, implying that PEA-15 contributes to selumetinib sensitivity. Both selumetinib and erlotinib significantly suppressed tumor growth (P < 0.0001) in a CCC xenograft model. However, selumetinib was better tolerated; erlotinib-treated mice exhibited significant toxic effects (marked weight loss, severe skin peeling) at high doses. Our findings indicate that the MEK/ERK pathway is a potential target for EGFR-overexpressing CCC and indicate that selumetinib and erlotinib are worth exploring as therapeutic agents for CCC.
Keywords: ovarian clear cell carcinoma, erlotinib, selumetinib, AZD6244, PEA-15, ERK
INTRODUCTION
Clear cell carcinoma (CCC) of the ovary accounts for 3.7% to 12.1% of epithelial ovarian cancers (1, 2). The clinical behavior of CCC of the ovary is distinctly different from that of other epithelial ovarian cancers (3). In the first large study to examine survival and treatment response in patients with ovarian CCC and serous adenocarcinoma, the most common subtype of epithelial ovarian cancer, patients with stage III CCC had significantly lower 3-year and 5-year survival rates than patients with stage III serous adenocarcinoma (23.5% vs. 54.1%, 23.5% vs. 34.1%). In addition, response rates for platinum-based chemotherapy were 11.1% for CCC and 72.5% for serous adenocarcinoma (4).
The epidermal growth factor receptor (EGFR) is frequently expressed in malignant ovarian tumors (48% of tumors on average in the studies reported to date) (5), and in a phase II trial for advanced ovarian carcinoma, an EGFR tyrosine kinase inhibitor (TKI), erlotinib, showed marginal activity (partial response, 6%; stable disease, 44%; progressive disease, 50%) but was generally well tolerated (6). All patients in that study had EGFR-positive disease by immunohistochemical analysis, but there was no information about the histological subtype. Therefore, the sensitivity of CCC to erlotinib is not known.
A major downstream target of the EGFR signaling cascade is ERK. Erlotinib inhibits the activation of the ERK pathway in several human non-small-cell lung cancer cell lines (7). Inhibition of ERK by EGFR-TKIs intercepts ERK’s upstream aberrant mitogenic signals. To date, no therapeutically useful direct ERK inhibitors have been adopted in clinical practice. MEK1/2 plays a central role in integration of mitogenic signals into the ERK pathway, as MEK has no downstream substrates except for ERK1/2. Thus, MEK is an excellent alternative therapeutic target with which to modulate ERK activity. The MEK inhibitor PD-0325901 selectively binds to and inhibits MEK, which results in inhibition of ERK phosphorylation and inhibition of tumor cell proliferation. However, PD-0325901 has been discontinued from Pfizer’s drug development pipeline because the phase II trial of PD-0325901 showed no objective tumor response (8, 9). However, selumetinib (AZD6244), a potent, selective, ATP-uncompetitive inhibitor of MEK1/2 kinases (10, 11), is currently in phase II clinical trial development for melanoma and non-small-cell lung cancer.
PEA-15 is a phosphoprotein that slows cell proliferation by binding to and sequestering ERK in the cytoplasm (12). The structure of PEA-15 suggests that phosphorylation could regulate PEA-15 binding to ERK (13). We previously showed that transfection of low-PEA-15-expressing ovarian cancer cells with PEA-15 inhibited cell proliferation (14). Analysis of PEA-15 expression in a tissue microarray of epithelial ovarian cancer samples revealed that PEA-15 expression is associated with prolonged overall survival in patients with epithelial ovarian cancer (15).
We hypothesized that inhibition of the EGFR-ERK pathway suppresses tumorigenicity in ovarian CCC. To evaluate this hypothesis, we tested the impact of erlotinib and selumetinib on cell proliferation. We found that erlotinib suppressed cell proliferation in EGFR-overexpressing CCC cells, partly because of suppression of ERK. Further, erlotinib-sensitive cells were also sensitive to selumetinib. This selumetinib sensitivity of CCC cells was partially dependent on the phosphorylation status of PEA-15. Erlotinib and selumetinib suppressed CCC tumor growth in a mouse intraperitoneal xenograft model. Our findings indicate that the MEK/ERK pathway may be a promising therapeutic target in EGFR-overexpressing CCC.
MATERIALS AND METHODS
Cell lines and cell cultures
Four human ovarian CCC cell lines (RMG-I, KOC-7c, SMOV-2, and OVTOKO) were used. RMG-I was obtained from Professor Shiro Nozawa, Keio University, Tokyo, Japan; KOC-7c from Dr. Toru Sugiyama, Kurume University, Japan; SMOV-2 from Dr. Tomohiro Iida, St. Marianna University, Kawasaki, Japan; and OVTOKO from Dr. Hiroshi Minaguchi, Yokohama City University, Japan. RMG-I cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM)/F12 medium (GIBCO). KOC-7c, SMOV-2, and OVTOKO cells were maintained in RPMI-1640 medium (GIBCO).
Plasmid constructs
The plasmid construct PEA-15-AA, the nonphosphorylated variant of PEA-15 (S104A/S116A), was a gift from Dr. Mark Ginsberg (University of California, San Diego) (13). To construct PEA-15-AD, we exchanged Ser 104 for Ala. pcDNA3-PEA-15-S116D served as a template using the primer 5’-CAAGCTAACCCGTATCCCCGCTGCCAAGAAGTACAAAGAC-3’ and its reverse complement GTCTTTGTACTTCTTGGCAGCGGGGATACGGGTTAGCTTG.
Drugs
Erlotinib was purchased from ChemieTek. Selumetinib was provided by AstraZeneca under the auspices of the National Cancer Institute’s Cancer Therapy Evaluation Program.
Western blot analysis
Cell pellets were lysed as described previously (16). Primary antibodies were rabbit anti-EGFR antibody (diluted 1:500) (Santa Cruz Biotechnology), rabbit anti-phospho-EGFR (Tyr1173) (1:200) (Santa Cruz), rabbit anti-phospho-p42/44 MAP kinase (Thr202/Tyr204) (1:500) (Cell Signaling), rabbit anti-PEA-15 polyclonal antibody (1:1000) (SynPep, Dublin, CA), rabbit anti-pPEA-15 (S104) (1:500) (Cell Signaling), rabbit anti-pPEA-15 (S116) (1:500) (Invitrogen), mouse anti-α-tubulin (1:5000) (Sigma-Aldrich), and mouse anti-β-actin (1:2000) (Sigma-Aldrich). Signals were detected using an Odyssey IR imaging system (LI-COR Biosciences).
WST-1 assay
Cell viability was assayed using cell proliferation reagent WST-1 (Roche Applied Science) as described previously (17). Ovarian CCC cells (RMG-I, 4 × 103/90 μl; SMOV-2, 3 × 103/90 μl; OVTOKO, 2 × 103/90 μl; or KOC-7c, 2 × 103/90 μl) were seeded into each well of a 96-well plate and treated the next day with erlotinib or selumetinib at a final concentration of 0.001, 0.01, 0.1, 1, or 10 μM for 72 h.
siRNA against EGFR
Cells were seeded in six-well culture plates at 3.0 × 105 cells/well (30-50% confluence). The next day, cells were transfected with ON-TARGET SMART pools against EGFR or a scrambled control (Dharmacon) at a final siRNA concentration of 100 nM using Dharmafect 1 (Dharmacon) following the manufacturer’s protocol.
Cell cycle distribution analysis
Briefly, cells were plated in a six-well plate, cultured overnight, and then treated or untreated with erlotinib for 48 h (final concentration 0.1 or 1 μM for RMG-I, 1 or 5 μM for SMOV-2) or selumetinib for 24 h (final concentration 0.01 or 0.1 μM for RMG-I). Cell cycle distribution was analyzed by flow cytometry as described previously (16).
Mutation screening
Mutation screening was performed as described previously (18, 19). Genomic DNA was purified from all four CCC cell lines using a Gentra Puregene Cell Kit (Qiagen). PCR primers used to amplify the sequence of interest were the same as previously reported (18, 19). Annealing temperatures were 59°C for EGFR exons 19 and 21 and KRAS exons 2 and 3. For BRAF exon 15, DNA was amplified in reactions of 30 seconds at 94°C; 30 seconds at 68°C to 55°C touchdown; followed by 1 minute at 72°C for 30 cycles. Then, PCR products were sequenced using a capillary-based modified heteroduplex method optimized to run on an ABI PRISM 3100 genetic analyzer (Applied Biosystems).
Constitutive active MEK1 transfection
Constitutive active MEK1 (MEK1CA)-expressing plasmid was a gift from Dr. M. C. Hung (MD Anderson) (20). For MEK1CA transfection, briefly, cells were suspended in electroporation buffer. The MEK1CA or control vector pcDNA3 plasmids were transfected by electroporation using Nucleofector (Amaxa Biosystems). Stable transfectants were established by G418 selection. pERK protein expression was used for evaluating efficacy of the gene transduction. The stable transfectant RMG-1-vector and RMG-I-MEK cells were plated on six-well plates (2 × 105 cells/well), and beginning the next day, cells were treated with erlotinib (2 or 5 μM) for 48 or 72 h and a trypan blue assay was performed. RMG-I-vector and RMG-I-MEK cells were also plated on 96-well plates (5 × 103 cells/well), and beginning the next day, cells were treated with erlotinib (1, 2, 5, or 10 μM) for 72 h and then subjected to WST-1 assay.
Nuclear/cytoplasmic fractionation
Cells were plated at 2 × 105 cells/well in six-well plates and pelleted, after which the nuclear and cytoplasmic fractions were separated using a fractionation kit according to the manufacturer’s protocol (Chemicon International).
PEA-15 transfection
OVTOKO cells (1 × 105 cells/well) were plated on 35-mm plates. The next day, cells were transfected using FuGene HD transfection reagent (Roche Applied Sciences) with PEA-15-AD (S104A, phosphoinhibitory at S104, S116D, phosphomimetic at S116), PEA-15-AA (S104A, phosphoinhibitory at S104, S116A, phosphoinhibitory at S116), or control vector pcDNA3 in serum-free medium. Twenty-four hours later, cells were washed with PBS, and selumetinib 10 μM was added to the culture medium. Cells were stained with 0.2% trypan blue 44 h after addition of drug, and living cells and dead cells were counted under a microscope.
siRNA against PEA-15
RMG-I cells were seeded in six-well culture plates at 6.0 × 105/well (70-80% confluence) in DMEM/F12 medium supplemented with 10% FBS. The next day, cells were transfected with ON-TARGET plus siRNA SMART pools against PEA-15 or a scrambled control (Dharmacon) at a final siRNA concentration of 100 nM using Dharmafect 2 (Dharmacon) following the manufacturer’s protocol. Six hours later, cells were washed with PBS, and the medium was replaced. The next day, selumetinib (2 μM) was added to the culture medium. Forty-eight hours later, cells were stained with 0.2% trypan blue, and living cells and dead cells were counted under a microscope.
ERK-targeted therapy in an ovarian cancer xenograft model
For xenograft experiments, luciferase-expressing RMG-I cells (RMG-I/luc) were established using firefly luciferase+ gene pGL3 lentivirus vector (Lentigen). These cells have a morphology and proliferation rate similar to those of parental RMG-I cells. RMG-I/luc cells in log-phase growth were trypsinized, washed twice with PBS, and centrifuged at 250 × g. Viable cells were counted, and 4 × 106 viable cells (in 0.5 ml of PBS) were injected under aseptic conditions into the peritoneal cavities of female athymic mice. Mice were subjected to whole-body luciferase imaging under an IVIS 100 Imaging System (Xenogen) 4 days after inoculation. Before imaging, mice were injected intraperitoneally with luciferin (Caliper Life Sciences) at 150 mg/kg body weight with a 25-gauge syringe. Then mice were kept under anesthesia with isoflurane. Starting 5.5 min after the luciferin injection, images were collected for 30 sec each in the ventral and dorsal positions. Images and amounts of bioluminescent signals were analyzed using Living Image Software (Xenogen).
We performed three sets of animal experiments. In the first set, mice were randomly divided into three groups (7 mice/group), and treatment was started 5 days after tumor cell inoculation and continued for 3 weeks. Group 1 was given vehicle (0.5% hydroxypropyl methyl cellulose and 0.1% Tween 80, 10 ml/kg/day); group 2, erlotinib 100 mg/kg/day; and group 3, selumetinib 25 mg/kg/day. In the second set of experiments, mice were randomly divided into four groups (10 mice/group), and treatment was started 5 days after tumor cell inoculation and continued for 5 weeks. Group 1 was given vehicle as in the first set of experiments; group 2, erlotinib 50 mg/kg/day; group 3, selumetinib 50 mg/kg/day; and group 4, selumetinib 100 mg/kg/day. All agents were administered by oral gavage. Bioluminescent imaging was performed weekly to assess intraperitoneal tumor growth.
In the third set of experiments, mice were randomly divided into three groups (4 mice/group), and treatment was started 5 days after tumor cell inoculation and continued for 2 weeks. Groups 1-3 received the same daily treatments as groups 1-3 in the second set of experiments, by oral gavage. At the end of the 2 weeks, tissue samples were collected, fixed in 10% neutral buffered formalin, and embedded in paraffin. Paraffin blocks were sliced in 4-μm sections and deparaffinized. Expression of ERK, pERK, PEA-15, pPEA-15 (S116), and Ki-67 in tumor tissue sections was detected with mouse monoclonal anti-p42/44 MAP kinase (3A7) (Cell Signaling), rabbit anti-phospho-p42/44 MAP kinase (Thr202/Tyr204), rabbit anti-PEA-15 polyclonal antibody, rabbit anti-pPEA-15 (S116), and rabbit monoclonal Ki-67 (SP6) antibody.
Statistical analysis
Statistical analyses were performed with Prism version 5 (GraphPad Software Inc). Data are presented as means ±1 standard error. Means for all data were compared by one-way analysis of variance with post hoc testing or by unpaired t-test. P values of <0.05 were considered statistically significant.
RESULTS
Expression of EGFR and ERK in CCC cell lines
We hypothesized that suppression of the EGFR-ERK pathway inhibits cell proliferation in human ovarian CCC cells. We first screened a panel of four human CCC cell lines, RMG-I, SMOV-2, OVTOKO, and KOC-7c, for EGFR expression. RMG-I and SMOV-2 cells expressed high levels of EGFR, and OVTOKO and KOC-7c cells expressed low levels of EGFR. In RMG-I and SMOV-2 cells, stimulation with EGF increased expression of pEGFR and pERK, which suggested that the EGFR-downstream pathway was activated in these two cell lines (Figure 1A). In contrast, in OVTOKO and KOC-7c cells, stimulation with EGF increased expression of pERK but not pEGFR.
Figure 1.
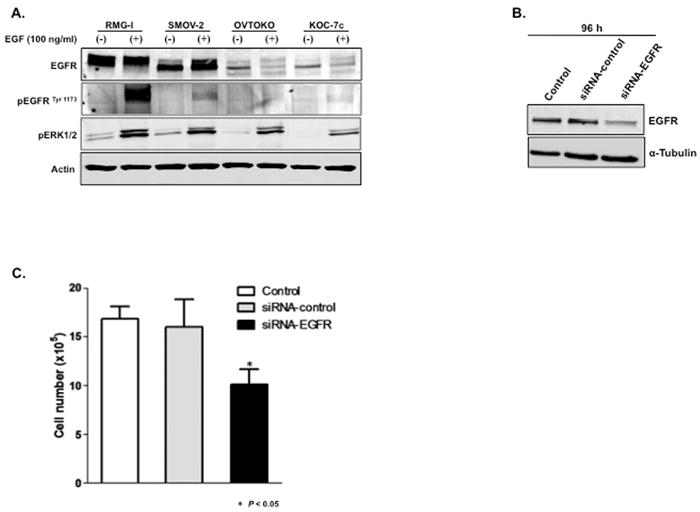
Downregulation of EGFR suppresses proliferation of CCC cells. A, Four CCC cell lines were cultured in serum-free medium for 24 h and then exposed to EGF in culture medium at a concentration of 100 ng/ml for 15 min. Western blot analysis was performed and β-actin was used as a loading control. B and C, RMG-I cells were treated with siRNA-control or siRNA-EGFR for 6 h, medium was changed, and 96 h after addition of siRNAs, (B) Western blot analysis was performed and (C) cell viability was measured by trypan blue exclusion test.
Next, we investigated whether proliferation of RMG-1 cells with a functional EGFR pathway is EGFR driven. Transfection of RMG-1 cells with siRNA specific for EGFR reduced EGFR protein levels to 25% of the levels in siRNA-control-treated cells (Figure 1B) and reduced the number of cells to 60% of the number of siRNA-control-treated cells (P < 0.05) (Figure 1C). These results suggest that RMG-I cell proliferation is dependent on the EGFR pathway. We observed similar results in SMOV-2 cells (data not shown).
Erlotinib suppresses proliferation of CCC cells via pERK inhibition and induction of G1 arrest
To determine if erlotinib could suppress proliferation of CCC cells, we treated cells with different concentrations of erlotinib and assessed cell viability by WST-1 assay 72 h later. RMG-I and SMOV-2 cells were sensitive to erlotinib (50% inhibitory concentration [IC50]: 0.1 μM and 1 μM, respectively), whereas OVTOKO and KOC-7c cells were resistant (IC50: 6 μM and 9 μM, respectively) (Figure 2A).
Figure 2.
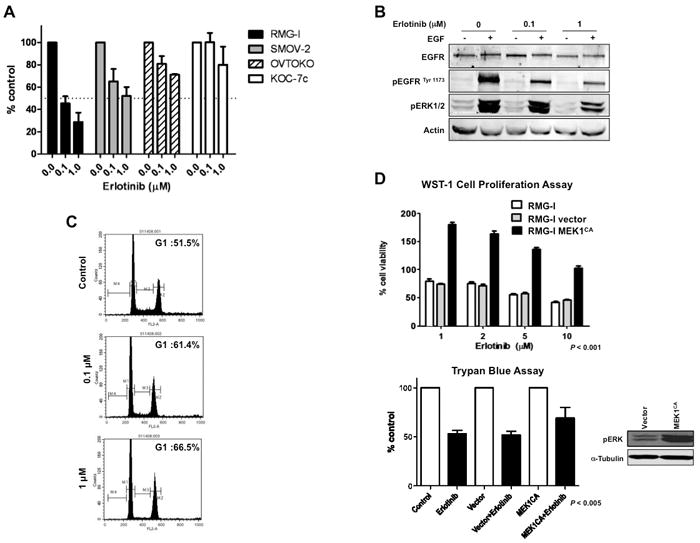
Erlotinib suppresses proliferation of CCC cells via pERK downregulation and G1 arrest. A, Four CCC cell lines were plated in 96-well plates and treated with 0.1 and 1 μM erlotinib for 72 h. WST-1 assay was performed. B, RMG-I cells were cultured in serum-free medium for 24 h and treated with 0.1 or 1 μM erlotinib for 3 h. Protein samples were collected after exposure to EGF in culture medium at a concentration of 100 ng/ml for 15 min. Western blot analysis was performed. C, RMG-I cells were treated with 0.1 or 1 μM erlotinib for 48 h, and FACScan analysis was performed to determine cell cycle distribution. D, RMG-I cells stably expressing MEK1CA were treated with erlotinib (1, 2, 5, or 10 μM), and WST-1 assay was performed at 72 h. Cells were also treated with erlotinib (2 μM), and trypan blue exclusion test was performed at 48 h to assess cell viability.
When RMG-I cells were treated with erlotinib, EGF-induced pEGFR and pERK expression were incompletely suppressed in a dose-dependent manner (Figure 2B). Erlotinib treatment also suppressed pAkt expression in RMG-I cells, but the baseline expression of pAkt and pERK did not correlate with sensitivity to erlotinib in CCC cell lines (data not shown). Similar results were observed in SMOV-2 cells (data not shown). These cell lines had no EGFR mutations at exon 19 or exon 21, which are known to correlate with response to EGFR-TKI in non-small cell lung cancer (18).
We then examined the effect of erlotinib on cell cycle distribution in erlotinib-sensitive CCC cell lines. RMG-I cells exhibited G1 arrest, but did not exhibit apoptosis (denoted by an increased proportion of cells in sub-G1), after 72 h of treatment with 0.1 and 1 μM erlotinib (Figure 2C). We observed similar G1 arrest in SMOV-2 cells after 72 h of treatment with 1 and 5 μM erlotinib (data not shown). In RMG-I cells (very sensitive to erlotinib), G1 arrest was associated with a marked increase in p27 expression at 48 and 72 h of erlotinib treatment, whereas in SMOV-2 cells (moderately sensitive to erlotinib), there was only a modest increase in p27 expression after 72 h of erlotinib treatment (data not shown).
After erlotinib treatment, p27 was located in both the cytoplasm and the nucleus of untreated cells but was sequestered in the nucleus in treated cells (data not shown).
MEK1CA transfection renders RMG-I cells resistant to erlotinib
As activated ERK is suppressed by erlotinib in a dose-dependent manner in erlotinib-sensitive cells, we hypothesized that overexpression of activated ERK reduces the sensitivity of these cells to erlotinib. Given that MEK has no downstream substrates except for ERK, we used MEK1CA to increase the expression of ERK. We examined cell viability after erlotinib treatment with or without MEK1CA. MEK1CA significantly reduced sensitivity to erlotinib in RMG-I cells (WST-1 assay, P < 0.001; trypan blue assay, P < 0.005) (Figure 2D), suggesting that erlotinib sensitivity is partially dependent on ERK.
Selumetinib suppresses proliferation in erlotinib-sensitive CCC cell lines
Given that MEK1CA reduced the sensitivity of RMG-I cells to erlotinib, we hypothesized that suppression of ERK inhibits proliferation of erlotinib-sensitive CCC cells. To date, no therapeutically nontoxic ERK inhibitors have been identified for ovarian cancer. Selumetinib is a highly selective MEK inhibitor that is in clinical trials and inhibits ERK activation, which is downstream of MEK. Consequently, we examined the sensitivity of erlotinib-sensitive and erlotinib-insensitive CCC cell lines to selumetinib. Similar to what we saw with erlotinib, RMG-I and SMOV-2 cells were sensitive to selumetinib (IC50: 0.078 μM and 0.85 μM, respectively), whereas OVTOKO and KOC-7c cells were resistant (IC50 >10 μM for both) (Figure 3A). These cell lines had no mutations at exon 2 or 3 of KRAS or exon 15 of BRAF, which are known to correlate with sensitivity to selumetinib in human cancer cells (21).
Figure 3.
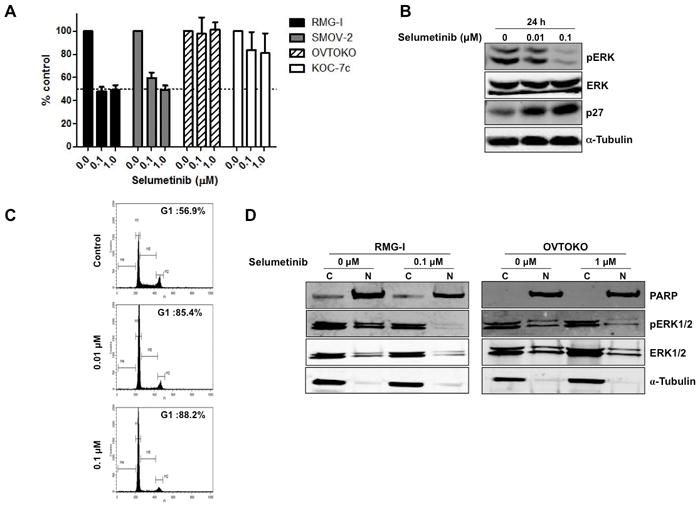
Selumetinib suppresses proliferation of CCC cells via pERK downregulation and G1 arrest. A, Four CCC cell lines were plated in 96-well plates and treated with 0.1 and 1 μM selumetinib for 72 h. WST-1 assay was performed to quantify the activity of erlotinib.B, RMG-I cells were treated with 0.01 or 0.1 μM selumetinib for 24 h. Western blot analysis was performed (α-tubulin was used as a loading control). C, RMG-I cells were treated with 0.01 or 0.1 μM selumetinib for 24 h, and FACScan analysis was performed. D, RMG-I and OVTOKO cells were treated with selumetinib (0.1 μM for RMG-I, 1 μM for OVTOKO) for 72 h and divided into cytoplasmic and nuclear fractions, and western blot analysis was performed.
Selumetinib suppresses proliferation of CCC cells via pERK inhibition and induction of G1 arrest
Treatment of RMG-I cells with selumetinib for 24 h reduced the expression of pERK and increased the expression of p27 (Figure 3B) concomitantly with the observed G1 arrest (G1 fraction; 56.9% for control, 85.4% for 24-h treatment with 0.01 μM selumetinib, 88.2% for 24-h treatment with 0.1 μM selumetinib (Figure 3C)). Similar results were observed in SMOV-2 cells (data not shown). In contrast, OVTOKO and KOC-7c cells did not show inhibition of ERK phosphorylation even after exposure to high selumetinib concentrations (data not shown). We did not observe apoptosis in these cell lines with selumetinib treatment.
Translocation of ERK from the cytoplasm to the nucleus is important for ERK-dependent transcription, which regulates cell proliferation by phosphorylating ERK’s nuclear substrates. We thus examined the nuclear and cytoplasmic expression level of ERK after selumetinib treatment in CCC cell lines. In RMG-I cells (selumetinib sensitive), the levels of nuclear and cytoplasmic pERK were suppressed after 72-h exposure to 0.1 μM selumetinib. In contrast, in OVTOKO cells (selumetinib resistant), there was a less significant suppression of nuclear pERK expression even though the cells were treated with a drug concentration 10 times as great (1 μM) (Figure 3D).
PEA-15 knockdown renders CCC cells resistant to selumetinib
Overall ERK activity in a cell could be defined not only by duration and magnitude of enzymatic activity but also by its location (cytoplasm vs. nucleus) (22). Therefore, we investigated the possible role of PEA-15 in selumetinib sensitivity. PEA-15 is known to sequester ERK in the cytoplasm by binding to ERK but does not directly change ERK kinase activity (12). After selumetinib treatment, pPEA-15 (S116) and total PEA-15 expression increased in RMG-I cells (selumetinib sensitive) (2.8 and 2.5 times of control, respectively) but did not change in OVTOKO cells (selumetinib resistant) (Figure 4A).
Figure 4.
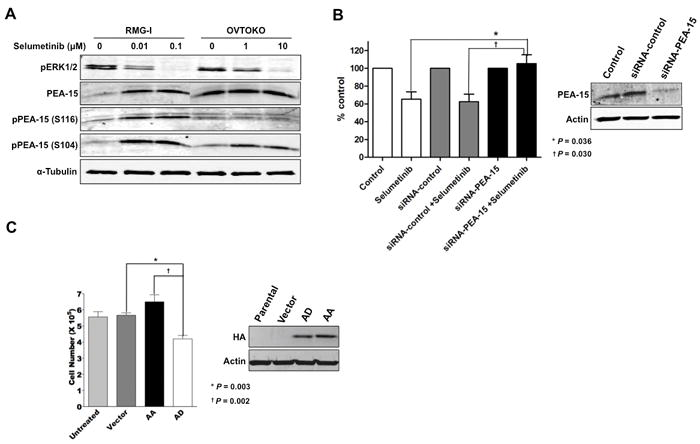
Phosphorylated PEA-15 sensitizes CCC cells to selumetinib. A, RMG-I and OVTOKO cells were treated with selumetinib (0.01 and 0.1 μM for RMG-I, 1 and 10 μM for OVTOKO) for 72 h. Western blot analysis was performed to detect pERK1/2, PEA-15, pPEA-15 (S116), and pPEA-15 (S104) expression. α-tubulin was used as a loading control. B, RMG-I cells were transfected with siRNA-PEA-15. Forty-eight hours later, cells were treated with 1 μM selumetinib for 48 h. Cell viability was examined by trypan blue exclusion test. C, OVTOKO cells were transfected with vector, nonphosphorylated mutant PEA-15-AA, or mutant PEA-15-AD. Twenty-four hours later, cells were treated with 10 μM selumetinib for 44 h. Cell viability was examined by trypan blue exclusion test.
To determine whether upregulation of PEA-15 by selumetinib contributes to the antiproliferative action of selumetinib, we knocked down PEA-15 by siRNA-PEA-15 in RMG-I cells and then examined their sensitivity to selumetinib. PEA-15 knockdown completely cancelled the growth-inhibitory effect of selumetinib (P < 0.05) (Figure 4B). To determine the role of PEA-15 phosphorylation at S116 in selumetinib sensitivity, we transfected OVTOKO cells (selumetinib resistant) with mutant PEA-15-AD (phosphoinhibitory at S104 and phosphomimetic at S116). OVTOKO cells transfected with PEA-15-AD were significantly more sensitive to selumetinib than OVTOKO cells transfected with PEA-15-AA (nonphosphorylated variant) (P = 0.002) (Figure 4C). These results suggested that when PEA-15 is phosphorylated at S116, cells are more sensitive to selumetinib.
Erlotinib and selumetinib suppress tumor growth in a CCC xenograft model
All mice injected with RMG-I/luc cells (luciferase expressing RMG-I cells) developed intraperitoneal tumors by 5 days after inoculation.
In our first set of xenograft experiments, all of the mice treated with a high dose of erlotinib (100 mg/kg/day) exhibited severe skin peeling with yellow discoloration on treatment day 6 (11 days after inoculation) and marked weight loss on treatment day 9 (14 days after inoculation) (Figure 5A). The mice continued to lose weight even after treatment was stopped for 2 days (P = 0.0015). Consequently, we reduced the dose of erlotinib to 50 mg/kg/day starting on day 11 (16 days after inoculation). In the mice treated with selumetinib 25/mg/kg/day, there were no signs of overt toxicity (skin toxicity, weight loss). Bioluminescent signals (to assess intraperitoneal tumor growth) of the erlotinib group (100 mg/kg/day and then 50 mg/kg/day) were significantly lower than those of the vehicle control group on days 18 and 25 after inoculation (P = 0.0072 and P = 0.0004, respectively) (data not shown). The bioluminescent signals of the selumetinib group (25 mg/kg/day) (data not shown) were lower (but not significantly lower) than those of the vehicle control group on days 11 and 18 after inoculation and increased to the same level as the vehicle control group on day 25. After treatment cessation, tumor regrowth was apparent within 6 days in the mice treated with selumetinib 25 mg/kg/day (data not shown). This dose was chosen because it effectively inhibited tumor growth in multiple MEK-inhibitor-sensitive models (21).
Figure 5.
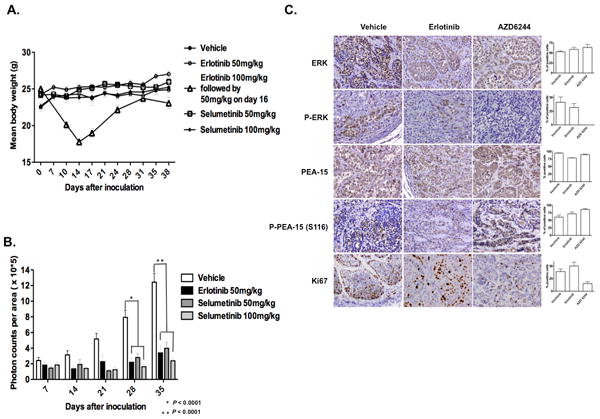
Selumetinib inhibits tumorigenicity in a CCC xenograft model without producing toxic effects. Female athymic nude mice were injected intraperitoneally with 4 × 106 luciferase-transfected RMG-I cells (RMG-I/luc). The experimental groups were treated with vehicle control (0.5% hydroxypropyl methyl cellulose and 0.1% Tween 80), 50 mg/kg/day erlotinib, or 50 or 100 mg/kg/day selumetinib for 5 weeks beginning 5 days after cell injection. A, Mean body weight in each treatment group. Data from an initial experiment in which mice were treated with 100 mg/kg/day erlotinib followed by 50 mg/kg/day erlotinib are included for reference. B, The photon counts per area (monitored by IVIS) of each treatment group were quantitatively analyzed on days 0, 7, 14, 21, 28, and 35 after inoculation. C, Immunohistochemical stains and quantification of ERK, pERK, PEA-15, pPEA-15 (S116D), and Ki-67 in representative tumor tissue samples from each treatment group.
In order to evaluate if higher doses of selumetinib could be more effective, we conducted a second RMG-I xenograft study to evaluate sensitivity to selumetinib 50 mg/kg/day and 100 mg/kg/day. Concurrently, we examined the effect of erlotinib 50 mg/kg/day (this time we used a fixed dose of 50 mg/kg/day). In our second set of xenograft experiments, no obvious toxicity was observed with higher doses of selumetinib (50 mg/kg/day and 100 mg/kg/day) or the fixed dose of erlotinib (50 mg/kg/day). Bioluminescent signals of the erlotinib group and of both selumetinib groups were significantly lower than those of the vehicle control group on days 21, 28, and 35 after inoculation, indicating significant inhibition of CCC tumor growth (P = 0.0001) (Figure 5B). Bioluminescent signals for the two selumetinib dose groups were similar (selumetinib 50 mg/kg vs. selumetinib 100 mg/kg: P = 0.67, P = 0.12, and P = 0.1434, respectively, on days 21, 28, and 35), and the signals for the two selumetinib dose groups were similar to the signals for the erlotinib group (selumetinib 50 mg/kg vs. erlotinib 50 mg/kg: P = 0.177, P = 0.4220, and P = 0.6241, respectively, on days 21, 28, and 35; selumetinib 100 mg/kg vs. erlotinib 50 mg/kg: P = 0.2242, P = 0.355, and P = 0.3827, respectively, on days 21, 28, and 35).
In the third set of xenograft experiments, tumor tissues were analyzed for ERK1/2, pERK1/2, PEA-15, and pPEA-15 (S116) expression and proliferation (Ki-67) in mice treated with erlotinib 50 mg/kg/day for 2 weeks and mice treated with selumetinib 50 mg/kg/day for 2 weeks. As expected, levels of pERK1/2 and Ki-67 expression were reduced in the erlotinib-treated mice and markedly reduced in the selumetinib-treated mice (Figure 5C). As in our in vitro experiments, PEA-15 and pPEA-15 (S116) expression were increased in selumetinib-treated groups in vivo.
DISCUSSION
Our results demonstrate that both selumetinib and erlotinib effectively suppress pERK expression in sensitive cell lines and suppress tumor growth in a CCC human xenograft model. However, mice treated with selumetinib 50 or 100 mg/kg/day showed significant tumor growth inhibition with no major toxicity, whereas mice treated with erlotinib 100 mg/kg/day showed skin peeling and marked weight loss. We speculate that the antitumorigenic effects of selumetinib and, to a lesser extent, erlotinib are due to targeting of ERK, which is downstream of EGFR. We also showed that in the selumetinib-sensitive cell lines, PEA-15, a protein that binds ERK and sequesters it in the cytoplasm, contributes to selumetinib’s effects. Through these findings, we have demonstrated that suppression of ERK pathway signaling by selumetinib can be effective in preclinical models of ovarian CCC.
In previous studies, it was shown that gefitinib, another EGFR-TKI, decreased the proliferation and invasion of CCC cells and inhibited tumor growth in a xenograft model in a dose-dependent manner (23). In our current study, we confirmed that overexpression of MEK1CA rendered erlotinib-sensitive CCC cells resistant to erlotinib. These findings suggest that sensitivity of ovarian CCC to erlotinib is partially dependent on ERK suppression, implying that ERK itself may be a potential therapeutic target in ovarian CCC.
In addition, erlotinib-insensitive cell lines were insensitive to the MEK inhibitor selumetinib. The low expression of EGFR and pEGFR and the lack of BRAF or KRAS mutation in these insensitive cell lines suggest possible involvement of other known pathways, such as the IGF-IR and PI3K-Akt pathways (24). Recently, it has been shown that patients with CCC have a high frequency of activating PIK3CA mutations (25). In our present study, the baseline expression of pAkt and pERK did not correlate with sensitivity to erlotinib or selumetinib in the CCC cell lines. Further studies are warranted on the IGF-IR and PI3K-Akt pathways to identify molecular markers that predict sensitivity or resistance of ovarian CCC to selumetinib.
Ovarian cancer patients with high-PEA-15-expressing tumors have been shown to survive longer than those with low-PEA-15-expressing tumors (15). In contrast, Stassi et al. reported that exogenous expression of a dominant-negative AKT cDNA or a PEA-15 antisense cDNA in breast cancer cells induced a significant down-regulation of PEA-15 and sensitized cells to chemotherapy-induced cell death (26). However, little is known about the relationship between PEA-15 and sensitivity to molecular targeted therapies related to the EGFR-ERK pathway. Here, we showed that expression of PEA-15 and pPEA-15 (S116) increased after selumetinib treatment only in the sensitive cell lines. Further, we demonstrated that siRNA knockdown of PEA-15 reduced sensitivity to selumetinib in selumetinib-sensitive CCC cells and that transfection with mutant PEA-15, which mimics phosphorylation at S116, significantly increased the sensitivity to selumetinib of selumetinib-insensitive CCC cells. Although further studies are needed to elucidate the role of pPEA-15 (S116) and the mechanism of its involvement in the antiproliferative action of selumetinib, our findings suggest that phosphorylated PEA-15 contributes to sensitivity to selumetinib and might be a useful marker to predict sensitivity to ERK-targeted therapy in CCC. Further studies are warranted to define the role of PEA-15 phosphorylation in selumetinib sensitivity.
Other investigators have shown that pPEA-15 (S116) binds to FADD and inhibits apoptosis whereas unphosphorylated PEA-15 binds to ERK and sequesters it in the cytoplasm (12, 13, 27). PEA-15 also binds to RSK2, which is a substrate of ERK, implying that PEA-15 acts as a scaffold for ERK and RSK2 (28). In this context, at low levels, PEA-15 expression enhances ERK binding to RSK2, whereas at very high levels, PEA-15 expression inhibits ERK binding to RSK2 (29, 30). Although whether pPEA-15 (S116) can bind to RSK2 is unknown, overexpression of S116D may cause a similar inhibitory effect.
In summary, we have shown that the ERK signaling pathway is a potential therapeutic target for CCC and that both selumetinib, which was less toxic than erlotinib in vivo, and erlotinib are worth exploring as therapeutic agents for patients with CCC. Further studies are warranted to determine if PEA-15 and/or phosphorylated PEA-15 may be useful to predict sensitivity to ERK pathway-targeted therapy in CCC.
Acknowledgments
We are grateful to Prof. Naoki Terakawa for his constant support and his discussions on clear cell carcinoma. We thank Dr. Paul D. Smith and AstraZeneca for providing technical support, the National Cancer Institute Cancer Therapy Evaluation Program for providing selumetinib, Sunita Patterson and Stephanie Deming of the Department of Scientific Publications at MD Anderson for their expert editorial assistance, Adam LaBaff of the Department of Molecular and Cellular Oncology at MD Anderson for providing technical advice, Wendy Schober of the Flow Cytometry and Cellular Imaging Facility at MD Anderson for assistance with cell cycle analysis, and the staff of the Research Histopathology Facility at MD Anderson.
Grant support: NIH grant R01 CA127562 (Naoto T. Ueno) and the National Institutes of Health through MD Anderson’s Cancer Center Support Grant, CA016672.
References
- 1.McGuire V, Jesser CA, Whittemore AS. Survival among U.S. women with invasive epithelial ovarian cancer. Gynecol Oncol. 2002;84:399–403. doi: 10.1006/gyno.2001.6536. [DOI] [PubMed] [Google Scholar]
- 2.Kennedy AW, Biscotti CV, Hart WR, Webster KD. Ovarian clear cell adenocarcinoma. Gynecol Oncol. 1989;32:342–9. doi: 10.1016/0090-8258(89)90637-9. [DOI] [PubMed] [Google Scholar]
- 3.Scully RE. World Health Organization classification and nomenclature of ovarian cancer. J Natl Cancer Inst Monogr. 1975;42:5–7. [PubMed] [Google Scholar]
- 4.Sugiyama T, Kamura T, Kigawa J, Terakawa N, Kikuchi Y, Kita T, et al. Clinical characteristics of clear cell carcinoma of the ovary: a distinct histologic type with poor prognosis and resistance to platinum-based chemotherapy. Cancer. 2000;88:2584–9. [PubMed] [Google Scholar]
- 5.Lafky JM, Wilken JA, Baron AT, Maihle NJ. Clinical implications of the ErbB/epidermal growth factor (EGF) receptor family and its ligands in ovarian cancer. Biochim Biophys Acta. 2008;1785:232–65. doi: 10.1016/j.bbcan.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 6.Gordon AN, Finkler N, Edwards RP, Garcia AA, Crozier M, Irwin DH, et al. Efficacy and safety of erlotinib HCl, an epidermal growth factor receptor (HER1/EGFR) tyrosine kinase inhibitor, in patients with advanced ovarian carcinoma: results from a phase II multicenter study. Int J Gynecol Cancer. 2005;15:785–92. doi: 10.1111/j.1525-1438.2005.00137.x. [DOI] [PubMed] [Google Scholar]
- 7.Adjei AA. Novel combinations based on epidermal growth factor receptor inhibition. Clin Cancer Res. 2006;12:4446s–50s. doi: 10.1158/1078-0432.CCR-06-0623. [DOI] [PubMed] [Google Scholar]
- 8.Garber K. Trials offer early test case for personalized medicine. J Natl Cancer Inst. 2009;101:136–8. doi: 10.1093/jnci/djn506. [DOI] [PubMed] [Google Scholar]
- 9.Haura EB, Ricart AD, Larson TG, Stella PJ, Bazhenova L, Miller VA, et al. A phase II study of PD-0325901, an oral MEK inhibitor, in previously treated patients with advanced non-small cell lung cancer. Clin Cancer Res. 2010;16:2450–7. doi: 10.1158/1078-0432.CCR-09-1920. [DOI] [PubMed] [Google Scholar]
- 10.Yeh TC, Marsh V, Bernat BA, Ballard J, Colwell H, Evans RJ, et al. Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin Cancer Res. 2007;13:1576–83. doi: 10.1158/1078-0432.CCR-06-1150. [DOI] [PubMed] [Google Scholar]
- 11.Adjei AA, Cohen RB, Franklin W, Morris C, Wilson D, Molina JR, et al. Phase I pharmacokinetic and pharmacodynamic study of oral, small-molecule mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers. J Clin Oncol. 2008;26:2139–46. doi: 10.1200/JCO.2007.14.4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Formstecher E, Ramos JW, Fauquet M, Calderwood DA, Hsieh J-C, Canton B, et al. PEA-15 mediates cytoplasmic sequestration of ERK MAP kinase. Dev Cell. 2001;1:239–50. doi: 10.1016/s1534-5807(01)00035-1. [DOI] [PubMed] [Google Scholar]
- 13.Krueger J, Chou FL, Glading A, Schaefer E, Ginsberg MH. Phosphorylation of phosphoprotein enriched in astrocytes (PEA-15) regulates extracellular signal-regulated kinase-dependent transcription and cell proliferation. Mol Biol Cell. 2005;16:3552–61. doi: 10.1091/mbc.E04-11-1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bartholomeusz C, Itamochi H, Nitta M, Saya H, Ginsberg MH, Ueno NT. Antitumor effect of E1A in ovarian cancer by cytoplasmic sequestration of activated ERK by PEA-15. Oncogene. 2006;25:79–90. doi: 10.1038/sj.onc.1209014. [DOI] [PubMed] [Google Scholar]
- 15.Bartholomeusz C, Rosen D, Wei C, Kazansky A, Yamasaki F, Takahashi T, et al. PEA-15 induces autophagy in human ovarian cancer cells and is associated with prolonged overall survival. Cancer Res. 2008;68:9302–10. doi: 10.1158/0008-5472.CAN-08-2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamasaki F, Johansen M, Zhang D, Krishnamurthy S, Felix E, Bartholomeusz C, et al. Acquired resistance to erlotinib in A-431 epidermoid cancer cells requires down-regulation of MMAC1/PTEN and up-regulation of phosphorylated Akt. Cancer Res. 2007;67:5779–88. doi: 10.1158/0008-5472.CAN-06-3020. [DOI] [PubMed] [Google Scholar]
- 17.Zhang D, Pal A, Bornmann WG, Yamasaki F, Esteva FJ, Hortobagyi GN, et al. Activity of lapatinib is independent of EGFR expression level in HER2-overexpressing breast cancer cells. Mol Cancer Ther. 2008;7:1846–50. doi: 10.1158/1535-7163.MCT-08-0168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–39. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 19.McIntyre AJ, Summersgill BM, Spendlove HE, Huddart RA, Houlston R, Shipley J. Activating mutations and/or expression levels of tyrosine kinase receptors GRB7, RAS, and BRAF in testicular germ cell tumors. Neoplasia. 2005;7:1047–52. doi: 10.1593/neo.05514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang JY, Zong CS, Xia W, Yamaguchi H, Ding Q, Xie X, et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat Cell Biol. 2008;10:138–48. doi: 10.1038/ncb1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davies BR, Logie A, McKay JS, Martin P, Steele S, Jenkins R, et al. AZD6244 (ARRY-142886), a potent inhibitor of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase 1/2 kinases: mechanism of action in vivo, pharmacokinetic/pharmacodynamic relationship, and potential for combination in preclinical models. Mol Cancer Ther. 2007;6:2209–19. doi: 10.1158/1535-7163.MCT-07-0231. [DOI] [PubMed] [Google Scholar]
- 22.Ebisuya M, Kondoh K, Nishida E. The duration, magnitude and compartmentalization of ERK MAP kinase activity: mechanisms for providing signaling specificity. J Cell Sci. 2005;118:2997–3002. doi: 10.1242/jcs.02505. [DOI] [PubMed] [Google Scholar]
- 23.Fujimura M, Hidaka T, Saito S. Selective inhibition of the epidermal growth factor receptor by ZD1839 decreases the growth and invasion of ovarian clear cell adenocarcinoma cells. Clin Cancer Res. 2002;8:2448–54. [PubMed] [Google Scholar]
- 24.Buck E, Eyzaguirre A, Rosenfield-Franklin M, Thomson S, Mulvihill M, Barr S, et al. Feedback mechanisms promote cooperativity for small molecule inhibitors of epidermal and insulin-like growth factor receptors. Cancer Res. 2008;68:8322–32. doi: 10.1158/0008-5472.CAN-07-6720. [DOI] [PubMed] [Google Scholar]
- 25.Kuo KT, Mao TL, Jones S, Veras E, Ayhan A, Wang TL, et al. Frequent activating mutations of PIK3CA in ovarian clear cell carcinoma. Am J Pathol. 2009;174:1597–601. doi: 10.2353/ajpath.2009.081000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stassi G, Garofalo M, Zerilli M, Ricci-Vitiani L, Zanca C, Todaro M, et al. PED mediates AKT-dependent chemoresistance in human breast cancer cells. Cancer Res. 2005;65:6668–75. doi: 10.1158/0008-5472.CAN-04-4009. [DOI] [PubMed] [Google Scholar]
- 27.Renganathan H, Vaidyanathan H, Knapinska A, Ramos JW. Phosphorylation of PEA-15 switches its binding specificity from ERK/MAPK to FADD. Biochem J. 2005;390:729–35. doi: 10.1042/BJ20050378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vaidyanathan H, Ramos JW. RSK2 activity is regulated by its interaction with PEA-15. J Biol Chem. 2003;278:32367–72. doi: 10.1074/jbc.M303988200. [DOI] [PubMed] [Google Scholar]
- 29.Vaidyanathan H, Opoku-Ansah J, Pastorino S, Renganathan H, Matter ML, Ramos JW. ERK MAP kinase is targeted to RSK2 by the phosphoprotein PEA-15. Proc Natl Acad Sci U S A. 2007;104:19837–42. doi: 10.1073/pnas.0704514104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ramos JW. The regulation of extracellular signal-regulated kinase (ERK) in mammalian cells. Int J Biochem Cell Biol. 2008;40:2707–19. doi: 10.1016/j.biocel.2008.04.009. [DOI] [PubMed] [Google Scholar]