

Abstract
Hereditary cerebral hemorrhage with amyloidosis–Dutch type is a disorder associated with a missense mutation (E693Q) in the β-amyloid (Aβ)-coding region of the amyloid precursor protein (APP). This familial disease is characterized by cognitive deficits secondary to intracerebral hemorrhage and, in some cases, progressive Alzheimer’s disease (AD)-like dementia. Although this mutation was the first ever reported in the human APP gene, little is known about the molecular mechanisms underlying the direct toxic effects of this mutated Aβ on central neurons. In the present study, we assessed the role of calpain-mediated toxicity in such effects using an AD primary culture model system. Our results showed that Dutch mutant Aβ (E22Q) induced calpain-mediated cleavage of dynamin 1 and a significant decrease in synaptic contacts in mature hippocampal cultures. These synaptic deficits were similar to those induced by wild-type (WT) Aβ. In contrast, calpain-mediated tau cleavage leading to the generation of a 17-kDa neurotoxic fragment, as well as neuronal death, were significantly reduced in E22Q Aβ–treated neurons when compared with WT Aβ–treated ones. This complex regulation of the calpain-mediated toxicity pathway by E22Q Aβ could have some bearing in the pathobiology of this familial AD form.
INTRODUCTION
Sporadic Alzheimer’s disease (AD) comprises more than 95% of the cases of this neurodegenerative disorder. Senile plaques and neurofibrillary tangles, abnormal deposits of β-amyloid (Aβ) and tau, respectively, are commonly seen in the brains of these patients. These lesions not only coexist in affected brain areas but also seem to be mechanistically linked. A growing body of evidence suggests that one of the mechanisms linking Aβ and tau pathologies involved enhanced calcium (Ca2+) influx. This deregulated Ca2+ influx triggered by aggregated Aβ is mediated by N-methyl-d-aspartic acid (NMDA) receptors and leads to the activation of calpain (1–6). The activation of this Ca2+-dependent protease plays an important role both in synapse dysfunction underlying early cognitive deficits and in neuronal degeneration associated with mid-stages and late stages of this disease (4–6). Research in our laboratory identified dynamin 1 and tau as two of the downstream effectors in these pathological pathways, respectively. Dynamin 1 is a neuron-specific mechanochemical GTPase responsible for vesicle scission from the synaptic terminal membrane (7,8). Aβ-induced calpain activation resulted in dynamin 1 cleavage altering synaptic vesicle recycling and, hence, synaptic transmission in hippocampal neurons (4,5,9). In addition, abnormally activated calpain cleaved the microtubule-associated protein tau leading to the generation of a 17-kDa neurotoxic fragment in hippocampal neurons (10–12).
No comparable information is available regarding the participation of these mechanisms in early-onset familial forms of AD. One of these autonomic-dominant familial forms, hereditary cerebral hemorrhage with amyloidosis–Dutch type, is associated with the missense mutation G to C at codon 693 (E693Q), resulting in a glutamic acid to glutamine substitution at position 22 (E22Q) in the Aβ-coding region of the amyloid precursor protein (13). Clinically, these patients suffer from cognitive deficits secondary to intracerebral hemorrhage and, in some cases, progressive AD-like dementia. These symptoms are associated with extensive amyloid deposits in the wall of small cerebral arteries as well as preamyloid diffuse plaques surrounded by some dystrophic neurites (14–17). Recently, several studies have shown that Aβ carrying the Dutch mutation was toxic to cultured human leptomeningeal smooth muscle and neuroblastoma cells (18–20). However, the effects of this mutant Aβ on central neurons are yet to be determined.
In the present study, we analyzed the contribution of the calpain-mediated mechanisms in such effects. Our results showed that the Dutch mutant Aβ was capable of inducing loss of synaptic contacts and calpain activation leading to dynamin 1 cleavage, as the wild-type (WT) form did in hippocampal neurons. On the other hand, calpain-mediated generation of a 17-kDa neurotoxic tau fragment and cell death were significantly decreased in E22Q Aβ–treated mature hippocampal neurons as compared with WT Aβ–treated ones. The diverse effects of the Dutch Aβ on the calpain-mediated mechanisms underlying pathological processes associated with AD could have some bearing on the clinical manifestations observed in patients carrying this Aβ mutation.
MATERIALS AND METHODS
Preparation of Primary Hippocampal Cultures
Timed-pregnant Sprague-Dawley rats were purchased from Taconic and euthanized by CO2 overdose at embryonic d 18. Embryos were used to prepare hippocampal neuron cultures as described previously (21). Briefly, the hippocampi were dissected and freed of meninges. The cells were dissociated using trypsin (0.25%) for 15 min at 37°C followed by trituration with a fire-polished Pasteur pipette. The cell suspension was plated in minimum essential medium (MEM) with 10% horse serum (MEM10) on poly-l-lysine–coated dishes at 800,000 cells per 60-mm dish. After 4 h, the medium was changed to glia-conditioned MEM containing 0.1% ovalbumin, 0.1 mmol/L sodium pyruvate and N2 supplements (N2 medium) (22). For immunocytochemical analyses, neurons were plated (150,000 cells per 60-mm dish) onto poly-l-lysine–coated coverslips in MEM10. After 4 h, the coverslips were transferred to dishes containing an astroglial monolayer and maintained in N2 medium. Experiments were performed using neurons cultured for 18–22 d.
The Northwestern University Animal Care and Use Committee approved the experimental protocol used in this study in accordance with U.S. Public Health Service regulations and applicable federal and local laws.
Preaggregation of Aβ
Synthetic WT and Dutch mutant (E22Q) Aβ1–40 peptides were dissolved in N2 medium at a final concentration of 0.5 mg/mL. These Aβ solutions were kept at 37°C for 3 d to preaggregate the peptides (23). Neurons were incubated in the presence of the peptides for 24 h at concentrations ranging from 2 to 20 μmol/L (10).
Cell Death Assay
Cell viability was assessed by the try-pan blue exclusion method and by using a Live/Dead Viability/Cytotoxicity kit (Invitrogen, Carlsbad, CA, USA) as previously described (6,23,24). Briefly, control hippocampal neurons cultured on coverslips for 21 d, and sister cultures treated for 24 h in the presence or absence of WT or E22Q Aβ, were incubated in 0.2% trypan blue stain (Sigma Aldrich, St. Louis, MO, USA) for 5 min at room temperature or in the ethidium homodimer/calcein acetoxy methyl ester combination of vital dyes (Live/Dead Viability/Cytotoxicity kit) for 30 min at 37°C. The cells were rinsed in phosphate-buffered saline (PBS) and immediately counted. Cells that did not exclude try-pan blue were counted as dead cells. Neurons stained using the Live/Dead kit were observed using a fluorescence microscope (Nikon Diaphot, Melville, NY, USA). Live (red) and dead (green) neurons were counted. Ten non-overlapping microscopic fields from three independent cultures were analyzed for each experimental condition. Cell death was expressed as a percentage of total cells in each field.
Apoptotic cell death was assessed using the In Situ Cell Death Detection Kit (Roche Applied Science, Indianapolis, IN, USA). Briefly, cells were fixed for 15 min with 4% paraformaldehyde in PBS containing 0.12 mol/L sucrose, permeabilized in 0.1% Triton X-100 in 0.1% sodium citrate for 2 min, and tetram-ethylrhodamine (TMR) fluorescein-labeled nucleotide was incorporated at 3′-OH DNA ends using the enzyme terminal deoxynucleotidyl transferase (TdT). Apoptotic cells (TUNEL+ [transferase-mediated dUTP nick-end labeling] cells) were counted using a fluorescence microscope (Nikon Diaphot) and expressed as a percentage of total number of cells in each field.
Immunocytochemistry
Hippocampal neurons were cultured on coverslips and treated with or without WT or E22Q Aβ peptides for 24 h. Neurons were then fixed in 4% paraformaldehyde in PBS containing 0.12 mmol/L sucrose for 15 min and permeabilized in 0.3% Triton X-100 in PBS for 4 min. Coverslips were then incubated in 10% bovine serum albumin in PBS at room temperature for 1 h before labeling with the primary antibody. The primary antibodies used were antitubulin (clone DM1A, 1:1,000; Sigma) and antisynaptophysin (1:500; Santa Cruz Biotechnology, Santa Cruz, CA, USA). Antimouse or antirabbit AlexaFluor secondary antibodies (1:200; Molecular Probes, Eugene, OR, USA) were used for protein detection. Images were taken using a Photometrics Cool Snap HQ2 camera coupled with a fluorescent microscope (Nikon Diaphot, Melville, NY, USA). Images were analyzed using MetaMorph Image Analysis software (Universal Imaging, Fryer Company, Huntley, IL, USA).
Synapse Counts
The number of synapses per neuron was determined by double labeling cultured hippocampal neurons with antisynaptophysin and antitubulin antibodies, as described above. The number of synapses per cell was calculated by counting the synaptophysin-immunoreactive puncta using MetaMorph Image Analysis software and dividing these values by the total number of cells as observed with tubulin labeling.
Electrophoresis and Immunoblotting
Twenty-one days in culture hippocampal neurons incubated in the presence or absence of WT or E22Q Aβ were homogenized in Laemmli buffer and boiled for 10 min. Lysates were then loaded and run on sodium dodecyl sulfate (SDS)–polyacrylamide gels as previously described (25). The proteins were transferred onto Immobilon membranes (Millipore, Billerica, MA, USA) and immunoblotted (26,27). Immunodetection was performed using a phosphorylation-independent anti-tau (clone tau5; 1:1,000; BioSource International, Foster City, CA, USA), anti–phosphorylated tau at Ser202/Thr205 (clone AT8; 1:1,000; Thermo Scientific, Rockford, IL, USA), at Thr217 (1:500; BioSource International), at Ser262 (1:500; BioSource International), at Ser356 (1:500; BioSource International) and at Ser409 (1:1,000; Invitrogen) and antispectrin (1:1,000; Chemicon, Temecula, CA), anti–dynamin 1 (1:2,000; Affinity BioRe-agents, Golden, CO, USA) and anti–α-tubulin (clone DM1A; 1:200,000; Sigma) antibodies. Secondary antibodies conjugated to horseradish peroxidase (1:1,000; Promega, Madison, WI, USA) were used followed by enhanced chemiluminescence for the detection of proteins (28). A ChemiDoc XRS system and Quantity One Software (Bio-Rad, Hercules, CA, USA) were used to image and analyze immunoreactive bands.
Cytoskeletal Fractioning and Tau In Vitro Cleavage by Calpain 1
Cytoskeletal fractions were prepared as described previously (29). In short, untreated hippocampal neurons and neurons incubated in the presence of WT or E22Q Aβ were rinsed in microtubule-stabilizing buffer (MTSB: 4 mmol/L magnesium chloride, 10 mmol/L ethylene glycol-bis (2-amino-ethylether)-N,N,N′,N′-tetraacetic acid (EGTA), 130 mmol/L HEPES, pH 6.9) followed by extraction with MTSB containing 0.2% Triton X-100 (Sigma) for 90 s. The remaining cytoskeleton was rinsed with detergent-free MTSB and harvested in Laemmli buffer for Western blotting. For in vitro calpain assays, cytosolic extracts and cytoskeleton preparations were collected from 21 d in culture hippocampal neurons in lysis buffer (150 mmol/L sodium chloride, 5 mmol/L EGTA, 5 mmol/L ethylene-diamine-tetraacetic acid, 20 mmol/L Tris–hydrochloric acid, pH 7.4, 1% Triton X-100) per 800,000 cells and centrifuged at 16,000g for 10 min. The tau-containing supernatant was removed and incubated with 0.55 U cal-pain per 35 μL lysate for 1 h at 30°C. An equivalent volume of Laemmli was added, and samples were boiled for 10 min to stop the reaction. The cleavage products were run on SDS gels as described above.
Statistical Analyses
All experiments performed in this study were conducted in at least three independent cultures. The compiled data were analyzed across the experimental conditions using one-way analysis of variance (ANOVA) followed by Fisher least significant differences (LSD) post hoc test. The values in the graphs represent the mean ± standard error of the mean (SEM). The statistical significance is indicated in the graphs.
RESULTS
Toxic Effects of the Dutch Mutant Aβ on Cultured Hippocampal Neurons
We have extensively characterized preaggregated Aβ-induced neurodegeneration in mature cultures of hippocampal neurons (4–6,9–11,23,27,30). In contrast, little is known regarding the effects of the Dutch mutant Aβ on central neurons. In the present study, we assessed whether preaggregated E22Q Aβ has similar toxic effects as those described in WT Aβ–treated neurons. First, we determined cell death induced by E22Q Aβ in mature hippocampal cultures using try-pan blue. Only viable neurons with an intact plasma membrane were able to exclude this large dye from their cytoplasm. Quantification of the percentage of trypan blue–positive (dead) neurons showed a significant increase in overall cell death in both WT- and E22Q Aβ–treated neurons when compared with untreated controls (70% ± 2%** versus 55% ± 2%** versus 31% ± 1%, respectively; **differs from untreated controls, P < 0.01). However, cell viability was significantly higher in cultures treated with the mutant Aβ compared with those incubated with the WT peptide (P < 0.01). Similar results were obtained using fluorescent dyes (63% ± 2%** versus 44% ± 2%** versus 25 % ± 1%, dead cells in WT-and E22Q Aβ–treated neurons and untreated controls, respectively; **differs from untreated controls, P < 0.01). Because we have previously shown that not all cell death induced by Aβ was due to apoptosis, we determined the percentage of apoptotic neurons under these experimental conditions. Quantitative analysis showed that apoptotic cell death was significantly higher in cultures treated with the WT peptide when compared with those incubated in the presence of the mutant Aβ and with untreated controls (44% ± 4%** versus 29% ± 4%** versus 19% ± 1% apoptotic cells, respectively; **differs from untreated controls, P < 0.01).
Next, we determined the effect of E22Q Aβ treatment on the number of synaptic contacts formed by mature hippocampal neurons using an antibody specific for synaptophysin as a synaptic marker (31). Synaptophysin immunoreactive puncta were easily detectable around cell bodies and along the neurite processes extended by mature hippocampal neurons (Figure 1). Quantitative analysis of synaptophysin-immunoreactive spots showed that the number of these synaptic contacts was significantly lower in E22Q Aβ–treated mature neurons when compared with untreated controls (see Figure 1). In contrast to cell death, no significant differences in the number of synaptic contacts/cell were detected when E22Q Aβ–treated neurons were compared with WT Aβ–treated ones (Figure 1G).
Figure 1.

Reduced number of synaptic contacts in mature hippocampal neurons treated with E22Q Aβ. (A–F) Twenty-one days after plating, untreated controls (A, B) and neurons incubated with WT (C, D) or Dutch (E22Q) (E, F) Aβ (10 μmol/L) for 24 h were immunolabeled with tubulin (A, C, E) and synaptophysin (B, D, F) antibodies. Note the similar decrease in the numbers of synaptophysin immunoreactive spots in neurons treated with either form of Aβ compared with untreated controls. (G) Quantification of the number of synaptophysin-positive puncta per cell in untreated neurons and in neurons treated as described in A–F. Values represent the mean ± SEM for five independent experiments per condition (n = 90 cells/condition). **Differs from untreated control, P < 0.01. Scale bar: 50 μm.
Dutch Mutant Aβ Had Differential Effects on Calpain-Mediated Dynamin 1 and Tau Cleavage in Cultured Hippocampal Neurons
We have previously shown that, in addition to synapse loss and cell death, the deposition of WT Aβ oligomers induced synaptic dysfunction and neurite degeneration in mature hippocampal neurons (4,5,9,10,23,27). The synaptic dysfunction observed in the presence of WT Aβ was mediated, at least in part, by a decrease in dynamin 1 levels due to calpain-mediated cleavage both in cultured hippocampal neurons and in an AD animal model system (4,5,9). To test whether E22Q Aβ has similar synaptic effects, we analyzed dynamin 1 levels in cultured hippocampal neurons incubated in the presence of increasing concentrations of this mutant Aβ form. Western blot analysis of whole cell extracts obtained from E22Q Aβ–treated neurons showed a dose-dependent decrease of full-length dynamin 1 (~100 kDa) and the appearance of an ~90 kDa cleaved immunoreactive band (Figure 2A). These results were similar to those obtained when WT Aβ–treated neurons were analyzed (see Figure 2A) (4). Quantitative analysis showed no significant differences in dynamin degradation in E22Q Aβ–treated neurons compared with WT Aβ–treated ones (Figure 2B).
Figure 2.

E22Q Aβ–induced dynamin 1 cleavage in mature hippocampal neurons. (A) Western blot analysis of whole cell extracts prepared from 21 d in culture hippocampal neurons incubated in the absence (C ) or presence of increasing concentrations of WT or Dutch (E22Q) Aβ and reacted with dynamin 1 and spectrin antibodies. Strong immunoreactive cleaved dynamin 1 (~90 kDa) bands were detected in either WT or E22Q Aβ–treated neurons. Tubulin was used as a loading control. (B) Graphs on the left show the levels of full-length (~100 kDa) and cleaved (~90 kDa) dynamin 1 bands, and the spectrin 150/240-kDa ratio in control and WT or Dutch Aβ–treated hippocampal neurons. Values are expressed as the percentage of untreated controls, considering the values obtained in these neurons as 100%. Each number represents the mean ± SEM from five different experiments. Charts on the right show the statistical significance of the differences among all experimental conditions calculated by one-way ANOVA followed by Fisher LSD post hoc test. *Differs from control, P < 0.05; **differs from control, P < 0.01.
We next studied the extent of calpain activation under these experimental conditions. For these experiments, we determined calpain activation by assessing spectrin cleavage (4,5,10). Spectrin degradation is highly sensitive to calpain activation and is considered an excellent marker for this protease activity (32). Western blot analysis of whole cell extracts obtained from E22Q Aβ–treated neurons and reacted with a specific spectrin antibody showed a significant decrease in full-length spectrin (240 kDa) and a concomitant increase in the 150-kDa degradation fragment (see Figure 2A) (4,5,10). Quantitative analysis of immunoreactive bands showed a significant increase in the spectrin 150/240 kDa ratio in hippocampal neurons treated with either E22Q or WT Aβ when compared with untreated controls (see Figure 2B). However, no differences were detected in E22Q Aβ–treated neurons compared with WT Aβ–treated ones.
Our previous findings show that Aβ-induced calpain activation also played a key role in neurite degeneration by cleaving tau into a neurotoxic fragment (10,11). Therefore, we next assessed whether the Aβ E22Q mutant was able to induce calpain-mediated tau cleavage leading to the generation of this 17-kDa neurotoxic fragment. Western blot analysis of whole cell extracts reacted with a non–phosphorylation-dependent tau antibody showed strong immunoreactive full-length tau bands and the absence of tau immunoreactivity at the 17-kDa molecular weight in untreated controls. On the other hand, a clear band at this molecular weight was detected in cultured hippocampal neurons treated with E22Q Aβ (Figure 3A). As previously described, the presence of this 17-kDa tau fragment was also detected in hippocampal neurons treated with WT Aβ (see Figure 3A) (10). Quantitative analysis of immunoreactive bands showed a dose-dependent increase in 17-kDa tau in neurons treated with either form of Aβ. However, the 17-kDa tau/full-length tau ratio was significantly lower in E22Q Aβ–treated neurons compared with WT ones when cultures were incubated with the aggregated peptides at a final concentration of at least 10 μmol/L (Figure 3B).
Figure 3.
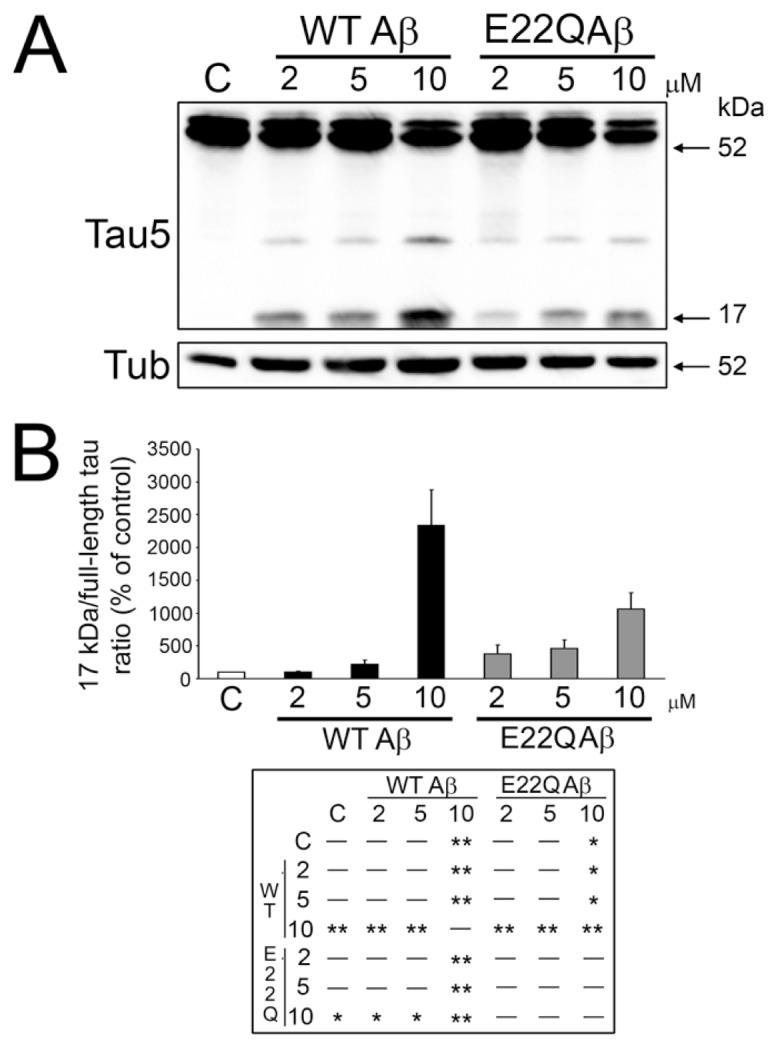
Dutch Aβ was less effective than WT Aβ in the generation of the 17-kDa tau fragment in cultured hippocampal neurons. (A) Western blot analysis of whole cell extracts prepared from 21 d in culture hippocampal neurons incubated in the absence (C ) or the presence of increasing concentrations of WT or Dutch (E22Q) Aβ. The membranes were reacted with a tau antibody (clone tau 5). Tubulin was used as a loading control. Note the decrease in the immunoreactive bands corresponding to full-length tau and the increase in the one corresponding to the cleaved 17-kDa tau fragment as the dose of either Aβ increased. (B) Graph showing the 17-kDa/full-length tau ratio in control and WT or Dutch Aβ–treated hippocampal neurons. Values are expressed as the percentage of untreated controls, considering the values obtained in these neurons as 100%. Each number represents the mean ± SEM from five different experiments. One-way ANOVA followed by Fisher LSD post hoc test were performed. *Differs from control, P < 0.05; **differs from control, P < 0.01.
Increased Levels of Cytoskeletal-Bound Tau Associated with Decreased Calpain-Mediated Degradation in E22Q Aβ–Treated Neurons
The results described above indicated that despite similar calpain activation, tau cleavage was significantly decreased in cultured hippocampal neurons incubated in the presence of E22Q Aβ (10 μmol/L) when compared with those incubated with WT Aβ. These differences in tau cleavage induced by either aggregated Aβ peptide could be due to a reduced access of active calpain to tau due to changes in the distribution of this microtubule-associated protein. It was shown that under normal conditions, ~75% of tau is associated with the cytoskeleton in cultured central neurons (29). This association was significantly reduced, and most of the tau content was recovered in cytosolic fractions, in mature neurons treated with WT Aβ (27). Therefore, we assessed both the distribution of tau in these two intracellular pools and the susceptibility to cytosolic and cytoskeletal tau to calpain cleavage. Quantitative Western blot analysis showed a significant decrease in cytoskeleton-bound tau in either E22Q Aβ–or WT Aβ–treated neurons compared with untreated controls. However, the levels of microtubule-bound tau were significantly higher in cytoskeletal fractions prepared from neurons incubated with E22Q Aβ compared with those incubated with WT Aβ (Figure 4A).
Figure 4.

Decreased susceptibility of cytoskeletal-bound tau to calpain cleavage. (A) Western blot analysis of cytoskeletal fractions prepared from 21 d in culture hippocampal neurons incubated in the absence (C in panels A and B) or in the presence of WT Aβ or Dutch (E22Q) Aβ. The membranes were reacted with tau and tubulin antibodies. (B) Graph showing the results of the quantitative analysis of immunoreactive bands. Values are expressed as a percentage of the tau/tubulin ratio of untreated controls, considering the values obtained in these neurons as 100%. Each number represents the mean ± SEM from five different experiments. **Differs from control, P < 0.01. (C) Western blot analysis of cytosolic and cytoskeletal fractions obtained from untreated hippocampal neurons and then incubated in the presence (+) or absence (–) of active calpain. Membranes were reacted with a tau antibody (clone tau 5) and then stripped and reprobed with a tubulin antibody as a loading control. Note that microtubule binding decreased the susceptibility of tau to calpain cleavage.
We assessed next whether cytoskeleton-bound tau was less susceptible to cal-pain-mediated proteolysis than soluble tau. For these experiments, cytosolic and cytoskeletal lysates were incubated in the presence of active calpain, and the final proteolytic products were analyzed by Western blotting. Tau present in cytosolic fractions prepared from 21 d in culture hippocampal neurons was almost completely degraded in the presence of active calpain (Figure 4C). On the other hand, a strong full-length tau immunoreactive band was detected in cytoskeletal fractions incubated with active calpain 1 (see Figure 4C).
Because tau binding to the cytoskeleton is regulated by phosphorylation, we analyzed next the extent of this tau post-translational modification in neurons incubated with E22Q Aβ and compared with those treated with WT peptide. Quantitative Western blotting using a series of phosphorylation-dependent antibodies (at Ser202/Thr205, Thr217, Ser262, Ser356 and Ser409) showed no significant differences in tau phosphorylation at any of these residues when neurons treated with either Aβ peptide were compared (data not shown).
DISCUSSION
Individuals affected with hereditary cerebral hemorrhage with amyloidosis–Dutch type show a continuous cognitive decline similar to that of AD patients. These deficits have been attributed to frequent strokes caused by the extensive Aβ deposits in the wall of cerebral vessels leading to the formation of stenotic vessels and microvasculopathies (14–17). Our results strongly suggest the existence of an alternative mechanism that might contribute to synaptic dysfunction and memory and behavioral abnormalities observed in these patients. This mechanism involved both a decrease in the number of synaptic contacts and the cleavage of dynamin 1 in hippocampal neurons. The E22Q Aβ–induced decrease in dynamin 1 levels could play a critical role in synaptic dysfunction in view of recent results obtained under experimental conditions that led to loss-of-function and/or dynamin cleavage like the one described in this study. Those reports showed that synapses lost their ability to successively release neurotransmitters as synaptic vesicles became trapped at the plasma membrane and the synaptic vesicle pool was depleted in neurons in which dynamin 1 had been downregulated and/or cleaved (4,5,7–9,33–35).
Our results showed no differences in the deleterious effects of E22Q Aβ on synapse number, dynamin 1 cleavage and calpain activation when compared with WT Aβ. However, milder cognitive defects than those associated with AD are described in patients suffering from this form of amyloidosis. One potential explanation for these milder effects of E22Q Aβ could be the extent of deposition of this peptide in affected brain areas. Thus, it has been shown that E22Q Aβ burden in the brain parenchyma of these patients is less extensive than the WT Aβ burden observed in AD patients (36,37). A contributing factor to this differential Aβ deposition might be the aggregation properties of E22Q Aβ. A series of reports have shown that, compared with WT peptide, the Dutch mutant Aβ aggregates more rapidly into fibrils that are less toxic than oligomeric forms (20,37,38). Alternatively, the subtler behavioral phenotype in patients suffering from hereditary cerebral hemorrhage with amyloidosis–Dutch type could be related to decreased cell death.
Interestingly, we observed a significant decrease in cell death in E22Q Aβ–treated hippocampal neurons when compared with those treated with WT Aβ under the experimental conditions used in this study. A similar result was obtained when the neurotoxic effects of the Dutch Aβ were compared with those of the Flemish mutant and WT Aβ in a neuroblastoma cell line incubated in the presence of these peptides for a very short time (2 h). In contrast, E22Q Aβ was more toxic than the Flemish mutant and WT Aβ with longer incubation times (24 h) (39). The enhanced survival observed in E22Q Aβ–treated hippocampal neurons could be due to the reduced rate of calpain-mediated tau cleavage leading to the generation of the 17-kDa tau fragment. We have previously shown that the expression of this fragment in hippocampal neurons results in progressive neurodegeneration followed by cell death (10,11). Conversely, experimental conditions that reduce tau cleavage prevented neuronal death (6,12). Therefore, it is tempting to speculate that a decrease in the generation of this toxic fragment could play a role in the regulation of neurodegeneration in the presence of E22Q Aβ. This differential effect of the mutant Aβ on tau cleavage is consistent with a decrease in other forms of tau pathology in affected brain areas in these patients. Thus, no tau aggregation into neurofibrillary tangles has been identified in patients suffering from this type of cerebral amyloid angiopathy (15,36,40–42).
The reduced levels of the 17-kDa tau fragment observed in E22Q Aβ–treated neurons could be the result of the decreased activation of the protease responsible for this cleavage. Our results showing no significant differences in calpain activation in neurons treated in the presence of WT or E22Q Aβ indicate that the decrease in calpain-mediated tau cleavage observed in the mutant Aβ–treated neurons was independent of the extent of calpain activation. Alternatively, this diminished production of 17-kDa tau fragment could be due to differences in calpain access to tau under these experimental conditions. Calpain is a soluble protease highly concentrated in the neuronal cytoplasm, whereas tau is both soluble and cytoskeletal bound (27,29,43). In the presence of E22Q Aβ, we detected a significantly higher ratio of tau/tubulin in cytoskeletal fractions than in WT Aβ–treated ones. This enhanced preservation of tau binding to microtubules in E22Q Aβ–treated neurons could prevent its cleavage by calpain. Our results showing that soluble tau was significantly more susceptible to calpain degradation than cytoskeletal-bound tau in in vitro calpain assays suggest that this might be the case in E22Q Aβ–treated neurons.
The differences in the tau/tubulin ratio in cytoskeletal fractions induced by each Aβ peptide could be due to differential tau phosphorylation, a posttranslational modification that regulates such binding. As previously described, WT Aβ induced a significant increase in this posttranslational modification of tau in cultured neurons (27,44). However, no differences in tau phosphorylation levels at several residues were detected in E22Q Aβ–treated neurons compared with WT Aβ–treated ones. Yet, we cannot rule out a cumulative effect of small variations in the phosphorylation of different tau epitopes resulting in an overall decrease in the phosphorylation of this microtubule-associated protein in the presence of E22Q Aβ.
CONCLUSION
Regardless of the mechanisms involved, the results presented here provide evidence of the direct toxic effects of the Dutch mutant Aβ on central neurons. The complement of molecular mechanisms underlying the neuronal pathobiology of this familial form of AD awaits further investigation.
ACKNOWLEDGMENTS
We thank Sara Kleinschmidt and Jessica Bernstein for their contributions to the early stages of this work. This study was supported by grants NIH/NS39080 to A Ferreira and Wellcome Trust grant 067660 and NIH AG027443 to DM Walsh.
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
REFERENCES
- 1.Saito K, Elce JS, Hamos JE, Nixon RA. Widespread activation of calcium-activated neutral proteinase (calpain) in the brain in Alzheimer disease: a potential molecular basis for neuronal degeneration. Proc Natl Acad Sci U S A. 1993;90:2628–32. doi: 10.1073/pnas.90.7.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tsuji T, Shimohama S, Kimura J, Shimizu K. m-Calpain (calcium-activated neutral proteinase) in Alzheimer’s disease brains. Neurosci Lett. 1998;248:109–12. doi: 10.1016/s0304-3940(98)00348-6. [DOI] [PubMed] [Google Scholar]
- 3.Veerana J, et al. Calpain mediates calcium-induced activation of the erk1,2 MAPK pathway and cytoskeletal phosphorylation in neurons: relevance to Alzheimer’s disease. Am J Pathol. 2004;165:795–805. doi: 10.1016/S0002-9440(10)63342-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kelly BL, Vassar R, Ferreira A. Beta-amyloid-induced dynamin 1 depletion in hippocampal neurons: a potential mechanism for early cognitive decline in Alzheimer disease. J Biol Chem. 2005;280:31746–53. doi: 10.1074/jbc.M503259200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kelly BL, Ferreira A. Beta-amyloid-induced dynamin 1 degradation is mediated by NMDA receptors in hippocampal neurons. J Biol Chem. 2006;281:28079–89. doi: 10.1074/jbc.M605081200. [DOI] [PubMed] [Google Scholar]
- 6.Nicholson A, Ferreira A. Increased membrane cholesterol might render mature hippocampal neurons more susceptible to beta-amy-loid-induced calpain activation and tau toxicity. J Neurosci. 2009;29:4640–51. doi: 10.1523/JNEUROSCI.0862-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Damke H, Baba T, Warnock DE, Schmid SL. Induction of mutant dynamin specifically blocks endocytic coated vesicle formation. J Cell Biol. 1994;127:915–34. doi: 10.1083/jcb.127.4.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clark SG, Shurland DL, Meyerowitz EM, Bargmann CI, van der Bliek AM. A dynamin GTPase mutation causes a rapid and reversible temperature-inducible locomotion defect in C. elegans. Proc Natl Acad Sci U S A. 1997;94:10438–43. doi: 10.1073/pnas.94.19.10438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kelly BL, Ferreira A. Beta-amyloid disrupted synaptic vesicle endocytosis in cultured hippocampal neurons. Neuroscience. 2007;146:60–70. doi: 10.1016/j.neuroscience.2007.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Park SY, Ferreira A. The generation of a 17 kDa neurotoxic fragment: an alternative mechanism by which tau mediates beta-amyloid-induced neurodegeneration. J Neurosci. 2005;25:5365–75. doi: 10.1523/JNEUROSCI.1125-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park SY, Tournell C, Sinjoanu RC, Ferreira A. Caspase-3- and calpain-mediated tau cleavage are differentially prevented by estrogen and testosterone in beta-amyloid-treated hippocampal neurons. Neuroscience. 2007;144:119–27. doi: 10.1016/j.neuroscience.2006.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sinjoanu RC, et al. The novel calpain inhibitor A-705253 potently inhibits oligomeric beta-amyloid-induced dynamin 1 and tau cleavage in hippocampal neurons. Neurochem Intl. 2008;53:79–88. doi: 10.1016/j.neuint.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levy E, et al. Mutation of the Alzheimer’s disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science. 1990;248:1124–6. doi: 10.1126/science.2111584. [DOI] [PubMed] [Google Scholar]
- 14.Mirravalle L, et al. Substitutions at codon 22 of Alzheimer’s abeta peptide induce diverse conformational changes and apoptotic effects in human cerebral endothelial cells. J Biol Chem. 2000;275:27110–6. doi: 10.1074/jbc.M003154200. [DOI] [PubMed] [Google Scholar]
- 15.Natte R, et al. Dementia in hereditary cerebral hemorrhage with amyloidosis-Dutch type is associated with cerebral amyloid angiopathy but is independent of plaques and neurofibrillary tangles. Ann Neurol. 2001;50:765–72. doi: 10.1002/ana.10040. [DOI] [PubMed] [Google Scholar]
- 16.Murakami K, et al. Neurotoxicity and physicochemical properties of Abeta mutant peptides from cerebral amyloid angiopathy. J Biol Chem. 2003;278:46179–87. doi: 10.1074/jbc.M301874200. [DOI] [PubMed] [Google Scholar]
- 17.Herzig MC, et al. Abeta is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nat Neurosci. 2004;7:954–60. doi: 10.1038/nn1302. [DOI] [PubMed] [Google Scholar]
- 18.Davis J, Cribbs DH, Cotman CW, Van Nostrand WE. Pathogenic amyloid beta protein induces apoptosis in cultured human cerebrovascular smooth muscle cells. Amyloid. 1999;6:157–64. doi: 10.3109/13506129909007321. [DOI] [PubMed] [Google Scholar]
- 19.Melchor JP, McVoy L, Van Nostrand WE. Charge alterations of E22 enhance the pathogenic properties of the amyloid-beta protein. J Neurochem. 2000;74:2209–12. doi: 10.1046/j.1471-4159.2000.0742209.x. [DOI] [PubMed] [Google Scholar]
- 20.Dahlgren KN, et al. Oligomeric and fibrillar species of amyloid-beta peptides differentially affect neuronal viability. J Biol Chem. 2002;277:32046–53. doi: 10.1074/jbc.M201750200. [DOI] [PubMed] [Google Scholar]
- 21.Goslin K, Asmussen H, Banker G. Rat hippocampal neurons in low-density culture. In: Banker G, Goslin K, editors. Culturing Nerve Cells. 2nd ed. Cambridge, MA: MIT Press; 1998. pp. 339–57. [Google Scholar]
- 22.Bottenstein JE, Sato GH. Growth of a rat neuroblastoma cell line in serum-free supplemented medium. Proc Natl Acad Sci U S A. 1979;76:514–7. doi: 10.1073/pnas.76.1.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rapoport M, Ferreira A. PD98059 prevents neurite degeneration induced by fibrillar beta amyloid in mature hippocampal neurons. J Neurochem. 2000;74:125–33. doi: 10.1046/j.1471-4159.2000.0740125.x. [DOI] [PubMed] [Google Scholar]
- 24.Black L, Berenbaum MC. Factors affecting the dye exclusion test for cell viability. Exp Cell Res. 1964;35:9–13. doi: 10.1016/0014-4827(64)90066-7. [DOI] [PubMed] [Google Scholar]
- 25.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–5. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 26.Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A. 1979;76:4350–4. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferreira A, Lu Q, Orecchio L, Kosik KS. Selective phosphorylation of adult tau isoforms in mature hippocampal neurons exposed to fibrillar A beta. Mol Cell Neurosci. 1997;9:220–34. doi: 10.1006/mcne.1997.0615. [DOI] [PubMed] [Google Scholar]
- 28.Yakunin AF, Hallenbeck PC. A luminol/iodophenol chemiluminescent detection system for Western immunoblots. Anal Biochem. 1998;258:146–9. doi: 10.1006/abio.1998.2571. [DOI] [PubMed] [Google Scholar]
- 29.Ferreira A, Busciglio J, Caceres A. Micro-tubule formation and neurite growth in cerebellar macroneurons which develop in vitro: evidence for the involvement of the microtubule-associated proteins, MAP-1a, HMW-MAP2 and Tau. Brain Res Dev Brain Res. 1989;49:215–28. doi: 10.1016/0165-3806(89)90023-0. [DOI] [PubMed] [Google Scholar]
- 30.Rapoport M, Dawson HN, Binder LI, Vitek M, Ferreira A. Tau is essential for beta-amy-loid induced neurotoxicity. Proc Natl Acad Sci U S A. 2002;99:6364–9. doi: 10.1073/pnas.092136199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fletcher TL, Cameron P, De Camilli P, Banker G. The distribution of synapsin I and synaptophysin in hippocampal neurons developing in culture. J Neurosci. 1991;11:1617–26. doi: 10.1523/JNEUROSCI.11-06-01617.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Czogalla A, Sikorski AF. Spectrin and cal-pain: a ‘target’ and a ‘sniper’ in the pathology of neuronal cells. Cell Mol Life Sci. 2005;62:1913–24. doi: 10.1007/s00018-005-5097-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koenig JH, Ikeda K. Disappearance and reformation of synaptic vesicle membrane upon transmitter release observed under reversible blockage of membrane retrieval. J Neurosci. 1989;9:3844–60. doi: 10.1523/JNEUROSCI.09-11-03844.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kosaka T, Ikeda K. Possible temperature-dependent blockage of synaptic vesicle recycling induced by a single gene mutation in Drosophila. J Neurobiol. 1983;14:207–25. doi: 10.1002/neu.480140305. [DOI] [PubMed] [Google Scholar]
- 35.van der Bliek AM, Meyerowitz EM. Dynamin-like protein encoded by the Drosophila shibire gene associated with vesicular traffic. Nature. 1991;351:411–4. doi: 10.1038/351411a0. [DOI] [PubMed] [Google Scholar]
- 36.Maat-Schieman ML, van Duinen SG, Bornebroek M, Haan J, Roos RA. Hereditary cerebral hemorrhage with amyloidosis-Dutch type (HCHWA-D): II. A review of histopathological aspects. Brain Pathol. 1996;6:115–20. doi: 10.1111/j.1750-3639.1996.tb00794.x. [DOI] [PubMed] [Google Scholar]
- 37.Demeester N, et al. Comparison of the aggregation properties, secondary structure and apoptotic effects of wild-type, Flemish and Dutch N-terminally truncated amyloid beta peptides. Eur J Neurosci. 2001;13:2015–24. doi: 10.1046/j.0953-816x.2001.01579.x. [DOI] [PubMed] [Google Scholar]
- 38.Baumketner A, Krone MG, Shea JE. Role of the familial Dutch mutation EE22Q in the folding and aggregation of the 15–28 fragment of the Alzheimer amyloid-beta protein. Proc Natl Acad Sci U S A. 2008;105:6027–32. doi: 10.1073/pnas.0708193105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kumar-Sing S, et al. In vitro studies of Flemish, Dutch, and Wild-type-amyloid provide evidence for two staged neurotoxicity. Neurobiol Dis. 2002;11:330–40. doi: 10.1006/nbdi.2002.0529. [DOI] [PubMed] [Google Scholar]
- 40.Watterndorff AR, Frangione B, Luyendijk W, Bots GT. Hereditary cerebral hemorrhage with amyloidosis, Dutch type (HCHWA-D): clinico-pathological studies. J Neurol Neurosurg Psych. 1995;58:699–705. doi: 10.1136/jnnp.58.6.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maat-Schieman ML, Radder CM, van Duinen SG, Haan J, Roos RA. Hereditary cerebral hemorrhage with amyloidosis (Dutch): a model for congophilic plaque formation without neurofibrillary pathology. Acta Neuroph. 1994;88:371–8. doi: 10.1007/BF00310382. [DOI] [PubMed] [Google Scholar]
- 42.Bornebroek M, et al. Hereditary cerebral hemorrhage with amyloidosis-Dutch type: better correlation of cognitive deterioration with advancing age than with number of focal lesions or white matter hyperintensities. Alz Dis Ass Dis. 1996;10:224–31. doi: 10.1097/00002093-199601040-00008. [DOI] [PubMed] [Google Scholar]
- 43.Goll DE, Thompson VF, Li H, Wei W, Cong J. The calpain system. Physiol Rev. 2003;83:731–801. doi: 10.1152/physrev.00029.2002. [DOI] [PubMed] [Google Scholar]
- 44.Busciglio J, Lorenzo A, Yeh J, Yankner BA. Beta-amyloid fibrils induce tau phosphorylation and loss of microtubule binding. Neuron. 1995;14:879–88. doi: 10.1016/0896-6273(95)90232-5. [DOI] [PubMed] [Google Scholar]