

Abstract
Objective:
To test efficacy and safety of atorvastatin in subjects with clinically isolated syndrome (CIS).
Methods:
Subjects with CIS were enrolled in a phase II, double-blind, placebo-controlled, 14-center randomized trial testing 80 mg atorvastatin on clinical and brain MRI activity. Brain MRIs were performed quarterly. The primary endpoint (PEP) was development of ≥3 new T2 lesions, or one clinical relapse within 12 months. Subjects meeting the PEP were offered additional weekly interferon β-1a (IFNβ-1a).
Results:
Due to slow recruitment, enrollment was discontinued after 81 of 152 planned subjects with CIS were randomized and initiated study drug. Median (interquartile range) numbers of T2 and gadolinium-enhancing (Gd) lesions were 15.0 (22.0) and 0.0 (0.0) at baseline. A total of 53.1% of atorvastatin recipients (n = 26/49) met PEP compared to 56.3% of placebo recipients (n = 18/32) (p = 0.82). Eleven atorvastatin subjects (22.4%) and 7 placebo subjects (21.9%) met the PEP by clinical criteria. Proportion of subjects who did not develop new T2 lesions up to month 12 or to starting IFNβ-1a was 55.3% in the atorvastatin and 27.6% in the placebo group (p = 0.03). Likelihood of remaining free of new T2 lesions was significantly greater in the atorvastatin group compared with placebo (odds ratio [OR] = 4.34, p = 0.01). Likelihood of remaining free of Gd lesions tended to be higher in the atorvastatin group (OR = 2.72, p = 0.11). Overall, atorvastatin was well tolerated. No clear antagonistic effect of atorvastatin plus IFNβ-1a was observed on MRI measures.
Conclusion:
Atorvastatin treatment significantly decreased development of new brain MRI T2 lesion activity, although it did not achieve the composite clinical and imaging PEP.
Classification of Evidence:
This study provided Class II evidence that atorvastatin did not reduce the proportion of patients with CIS meeting imaging and clinical criteria for starting immunomodulating therapy after 12 months, compared to placebo. In an analysis of a secondary endpoint (Class III), atorvastatin was associated with a reduced risk for developing new T2 lesions.
In vitro and in vivo studies have indicated that statins have immunomodulatory properties.1–4 In the multiple sclerosis (MS) model, experimental autoimmune encephalomyelitis (EAE), oral statins prevented or reversed relapsing paralysis, suppressed proinflammatory Th1 responses, and promoted Th2 deviation through inhibition of isoprenylation of ras and rho, GTP-binding proteins that activate molecules involved in T-cell differentiation.2,4 These findings suggested statins may be beneficial in treatment of MS.
In one crossover MS study, simvastatin decreased the number of new T2 hyperintense lesions on brain MRI scans compared to pretherapy.5 MS studies subsequently tested statins in combination with interferon-β (IFNβ).6–10 Whether a statin medication alone is beneficial has never been tested in a placebo-controlled trial. We hypothesized that the potential anti-inflammatory effects of statins would be beneficial in the earliest phase of clinical MS. When this study was designed, one published trial had shown benefit of IFNβ-1a in clinically isolated syndromes (CIS).11 The objective of the STAyCIS study was to test the efficacy and safety of atorvastatin (Lipitor, Pfizer, New York, NY) 80 mg daily vs placebo in subjects with CIS. Our primary endpoint (PEP) was the proportion of subjects who developed 3 or more new brain MRI T2 lesions or a clinical relapse within 12 months. Secondary endpoints included proportion of subjects who remained free of new brain MRI T2 lesions, cumulative number of new brain MRI T2 lesions, and proportion of subjects who remained free of new gadolinium-enhancing (Gd) lesions.
METHODS
Standard protocol approvals, registrations, and patient consents.
The study was approved by institutional review boards at 14 centers in the United States and Canada. Written informed consent was obtained from subjects prior to enrollment in the STAyCIS study (NCT00094172).
Study design.
STAyCIS is a 12-month phase II, randomized, multicenter, controlled double-blind trial of atorvastatin 80 mg once daily vs placebo. The PEP was chosen based upon data from the CHAMPS study.11 It was determined that a 26% treatment effect would have been found at 12 months with a composite outcome of ≥3 new brain MRI lesions or ≥1 MS relapse, and that 69% of the placebo group would have met that endpoint by 12 months. This outcome was chosen for the PEP as in clinical practice it would likely have led to initiation of an approved DMT. With the assumption that 69% of placebo subjects would meet the PEP by month 12, we determined that 152 subjects would be required using a 3:2 randomization (atorvastatin: placebo) to detect a 39% therapeutic effect, with a power of 0.80 at a significance level (2-tailed) of 0.05, assuming a 10% dropout rate. Subjects who met the PEP were offered IFNβ-1a 30 μg IM weekly initiated after their month 6 visit and remained in the study until month 12. Secondary endpoints included incidence of adverse events, proportion of subjects free of new T2 and Gd lesions up to month 12 or to IFNβ-1a initiation, cumulative number of T2 and Gd lesions to month 12 or to IFNβ-1a initiation, time to first relapse, changes in Expanded Disability Status Scale (EDSS),12 Multiple Sclerosis Functional Composite (MSFC),13 visual analog scale (VAS) of well-being, and T1 and T2 lesion volumes.
Brain MRI scans and VAS were obtained at baseline, months 3, 6, 9, and 12. Clinical (EDSS, physical, MSFC) and laboratory evaluations occurred at screening, baseline, months 1, 2, 3, 6, 9, and 12. All subjects were required to complete a 3-day course of IV methylprednisolone 1 g daily or equivalent, started within 90 days of CIS onset. Steroids were discontinued ≥28 days before any MRI scan. Subjects experiencing new symptoms suggestive of an MS relapse were examined within 7 days. Relapses were defined as new or recurrent neurologic symptoms not associated with fever or infection, persisting >48 hours in subjects who were neurologically stable or improving for at least 30 days following CIS. Relapses were confirmed only when symptoms were accompanied by new objective changes on clinical examination. Recurrent neurologic symptoms of the same nature and severity as the prior one or change in sphincter or cognitive function alone did not qualify as relapses. Subjects started on atorvastatin 80 mg once daily or placebo at baseline.14 Subjects exhibiting aspartate aminotransferase or alanine aminotransferase elevation (>3× normal upper limit [NUL] but <10× NUL) on 2 consecutive draws decreased study medication to 40 mg. Subjects who experienced low low-density lipoprotein cholesterol levels (<25 mg/dL) discontinued medication for 1 month. If upon retesting levels were ≥25 mg/dL but ≤30 mg/dL study drug was restarted at 40 mg.
Subjects returned for follow-up visits at months 15 and 18. Patients who did not meet the PEP during the 12-month treatment phase and developed no new MRI lesions after month 3 received brain MRI scans at months 15 and 18 to address whether there could be sustained effects after atorvastatin discontinuation (“exploratory tolerance phase”).
Inclusion criteria.
Subjects between 18 and 55 years with a CIS lasting ≥48 hours, seen within 90 days of symptom onset, were offered participation provided they had ≥2 silent MRI T2-hyperintense foci ≥3 mm in diameter (2 brain or 1 brain and 1 spinal cord). Subjects were naive to approved and off-label MS DMT. They could not have received cholesterol-lowering agents for 3 months prior to screening, and must have had normal hepatic function.
Randomization.
The randomization sequence was computer-generated in balanced block sizes of 5, stratified by center. Study drug kits were provided to the site pharmacist labeled with site and subject number. The study coordinator contacted the pharmacist to receive the subject number and corresponding study drug. The masked statistician and drug distributor maintained the randomization list containing treatment groups. Site staff were masked to treatment assignment.
MRI scanning and analysis.
Brain MRI (whole brain T2/T1-weighted images yielding 1 × 1 × 3 mm3 resolution without gap) with injection of single-dose Gd was acquired according to a standardized protocol at each site on a 1.5-T magnet. A central MRI reading unit (UCSF, San Francisco) evaluated MRI scans for quality and measurement of study endpoints without knowledge of treatment assignments.
T2 lesion volume analysis was performed on all scans using a semiautomated thresholding method and manual editing with simultaneous view access to T2 and PD-weighted slices. An automated coregistration procedure was applied on subsequent timepoints onto each subject baseline scan.
Volumetric high-resolution (1 mm3, 124 slices) T1-weighted gradient-echo images were used to measure annual percent brain volume changes derived from SIENA,15 and normalized gray and white matter volumes generated by SIENAX (Image Analysis Group, Oxford, UK). T1-lesion masks were incorporated into the SIENAX program to correct for misclassifications of parenchymal tissue.
Statistical analyses.
The primary analysis tested equality of survival distributions between the 2 groups using a log-rank test. Subjects were censored at initiation of IFNβ-1a if they started prior to meeting the endpoint. Secondary analyses were performed using Fisher exact test. For secondary analyses, subjects terminating the study before month 12 due to voluntary withdrawal were considered to not have met PEP.
Subjects who began IFNβ-1a before month 12 were excluded from change-from-baseline secondary endpoints. For secondary time-to-event endpoints, subjects were censored at initiation of IFNβ-1a if they did not experience the event. Count data were truncated at the IFNβ-1a start date if subjects started IFNβ-1a before month 12. During the first 12 months of treatment, IFNβ-1a has no proven effect on atrophy measures16; therefore month 12 atrophy measures were included for all subjects with data, regardless of whether they initiated IFNβ-1a. Secondary endpoint analysis methods included Fisher exact tests for proportions, log-rank tests for time-to-event data, Wilcoxon rank sum test or t tests for continuous data, and zero-inflated Poisson models. For analysis of T2 and Gd lesions, the model was adjusted for baseline lesion count and time to starting IFNβ-1a. The model for rate of lesions per month after starting IFNβ-1a was adjusted for baseline lesion counts and age.
All analyses were performed on the intent-to-treat sample, which included all randomized subjects who received any study drug. No data for secondary endpoints were imputed.
RESULTS
Baseline characteristics.
Due to slow recruitment, enrollment was halted after 82 of 152 planned subjects were randomized between May 2005 and September 2007, of whom 81 received study drug (49 atorvastatin, 32 placebo). Baseline characteristics are provided in table 1. Groups were well balanced overall except for higher T1 volumes in the atorvastatin group.
Table 1.
Baseline characteristicsa
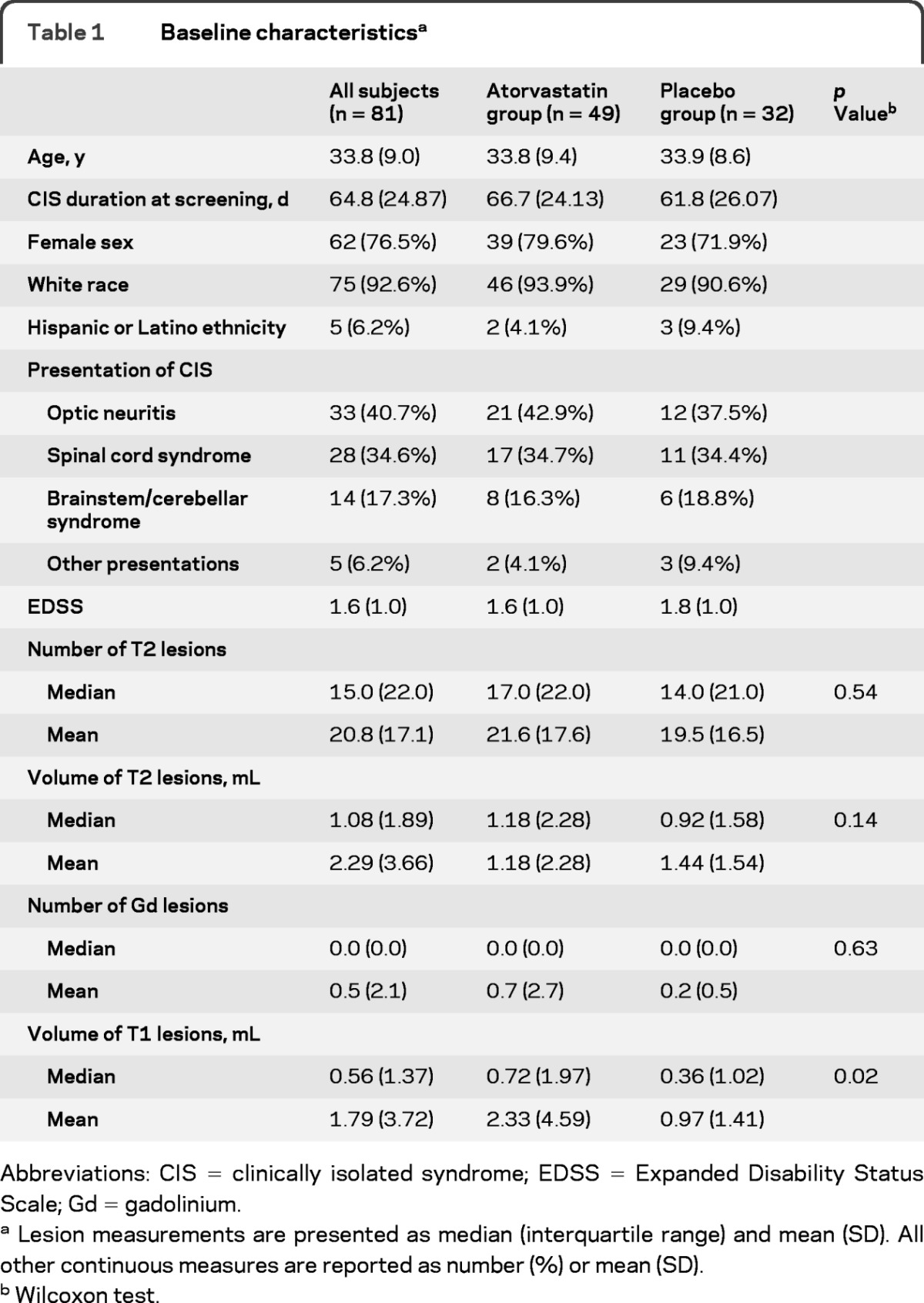
Abbreviations: CIS = clinically isolated syndrome; EDSS = Expanded Disability Status Scale; Gd = gadolinium.
Lesion measurements are presented as median (interquartile range) and mean (SD). All other continuous measures are reported as number (%) or mean (SD).
Wilcoxon test.
Dropout and treatment discontinuation.
Dropout and treatment discontinuation counts are provided in figure 1. Dosage of atorvastatin was reduced to 40 mg in 11 subjects, while none in the placebo group required modification.
Figure 1. CONSORT diagram.

IFNβ-1a = interferon β-1a.
Primary endpoint.
The proportion of subjects meeting the PEP was 53.1% (n = 26/49) in the atorvastatin group and 56.3% (n = 18/32) in the placebo group (p = 0.82, Fisher exact test). Median time to PEP was 275 days in the atorvastatin group and 284 days in the placebo group. Survival distributions were similar between both groups (log-rank test, p = 0.93). Eleven subjects (22.4%) on atorvastatin and 7 (21.9%) on placebo met PEP by clinical criteria.
Thirty-six subjects (20 in the atorvastatin group and 16 in the placebo group) received IFNβ-1a. Mean duration subjects were on IFNβ-1a was 151.3 ± 62.1 days for the atorvastatin group and 143.2 ± 76.9 days for the placebo group.
Imaging secondary endpoints.
Proportion of lesion-free subjects.
The proportion of subjects who did not develop new T2 lesions to month 12 or to starting IFNβ-1a was 55.3% in the atorvastatin and 27.6% in the placebo group (p = 0.03) (table 2, figure 2), which represents a 50% relative reduction favoring atorvastatin treatment. Odds of remaining T2-free was higher in the atorvastatin group compared with placebo: odds ratio (OR) = 4.34 (p = 0.01). For subjects who exhibited new T2 lesions, there was no difference between groups (p = 0.73). The odds of remaining Gd-free tended to be higher in the atorvastatin group compared with placebo: OR = 2.72 (p = 0.11). For subjects who exhibited new Gd lesions, there was no difference between groups (p = 0.65).
Table 2.
Effect of atorvastatin therapy on brain MRI metricsa
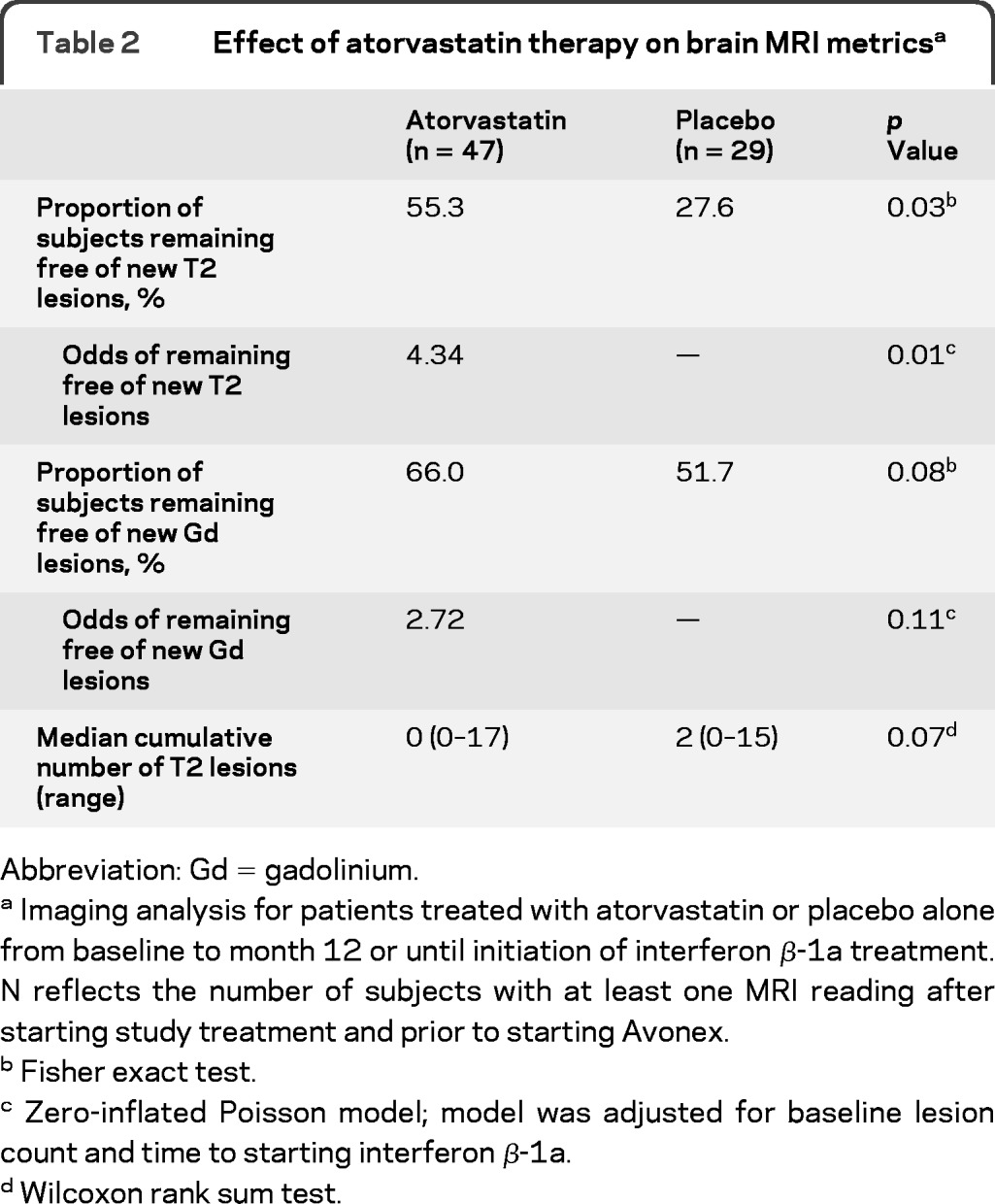
Abbreviation: Gd = gadolinium.
Imaging analysis for patients treated with atorvastatin or placebo alone from baseline to month 12 or until initiation of interferon β-1a treatment. N reflects the number of subjects with at least one MRI reading after starting study treatment and prior to starting Avonex.
Fisher exact test.
Zero-inflated Poisson model; model was adjusted for baseline lesion count and time to starting interferon β-1a.
Wilcoxon rank sum test.
Figure 2. Proportion of subjects with cumulative T2 lesion counts up to 12 months or prior to starting interferon β-1a (IFNβ-1a).

Data represent the proportion of subjects (y-axis) and the cumulative T2 lesion counts (x-axis) up to 12 months or prior to starting IFNβ-1a according to treatment group (blue = atorvastatin, red = placebo). Fifty-five percent of subjects receiving atorvastatin compared to 27% of subjects receiving placebo (p = 0.03) remained free of new T2 hyperintense lesions during the study period.
Median cumulative number of new T2 and gadolinium-enhanced lesions.
Median cumulative numbers of new T2 lesions to month 12 or to starting IFNβ-1a were 0 (range 0–17, mean 2.2 ± 3.6) and 2 (range 0–15, mean 3.0 ± 3.3) in atorvastatin and placebo groups. This corresponds to a 27% reduction in the mean cumulative number of T2 lesions (p = 0.08). Median cumulative numbers of Gd lesions to month 12 or before starting IFNβ-1a were 0 in both groups (atorvastatin range 0–12, mean 1.1 ± 2.3; placebo range 0–8, mean 1.2 ± 2.0).
Changes in T1 or T2 lesion volume from baseline to month 12.
No difference between groups was seen for changes in T1 and T2 lesion volumes between baseline and month 12 for subjects not initiating IFNβ-1a (median T1 lesion volume change: −0.01 [range −0.89 to 0.09] mL in the atorvastatin group and −0.02 [range −0.76 to 0.77] mL in the placebo group, p = 0.80; median T2 volume change: 0.02 [range −0.22 to 0.63] mL in the atorvastatin group and −0.03 [−0.55 to 2.18] mL in the placebo group, p = 0.49). Brain atrophy data are reported in table e-1 on the Neurology® Web site at www.neurology.org.
Clinical secondary endpoints.
No difference between groups was found for changes on MSFC scores (p = 0.70), EDSS (p = 0.67), and VAS (p = 0.92) between baseline and month 12 visits for subjects not initiating IFNβ-1a before month 12. Eleven subjects (22.4%) in the atorvastatin group and 7 (21.9%) in the placebo group had a clinical event before month 12 (log-rank test, p = 0.77).
Effect of the combination of atorvastatin and IFNβ-1a on MRI activity.
No clear antagonistic effect of combination of atorvastatin and IFNβ-1a compared to placebo and IFNβ-1a was found on the number of new T2 and Gd lesions (table e-2).
Safety.
A total of 961 treatment-emergent (TE) adverse events (AEs) were reported (table 3). The majority (82.7%) of subjects had AEs that were judged grade 1 (mild) or grade 2 (moderate) in severity.
Table 3.
Summary of treatment-emergent adverse eventsa
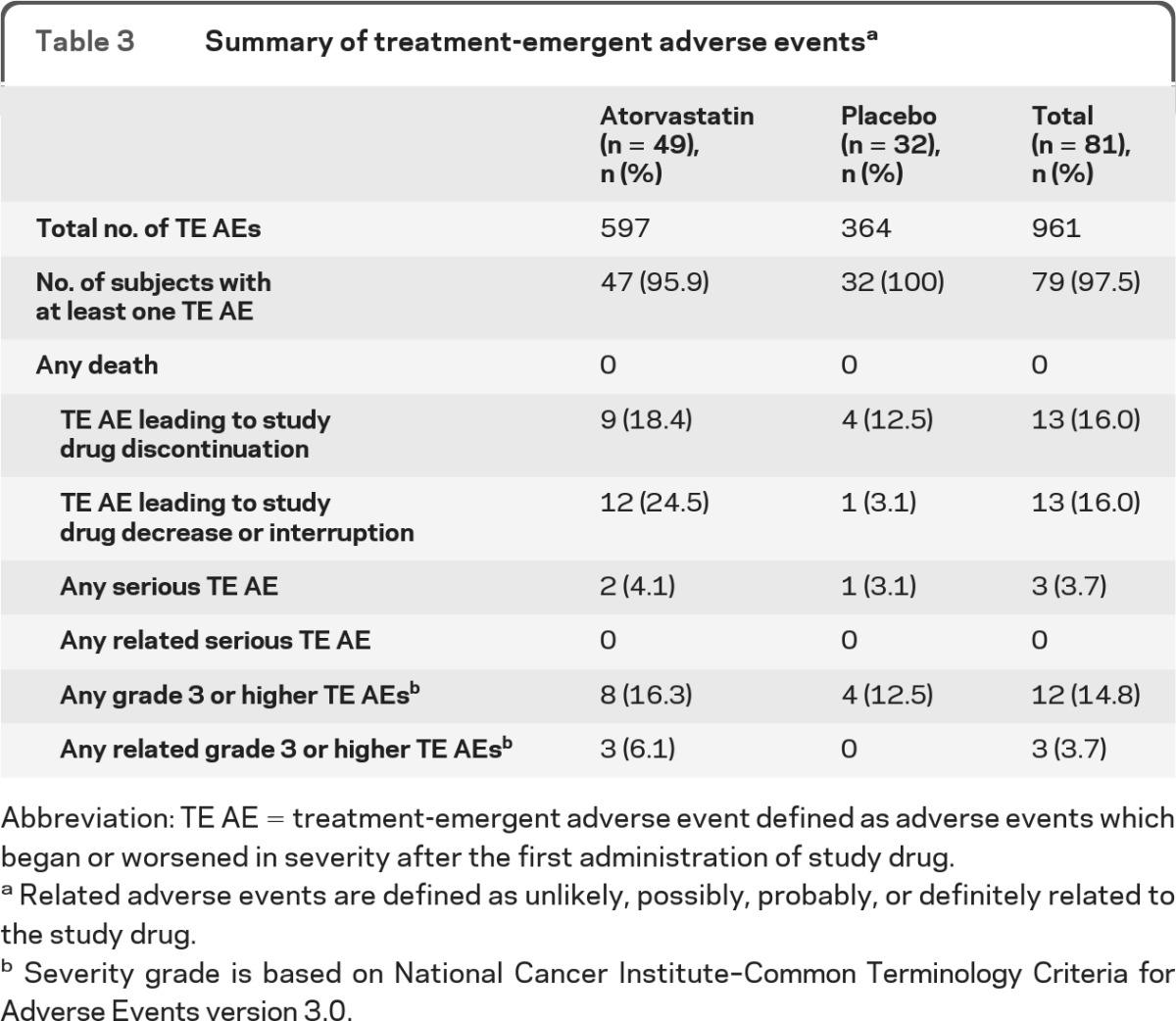
Abbreviation: TE AE = treatment-emergent adverse event defined as adverse events which began or worsened in severity after the first administration of study drug.
Related adverse events are defined as unlikely, possibly, probably, or definitely related to the study drug.
Severity grade is based on National Cancer Institute–Common Terminology Criteria for Adverse Events version 3.0.
A total of 213 TE AEs occurred after IFNβ-1a was started for subjects meeting the PEP, including 125 in the atorvastatin group and 88 in the placebo group. The 5 most common TE AEs experienced were disease progression (51.9%), hypoesthesia (43.2%), paraesthesia (25.9%), fatigue (24.7%), and headache (23.5%). Disease progression and hypoesthesia were reported in a greater percentage of subjects in the placebo group (59.4% and 50.0%) than the atorvastatin group (46.9% and 38.8%). Paraesthesia, fatigue, and headache were reported nearly equally across groups (atorvastatin: 26.5%, 26.5%, and 24.5%; placebo: 25.0%, 21.9%, and 21.9%).
The following AEs occurred more frequently in subjects who received IFNβ-1a in combination with atorvastatin than placebo: headache (27.8% vs 6.7%), elevation of alanine aminotransferase (27.8% vs 0%) or aspartate aminotransferase (27.8% vs 0%), and nausea (16.7% vs 0%).
Blinding analysis.
When EDSS physicians were asked the treatment assignment for atorvastatin-treated subjects, 33 (89.2%) responded “do not know,” 2 (5.4%) guessed correctly, and 2 (5.4%) incorrectly. For placebo subjects, 24 (88.9%) EDSS physicians responded “do not know,” 2 (7.4%) guessed correctly, and 1 (3.7%) incorrectly. When treating physicians were asked the treatment assignment for atorvastatin-treated subjects, 23 (59%) responded “do not know,” 14 (35.9%) guessed correctly, and 2 (5.1%) incorrectly. For placebo subjects, 20 (69%) treating physicians responded “do not know,” 7 (24.1%) guessed correctly, and 2 (6.9%) incorrectly. When atorvastatin-treated subjects were asked to guess their treatment assignment, 8 (20.5%) responded “do not know,” 23 (59%) guessed correctly, and 8 (20.5%) incorrectly. When placebo subjects were asked to guess their treatment assignment, 9 (31%) responded “do not know,” 14 (48.3%) guessed correctly, and 6 (20.7%) incorrectly.
Exploratory tolerance phase.
Twenty-six patients (18 atorvastatin, 8 placebo) who did not meet the PEP and developed no new MRI lesions after month 3 entered a “tolerance phase” to determine whether there could be sustained clinical and imaging effects in the 6 months after discontinuation of atorvastatin. These subjects received brain MRIs at month 15 and 18. Ten atorvastatin subjects (55.6%), but none of the placebo subjects, developed new MRI lesions or experienced an exacerbation during those 6 months (p = 0.03), findings that did not support a tolerogenic effect of atorvastatin.
DISCUSSION
We report the only randomized double-blind controlled phase II trial that has tested whether a statin alone can reduce MS activity against placebo. The PEP was not met. However, secondary imaging endpoints were positive, suggesting a beneficial effect of atorvastatin compared with placebo. In this regard, there was a significant reduction in the risk to develop new T2 hyperintense foci in the atorvastatin group to month 12 or to initiation of IFNβ-1a (OR = 4.34; table 2). This was observed despite early discontinuation of enrollment and some randomization imbalance, namely higher T1 disease burden in the atorvastatin group (table 1). In addition, the exploratory tolerance phase suggested a higher disease activity in stable patients after discontinuing atorvastatin vs placebo (p = 0.03).
Other investigations have examined the role of various statins as MS therapy.5–10 Our imaging findings in CIS are comparable to those reported in the only other trial that tested statin monotherapy in MS. In that earlier study, 30 patients with relapsing-remitting MS were treated with simvastatin in an open-label single crossover design. Simvastatin treatment was associated with a 30% reduction in number of new T2 hyperintense foci, suggesting a beneficial effect.5 That design, however, was subject to regression to the mean,17 a concern averted in a placebo-controlled trial.
Our study has limitations. Beside underenrollment, the placebo group exhibited less clinical and radiologic activity than subjects included in the CHAMPS dataset used for our power calculation.11 Further, the composite PEP had never been tested as an endpoint in previous MS trials. Its sensitivity to detect changes in disease activity and treatment effects will require further clarification. As is typical for MS trials, this study was relatively short (1 year), which may limit the generalizability of our findings. In addition, the number of subjects who reduced study medication dosage due to unwanted clinical or laboratory-documented side effects was greater than anticipated, and may have decreased our ability to detect clinical effects.
For ethical reasons, subjects were offered to start on an approved DMT if they exhibited clinical or MRI activity (i.e., after meeting the PEP) so that all subjects contributed to the primary analysis. Such a study design impacts analyses of secondary endpoints not based on time to meet PEP, as data acquired at later time points could be confounded by concomitant IFNβ-1a. Therefore, our secondary analyses, except for atrophy measures, only used endpoints collected to month 12 for those subjects who did not start on DMT, and to initiation of IFNβ-1a for subjects who met the PEP before month 12.
As statins have a different mechanism of action than approved MS medications, they may have added benefit when used in combination.18 Statins have been tested in subjects with MS in combination with IFNβ. Two studies have suggested that the addition of oral statin to IFNβ therapy may result in a paradoxical increase in MS activity compared to subjects receiving IFNβ-1a alone.6,10 One theoretical possibility for such potential antagonism may relate to the opposing activity of type I interferon on STAT1 phosphorylation,19 i.e., statins inhibit STAT1 phosphorylation,19–21 whereas activation of the type I IFN receptor induces STAT1 phosphorylation.22 However, not all MS clinical studies have detected antagonism of IFNβ therapy by statins.8,9,23 We did not observe that addition of weekly IM IFNβ-1a 30 μg to atorvastatin in the rescue phase significantly reduced the treatment effect of IFNβ-1a on new T2 or Gd lesions. The degree of antagonism may be related to individual doses of these medications. Our study may also have been too small or the rescue phase too short to rule out such an effect. Given the concern for antagonism between a statin and IFNβ, one should be cautious in judging the potential benefit provided by a statin in MS therapy, based upon trials that have only tested a statin in combination with IFNβ.
Atorvastatin did not impact the clinical endpoint in this trial. However, as positive MRI results were observed, future studies to confirm the effect of statins in MS are warranted. Such studies may require enrolling greater numbers of subjects to provide sufficient power to assess the influence on clinical measures.
Supplementary Material
ACKNOWLEDGMENT
The authors thank members of the Central MRI unit (Dr. Okuda and project coordinator Alan Evangelista), site coordinators, and patients for their contributions to this study; and Dr. Gary Cutter for discussions. The centralized MRI reading was performed by the Advanced Imaging in Multiple Sclerosis (AIMS) Laboratory at UCSF (Dr. Daniel Pelletier, Director). The Data Safety Monitoring Board was sponsored by NIAID, and provided clinical, statistical, and ethical support.
GLOSSARY
- AE
adverse event
- CIS
clinically isolated syndrome
- DMT
disease-modifying therapy
- EAE
experimental autoimmune encephalomyelitis
- EDSS
Expanded Disability Status Scale
- Gd
gadolinium
- IFNβ
interferon-β
- IFNβ-1a
interferon β-1a
- MS
multiple sclerosis
- MSFC
Multiple Sclerosis Functional Composite
- NUL
normal upper limit
- OR
odds ratio
- PEP
primary endpoint
- TE
treatment-emergent
- VAS
visual analog scale
Footnotes
Supplemental data at www.neurology.org
Coinvestigators are listed on the Neurology® Web site at www.neurology.org.
AUTHOR CONTRIBUTIONS
All authors were involved in the study concept and design, data collection, standardized assessments of subjects, study conduct, data summary and analysis, literature search, and writing of the report. Dr. Waubant was involved in the study concept and design, data collection, standardized assessments of subjects, study conduct, data summary and analysis, literature search, and writing of the report. Dr. Pelletier was involved in the study design, data collection, standardized assessments of subjects, study conduct, MRI central coordination and analysis, data summary and analysis, and writing of the report. Dr. Zamvil was involved in the study concept and design, data collection, standardized assessments of subjects, study conduct, data summary and analysis, literature search, and writing of the report. Drs. Mass, Cohen, Kita, Cross, Bar-Or, Vollmer, Racke, Stüve, Schwid, Goodman, Kachuck, Preiningerova, Weinstock-Guttman, Calabresi, and Miller were involved in the study design and conduct, standardized assessment of subjects, and writing of the report. Dr. Mokhtarani, Dr. Iklé, Dr. Ding, and C. Spencer were involved in the study design and conduct, data collection and analysis, and writing of the report. S. Murphy and H. Kopetskie were involved in data collection and analysis, and writing of the report. E. Rosenberg and the ITN020AI Study Management Team were involved in the study design and conduct, data collection and analysis, and writing of the report.
STUDY FUNDING
This research was performed as a project of the Immune Tolerance Network (ITN020AI, ITN contract number N01-AI-15416), a clinical research consortium sponsored by the National Institute of Allergy and Infectious Diseases. Pfizer provided atorvastatin, placebo, and grant support. Biogen Idec provided IFNβ-1a IM and grant support. E.W. is also supported by the Nancy Davis Foundation. S.S.Z. was also supported for this study by the National Institutes of Health (RO1 AI059709) and the Maisin Foundation.
DISCLOSURE
Dr. Waubant serves on a data safety monitoring board for the NIH and on a scientific advisory board for Actelion Pharmaceuticals Ltd; has received speaker honoraria from Teva Pharmaceutical Industries Ltd.; served as a consultant for Actelion Pharmaceuticals Ltd, Roche, and sanofi-aventis; and receives research support from sanofi-aventis, Biogen Idec, the NIH, the National MS Society, and the Nancy Davis Foundation. Dr. Pelletier serves/has served as a consultant for Synarc Inc., Biogen Idec, Bayer Schering Pharma, Genentech, Inc., Teva Pharmaceutical Industries Ltd., and CNS Imaging Consultant, LLC; serves on the speakers' bureau for Biogen Idec; and receives research support from Biogen Idec, the NIH, and the National Multiple Sclerosis Society. Dr. Mass has received speaker honoraria from Biogen Idec, Novartis, Teva Pharmaceutical Industries Ltd., and Bayer Schering Pharma. Dr. Cohen has served on scientific advisory boards for Biogen Idec, Novartis, EMD Serono, Inc., and Teva Pharmaceutical Industries Ltd.; has received funding for travel from Biogen Idec, sanofi-aventis, and EMD Serono, Inc.; receives publishing royalties for Multiple Sclerosis Therapeutics (Informa Healthcare, 2007); has received speaker honoraria from sanofi-aventis; and receives research support from Biogen Idec, Novartis, Genzyme Corporation, Teva Pharmaceutical Industries Ltd., the Immune Tolerance Network, and the Nancy Davis Center Without Walls. Dr. Kita has received funding for travel from Teva Pharmaceutical Industries Ltd. and Biogen Idec; has received speaker honoraria (paid to institution) from Teva Pharmaceutical Industries Ltd. and Biogen Idec; and receives research support from Biogen Idec, Novartis, Genentech Inc., and Ono Pharmaceutical Co. Ltd. Dr. Cross serves/has served on scientific advisory boards for Eli Lilly and Company, Genentech, Inc., Biogen Idec, Roche, and Coronado BioSciences; serves on the editorial boards of Brain Pathology and the Journal of Neuroimmunology; has received speaker or consulting honoraria from Pfizer Inc, Biogen Idec, sanofi-aventis, Bayer Schering Pharma, Teva Pharmaceutical Industries Ltd., Gerson Lehrman Group, and Guidepoint Global, LLC; and receives research support from sanofi-aventis, Acorda Therapeutics Inc., Genentech, Inc., Biogen Idec, Roche, the NIH/NINDS, ICTS Washington University, the National MS Society USA, Consortium of MS Centers, and the Barnes-Jewish Hospital Foundation. Dr. Bar-Or serves on scientific advisory boards for BioMS Medical, DioGenix, Inc., Ono Pharmaceutical Co. Ltd., GlaxoSmithKline, Roche, Guthy Jackson Greater Good Foundation, and NMO Research and Clinical Care Consortium; serves on the editorial boards of Neurology® and Clinical and Experimental Neuroimmunology; has received speaker honoraria from Biogen Idec, Bayhill Therapeutics, Bayer Schering Pharma (Berlex), Eli Lilly and Company, Genentech, Inc., GlaxoSmithKline, Merck Serono, Novartis, Wyeth, sanofi-aventis, and Teva Pharmaceutical Industries Ltd.; and receives/has received research support from BioMS Medical, Merck Serono, Bayhill Therapeutics, Biogen Idec, Genentech, Inc., and Teva Pharmaceutical Industries Ltd. Dr. Vollmer has served on scientific advisory boards for Roche, Elan Corporation, Teva Pharmaceuticals Industries Ltd., Novartis, GlaxoSmithKline, EMD Serono, Inc., Biogen Idec, Abbott, Acorda Therapeutics Inc., Bayhill Therapeutics, Metabolic Solutions Development Co., and Genentech, Inc.; has received speaker honoraria from EMD Serono, Inc., Teva Pharmaceuticals Industries Ltd., Biogen Idec, XenoPort, Inc., and Ono Pharmaceutical Co. Ltd.; has served as a consultant for Biogen Idec, Acorda Therapeutics Inc., Teva Pharmaceutical Industries Ltd., EMD Serono, Inc., Eli Lilly and Company, XenoPort, Inc., IMPAX Laboratories, Inc., GlaxoSmithKline, Daiichi Sankyo, Guidepoint Global, Eisai Inc., Novartis, Ono Pharmaceutical Co. Ltd., and Elan Corporation; has served on speakers' bureaus for Biogen Idec, Teva Pharmaceuticals Industries Ltd., and Athena Diagnostics, Inc.; is listed as author on a pending patent re: Nicotinic attenuation of CNS inflammation and autoimmunity; and has received research support from Teva Pharmaceuticals Industries Ltd., Acorda Therapeutics Inc., Biogen Idec, Daiichi Sankyo, Elan Corporation, Eli Lilly and Company, EMD Serono, Inc., Genzyme Corporation, Biosite, Eisai Inc., Genentech, Inc., Novartis, Ono Pharmaceutical Co. Ltd., Accelerated Cure Project, IMPAX Laboratories, Inc., sanofi-aventis, BioMS Medical, and the Rocky Mountain Multiple Sclerosis Society. Dr. Racke serves on scientific advisory boards for the Accelerated Cure Project and Diogenix, Inc.; serves on the editorial boards of Archives of Neurology, The Neurologist, PPAR Research, and the Journal of Neuroimmunology; serves/has served as a consultant for Eli Lilly and Company, Novartis, Biogen Idec, and Relavesio, Inc.; receives research support from the NIH/NINDS and the National Multiple Sclerosis Society; and research support paid to his institution from Biogen Idec, Novartis, Merck Serono and Teva Pharmaceutical Industries Ltd. Dr. Stüve serves on scientific advisory boards for Novartis, EMD Serono, Inc., sanofi-aventis, and Teva Pharmaceutical Industries Ltd.; serves on editorial boards for Archives of Neurology and Therapeutic Advances in Neurological Disorders; has received honoraria from Teva Pharmaceutical Industries Ltd., Roche, Genzyme Corporation, and Bayer Schering Pharma; and has received research support from Teva Pharmaceutical Industries Ltd., the US Department of Veterans Affairs (Merit Review Grant), and the Doris Duke Charitable Foundation. Dr. Schwid is deceased; disclosures are not included for this author. Dr. Goodman serves on a scientific advisory board for Biogen Idec; has received funding for travel and speaker or consulting honoraria from Acorda Therapeutics Inc., Actelion Pharmaceuticals Ltd, Avanir Pharmaceuticals, Bayer Schering Pharma, Biogen Idec, EMD Serono, Inc., Genentech, Inc., Genzyme Corporation, Novartis, Pfizer Inc, and Teva Pharmaceutical Industries Ltd.; and receives research support from Acorda Therapeutics Inc., Bayer Schering Pharma, Biogen Idec, EMD Serono, Inc., Genentech, Inc., Genzyme Corporation, Novartis, Teva Pharmaceutical Industries Ltd., Takeda Pharmaceutical Company Limited, Ono Pharmaceutical Co. Ltd., the NIH, and the Montel Williams Foundation. Dr. Kachuck serves on scientific advisory boards for Acorda Therapeutics Inc., Teva Pharmaceutical Industries Ltd., EMD Serono, Inc., and Glycominds, Ltd.; has received funding for travel or speaker honoraria from Acorda Therapeutics Inc., Teva Pharmaceutical Industries Ltd., and Glycominds, Ltd.; serves as a consultant for Glycominds, Inc. and Acorda Therapeutics Inc.; serves on the speakers' bureaus for Acorda Therapeutics Inc., Teva Pharmaceutical Industries Ltd., and EMD Serono, Inc.; and receives research support from Teva Pharmaceutical Industries Ltd. and EMD Serono, Inc. Dr. Preiningerova has received speaker honoraria from Biogen Idec, Novartis, Teva Pharmaceutical Industries Ltd. and Acorda Therapeutics Inc.; and has received research support paid to her institution by Acorda Therapeutics Inc., sanofi-aventis, Teva Pharmaceutical Industries Ltd., Biogen Idec, and Novartis. Dr. Weinstock-Guttman serves on a medical advisory board for the National Multiple Sclerosis Society; serves on speakers' bureaus and as a consultant for Biogen Idec, Teva Pharmaceutical Industries Ltd., EMD Serono, Inc, Novartis, Acorda Therapeutics Inc., and Pfizer Inc; serves on the editorial boards of aan.com and Multiple Sclerosis International; and receives research support from Novartis, Acorda Therapeutics Inc., Biogen Idec, EMD Serono, Inc., Teva Pharmaceutical Industries Ltd., Shire plc, Questcor, Cyberonics, Inc., the NIH, and the NMSS. Dr. Calabresi serves on scientific advisory boards for Biogen Idec, Teva Pharmaceutical Industries Ltd., Vertex Pharmaceuticals, Vaccinex, Genzyme Corporation, Abbott, and Novartis; has received funding for travel from Biogen Idec, Teva Pharmaceutical Industries Ltd., Vertex Pharmaceuticals, EMD Serono, Inc., Novartis, and Novo Nordisk; serves on the editorial board of Neurology®; is an author on a patent re: Role of Kv1.3 as neuroprotective; serves/has served as a consultant for Biogen Idec, Teva Pharmaceutical Industries Ltd., Vertex Pharmaceuticals, and Novartis; and receives research support from Vertex Pharmaceuticals, Abbot, Bayer Schering Pharma, EMD Serono, Inc., Teva Pharmaceutical Industries Ltd., Genentech, Inc., Biogen Idec, the NIH/NINDS, the Nancy Davis Foundation, and the National Multiple Sclerosis Society. Dr. Miller has served on scientific advisory boards for sanofi-aventis, Biogen Idec, GlaxoSmithKline, EMD Serono, Inc., Teva Pharmaceutical Industries Ltd., Daiichi Sankyo, Merck Serono, Novartis, Ono Pharmaceutical Co. Ltd., and Acorda Therapeutics Inc.; has served on speakers' bureaus for and received speaker honoraria from Biogen Idec and EMD Serono, Inc.; has received funding for travel or speaker honoraria from sanofi-aventis, Biogen Idec, Avanir, Biomarin, Chelsea Therapeutics, Ono Pharmaceutical Co. Ltd., and Acorda Therapeutics Inc.; serves on the editorial board of Continuum; and has received research support paid to his institution from Acorda Therapeutics Inc., Biogen Idec, Teva Pharmaceutical Industries Ltd., Novartis, Genentech, Inc., Genzyme Corporation, and sanofi-aventis. Dr. Mokhtarani received compensation as an employee of the Immune Tolerance Network. Dr. Iklé has received support paid to his institution through a contract with the NIH/NIAID. S. Murphy has received support paid to her institution through a contract with the NIH/NIAID. H. Kopetskie has received support paid to her institution through a contract with the NIH/NIAID. Dr. Ding, E. Rosenberg, and C. Spencer report no disclosures. Dr. Zamvil serves as Consulting Editor of the Journal of Clinical Investigation, on the editorial board of Neurotherapeutics, and as an Associate Editor for the Journal the Neurological Sciences; has served as a consultant for and received speaker honoraria from Biogen Idec, Teva Pharmaceutical Industries Ltd., Genentech, Inc., and EMD Serono, Inc.; has served on speakers' bureaus for Advanced Health Media and Health Logix; and receives research support from the NIH, the National Multiple Sclerosis Society, the Guthy Jackson Charitable Foundation, the Maisin Foundation, and Teva Pharmaceutical Industries Ltd.
REFERENCES
- 1. Zamvil SS, Steinman L. Cholesterol-lowering statins possess anti-inflammatory activity that might be useful for treatment of MS. Neurology 2002; 59: 970– 971 [DOI] [PubMed] [Google Scholar]
- 2. Youssef S, Stuve O, Patarroyo JC, et al. The HMG-CoA reductase inhibitor, atorvastatin, promotes a Th2 bias and reverses paralysis in central nervous system autoimmune disease. Nature 2002; 420: 78– 84 [DOI] [PubMed] [Google Scholar]
- 3. Aktas O, Waiczies S, Smorodchenko A, et al. Treatment of relapsing paralysis in experimental encephalomyelitis by targeting Th1 cells through atorvastatin. J Exp Med 2003; 197: 725– 733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dunn SE, Youssef S, Goldstein MJ, et al. Isoprenoids determine Th1/Th2 fate in pathogenic T cells, providing a mechanism of modulation of autoimmunity by atorvastatin. J Exp Med 2006; 203: 401– 412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vollmer T, Key L, Durkalski V, et al. Oral simvastatin treatment in relapsing-remitting multiple sclerosis. Lancet 2004; 363: 1607– 1608 [DOI] [PubMed] [Google Scholar]
- 6. Birnbaum G, Cree B, Altafullah I, Zinser M, Reder AT. Combining beta interferon and atorvastatin may increase disease activity in multiple sclerosis. Neurology 2008; 71: 1390– 1395 [DOI] [PubMed] [Google Scholar]
- 7. Paul F, Waiczies S, Wuerfel J, et al. Oral high-dose atorvastatin treatment in relapsing-remitting multiple sclerosis. PLoS ONE 2008; 3: e1928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lanzillo R, Orefice G, Quarantelli M, et al. Atorvastatin combined to interferon to verify the efficacy (ACTIVE) in relapsing-remitting active multiple sclerosis patients: a longitudinal controlled trial of combination therapy. Mult Scler 2010; 16: 450– 454 [DOI] [PubMed] [Google Scholar]
- 9. Togha M, Karvigh SA, Nabavi M, et al. Simvastatin treatment in patients with relapsing-remitting multiple sclerosis receiving interferon beta 1a: a double-blind randomized controlled trial. Mult Scler 2010; 16: 848– 854 [DOI] [PubMed] [Google Scholar]
- 10. Sorensen PS, Lycke J, Eralinna JP, et al. Simvastatin as add-on therapy to interferon beta-1a for relapsing-remitting multiple sclerosis (SIMCOMBIN study): a placebo-controlled randomised phase 4 trial. Lancet Neurol 2011; 10: 691– 701 [DOI] [PubMed] [Google Scholar]
- 11. Jacobs LD, Beck RW, Simon JH, et al. Intramuscular interferon beta-1a therapy initiated during a first demyelinating event in multiple sclerosis. CHAMPS Study Group. N Engl J Med 2000; 343: 898– 904 [DOI] [PubMed] [Google Scholar]
- 12. Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology 1983; 33: 1444– 1452 [DOI] [PubMed] [Google Scholar]
- 13. Cutter GR, Baier ML, Rudick RA, et al. Development of a multiple sclerosis functional composite as a clinical trial outcome measure. Brain 1999; 122: 871– 882 [DOI] [PubMed] [Google Scholar]
- 14. Newman CB, Palmer G, Silbershatz H, Szarek M. Safety of atorvastatin derived from analysis of 44 completed trials in 9,416 patients. Am J Cardiol 2003; 92: 670– 676 [DOI] [PubMed] [Google Scholar]
- 15. Smith SM, De Stefano N, Jenkinson M, Matthews PM. Normalized accurate measurement of longitudinal brain change. J Comput Assist Tomogr 2001; 25: 466– 475 [DOI] [PubMed] [Google Scholar]
- 16. Rudick RA, Fisher E, Lee JC, Simon J, Jacobs L. Use of the brain parenchymal fraction to measure whole brain atrophy in relapsing-remitting MS: Multiple Sclerosis Collaborative Research Group. Neurology 1999; 53: 1698– 1704 [DOI] [PubMed] [Google Scholar]
- 17. Zhao Y, Traboulsee A, Petkau AJ, Li D. Regression of new gadolinium enhancing lesion activity in relapsing-remitting multiple sclerosis. Neurology 2008; 70: 1092– 1097 [DOI] [PubMed] [Google Scholar]
- 18. Stüve O, Youssef S, Weber MS, et al. Immunomodulatory synergy by combination of atorvastatin and glatiramer acetate in treatment of CNS autoimmunity. J Clin Invest 2006; 116: 1037– 1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zamvil SS, Steinman L. Combining statins with interferon beta in multiple sclerosis: think twice, it might not be all right. Lancet Neurol 2011; 10: 672– 673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dhawan N, Reder AT. Statins block interferon signaling in human immune cells: potential loss of the therapeutic effect of IFN-beta in multiple sclerosis. Neurology 2007; 68 (suppl 1): A364 Abstract. [Google Scholar]
- 21. Lee SJ, Qin H, Benveniste EN. Simvastatin inhibits IFN-gamma-induced CD40 gene expression by suppressing STAT-1alpha. J Leukoc Biol 2007; 82: 436– 447 [DOI] [PubMed] [Google Scholar]
- 22. Darnell JE, Jr, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994; 264: 1415– 1421 [DOI] [PubMed] [Google Scholar]
- 23. Rudick RA, Pace A, Rani MR, et al. Effect of statins on clinical and molecular responses to intramuscular interferon beta-1a. Neurology 2009; 72: 1989– 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.