

Abstract
The fundamental mechanisms that underlie platelet activation in atherothrombosis are still obscure. Oxidative stress is involved in central features of atherosclerosis. Platelet-derived microvesicles (PMVs) could be important mediators between oxidative stress and platelet activation. CD36 could be a receptor of PMVs, thus generating a PMV–CD36 complex. We aimed to investigate the detailed pathway by which oxidative damage contributes to platelet activation by the PMV–CD36 complex. We found that oxidized low-density lipoprotein stimulated the generation of PMVs. PMVs enhanced normal platelet activation, as assessed by the expression of integrin αIIbβ3, secretion of soluble P-selectin and platelet aggregation, but CD36-deficient platelets were not activated by PMVs. The function of the PMV–CD36 complex was mediated by the MKK4/JNK2 signaling axis. Meanwhile, PMVs increased the level of 8-iso-prostaglandin-F2α, a marker of oxidative stress, in a CD36- and phosphatidylserine-dependent manner. We concluded that PMVs are important mediators between oxidative stress and platelet activation. PMVs and CD36 may be effective targets for preventing platelet activation in cardiovascular diseases.
INTRODUCTION
Thromboembolic events are lethal consequences of atherosclerosis (AS) and are still a challenge. The activation of platelets has a critical role in thrombosis because it participates in the final and beginning stages. The fundamental mechanisms responsible for platelet activation in AS are still incompletely understood but may involve the etiology of AS. The pathogenesis of AS is multifactorial, and oxidative damage has aroused much attention. AS-related diseases such as coronary heart disease, hypertension and diabetes mellitus are characterized by oxidative stress, which results in a hypercoagulable or prothrombotic state. To find the source (such as oxidative stress) of platelet activation in AS, we can study the potential mechanism of the prothrombotic state and explore new, effective targets for preventing atherothrombosis.
Studies of microvesicles have offered a hint for the activation of platelets (1–3). Microvesicles are exosomes or small particles released by many cells on stimulation (4–6). The nomenclature of these vesicles lacks consensus. In general, exosomes are vesicles contained in multivesicular bodies. Ectosomes refer to shedding vesicles of neutrophils and monocytes. Microparticles are vesicles shedding from the surface of monocytes and platelets. The term “microvesicles” used here indicates both exosomes and shedding vesicles (1).
Advances in proteomics have revealed the importance of microvesicles in thrombosis. Platelet-derived microvesicles (PMVs) are the main culprit in the development of thrombosis. Two-thirds of microvesicles in peripheral blood are derived from platelets (7). The thromboplastic activity of PMVs is 50 to 100 times higher than that of platelets (8). PMVs carry multiple glycoproteins and phospholipids, including GPIb, P-selectin and phosphatidylserine (PS). Clinical study has revealed increased levels of oxidized low-density lipoprotein (oxLDL) highly related to elevated numbers of microvesicles in plasma (9). Thus, PMVs may be important sources of platelet activation by oxidative stress, which initiates the coagulation process in atherothrombosis.
CD36, a class B scavenger receptor, promotes platelet activation (10,11). It is expressed in platelets, monocytes and several other cells and mediates many pathophysiological processes by binding with various oxidized phospholipids, apoptotic cells and certain pathogens. The PS on the surface of PMVs could be a ligand of CD36, thus generating a PMV–CD36 complex. CD36 can be a signaling molecule, but the detailed signal pathway of the PMV–CD36 complex is unknown. Chen et al. (12) showed that platelet CD36 mediates mitogen-activated protein kinase (MAPK) activation induced by oxLDL. The modified lipids on oxLDL mediate its combination with CD36, and the surface of microvesicles contains multiple modified lipids (such as PS). Therefore, we speculated that the PMV–CD36 complex could activate MAPKs, thereby activating platelets.
We hypothesized that oxidative damage increases the generation of PMVs in AS. We aimed to investigate whether PMVs bearing the oxidative stress information amplify and disseminate this information by mediating communication among cells and whether the PMV–CD36 complex triggers MAPK signaling, thereby activating platelets.
MATERIALS AND METHODS
Materials
Biotin-conjugated anti-CD41 monoclonal antibody was from Abcam (Cambridge, UK). Recombinant human annexin V was from Enzo Life Sciences (Exeter, UK). SP600125 was from Calbiochem (San Diego, CA, USA). The antibodies rabbit anti-MKK4, p-MKK4, c-Jun NH2-terminal kinase (JNK) and mouse anti–p-JNK were from Cell Signaling Technology (Danvers, MA, USA). Phycoerythrin (PE)-conjugated anti-CD36 (clone CB38), PE-conjugated CD36 isotype control (clone G155-228), PE-cy™5-conjugated mouse anti-CD41a antibody (clone HIP8) and fluorescein isothiocyanate (FITC)-conjugated PAC-1 were from BD Biosciences/Pharmingen (San Jose, CA, USA). The 8-iso-prostaglandin-F2α (8-iso-PG-F2α) enzyme immunoassay (EIA) kit was from Cayman Chemical (Ann Arbor, MI, USA). The p-selectin enzyme-linked immunosorbent assay (ELISA) kit was from R&D Systems (Oxfordshire, UK). The monoclonal antibody AD-1 for microvesicles was obtained as described (13,14).
Sources of OxLDL
OxLDL (YB-002) was obtained from Yiyuan Biotechnology (Guangzhou, China). It was purified by ultracentrifugation (1.019–1.063 g/mL) and oxidized with 5 μmol/L Cu2SO4 at 37°C for 20 h. Oxidation was terminated by adding excess EDTA-Na2. Each lot was analyzed on agarose gel electrophoresis for migration versus LDL. This oxLDL was 1.0 mg protein/mL. Thiobarbituric acid–reactive substances were determined colorimetrically with malondialdehyde used as a standard. The oxLDL was stored at 4°C and used within 1 wk after receipt.
Platelet Isolation
Blood was collected in 0.109 mol/L sodium citrate (ratio 1:9) from healthy volunteers who were free of any medication for 2 wks. Written consent was obtained from all donors, and protocols were approved by the institutional ethics committee. Platelet-rich plasma (PRP) was prepared by centrifugation at 120g for 10 min at room temperature. Platelets were isolated from PRP after centrifugation at 800g for 10 min in the presence of acid citrate dextrose. Then platelets were washed and resuspended in modified Tyrode’s buffer (137 mmol/L NaCl, 2.7 mmol/L KCl, 12 mmol/L NaHCO3, 0.4 mmol/L NaH2PO4, 5 mmol/L HEPES, 0.1% glucose and 0.35% bovine serum albumin, pH7.2) in the presence of 100 nmol/L PG-E1 (Sigma-Aldrich, St. Louis, MO, USA). Platelet concentration was adjusted to 1 × 106/mL by use of a Z2 particle counter (Beckman Coulter, Fullerton, CA, USA), then used at once in all experiments.
Identification of PMVs
Assessment of PMVs by flow cytometry
Resting platelets (1×106/mL) were incubated with buffer control, 50 μg/mL native LDL (nLDL), 50 μg/mL oxLDL or 10 μmol/L adenosine diphosphate (ADP) for 15 min at 37°C. Samples (2.5 μL) were incubated with PEcy5-conjugated anti-CD41a antibody (5 μL) for 15 min. Only cells and particles positive for CD41a were gated. The lower limit of the plate let gate was set at the left-hand border for resting platelets to distinguish between platelets and microvesicles (15,16). For each sample, 10,000 positive events in the platelet gate were acquired by use of FACS Calibur cytometry (BD Biosciences).
ELISA detection of total amount of PMVs
After incubation with oxLDL or ADP, the platelet suspension was centrifuged at 3000g for 10 min. Then the supernatant was centrifuged again at 13,000g for 2 min to avoid platelet contamination. In total, 100 μL supernatant was stored at −80°C for ELISA.
Quantification of total PMVs in platelet supernatant involved use of the homemade monoclonal antibody AD-1 as described (13), with minor modification. Briefly, after specific capture for microvesicles, the plate coated with AD-1 was incubated with 50 μL/well of diluted biotin-conjugated mouse anti–human CD41 monoclonal antibody (specific platelet marker) at 37°C for 2 h, then with 50 μL/well horseradish peroxidase (HRP)-conjugated streptavidin (1:200 dilution) for 30 min at room temperature in the dark. After 6 washings, 50 μL of HRP substrate was added to each well and incubated for 5 min at room temperature. Then, the enzymatic reaction was stopped and the plate was read at 450 nm by use of a microplate reader (Thermo Scientific, Waltham, MA, USA). Intra- and interassay coefficients of variation were approximately 4.9% and 12.4%, respectively.
ELISA detection of annexin V–positive PMVs
ELISA was used for quantification of annexin V–positive PMVs in platelet supernatant stimulated with oxLDL and other controls. After staining for PMVs on annexin V–coated wells (for 2 h at 37°C), to take advantage of the strong affinity of annexin V for PS present on PMVs, samples were washed for 3 steps with phosphate buffered saline containing 0.05% Tween 20. Diluted biotin-conjugated CD41 monoclonal antibody (50 μL/well) was added as the detection antibody. After incubation for 2 h at 37°C, 50 μL/well diluted HRP-conjugated streptavidin was added and incubated for 30 min at room temperature in the dark. The linear absorbance was recorded at 450 nm after the addition of substrate solution. Intra-and interassay coefficients of variation were approximately 5.8% and 14.5%, respectively.
Platelet Functional Studies
Collection of PMVs derived from platelets stimulated by oxLDL
Platelets (1× 106/mL) were treated with oxLDL (50 μg/mL) for 15 min at 37°C, then sedimented at 3000g for 10 min. An amount of 2.0 mmol/L phenylmethylsulfonyl fluoride was added to the PMV-enriched supernatants, which were then centrifuged at 15,000g for 1 h at 4°C. Then PMV pellets were washed twice (to avoid contamination of oxLDL) and re-suspended in Modified Tyrode buffer and stored at −80°C. Freezing had no adverse effect on microvesicles (11). The protein content was measured by the Bradford Protein Assay Kit (Beyotime, Jiangsu, China).
Flow cytometry detection of platelet activation and CD36 expression
Platelet integrin αIIbβ3 and CD36 expression was quantified by flow cytometry. After treatment with PMVs (30 μg/mL), the platelet suspension (2.5 μL) was incubated in the dark at room temperature for 15 min with 5 μL PEcy5-conjugated anti-CD41a antibody and 5 μL FITC-conjugated PAC-1 antibody (for the activated conformation of the platelet integrin αIIbβ3). For CD36 detection, the platelet suspension was incubated with 5 μL PEcy5-conjugated anti-CD41a antibody and 5 μL PE-conjugated anti-CD36 antibody or isotype-matched control IgG, then pelleted, washed and analyzed. In some studies, before stimulation with PMVs, platelets were incubated with anti–human CD36 antibody (clone FA6; invitrogen), isotype-matched control IgG (Sigma) or JNK inhibitor SP600125 or preincubated with annexin V to mask PS.
Platelet aggregation studies
Aggregation was assessed by turbidimetry with a dual channel aggregometer (Chrono-log Corp., Havertown, PA, USA), with 2 μmol/L ADP used as an agonist. PRP was obtained by centrifuging whole blood at 120g for 10 min at 22°C, and platelet-poor plasma (PPP) was obtained by centrifuging PRP at 3000g for 10 min. The platelet concentration of PRP was adjusted to 2.5 × 108/mL by the addition of PPP. An amount of 100% aggregation was defined as the light transmission of PPP, and 0% was defined as the light transmission of PRP before the addition of agonists. Then the PRP was stimulated with 2 μmol/L ADP, and the change in light transmission was recorded.
Immunoassay for soluble P-selectin and 8-iso-PG-F2α
After the treatment described above, the platelet-PMV mixtures were centrifuged at 3000g for 10 min. Then the supernatant was centrifuged again at 13,000g for 2 min to avoid platelet contamination. The last supernatant was used for detection of soluble P-selectin (R&D Systems) and 8-iso-PG-F2α (Cayman Chemical, Ann Arbor, MI, USA) with use of commercially available immunoassay kits. The lower limit of sensitivity of the assay was 0.5 ng/mL for P-selectin and 2.7 pg/mL for 8-iso-PG-F2α.
Immunoblot Analysis of MKK4 and JNK
Platelets treated with PMVs or controls were lysed in 2 mmol/L Tris-HCl (pH 7.5), 150 mmol/L NaCl, 1 mmol/L EGTA, 1 mmol/L EDTA, 1% Triton X-100, 2.5 mmol/L sodium pyrophosphate, 1 mmol/L Na3VO4, 1 mmol/L phenylmethylsulfonyl fluoride and 1 μg/mL leupeptin, and protein concentrations were measured with use of a Bradford protein assay kit (Beyotime). Lysate protein (40 μg) was separated on 10% polyacrylamide gel and transferred onto polyvinylidene fluoride membranes. After a blocking for 1 h at room temperature in 5% nonfat milk, the membranes were incubated with mouse anti–phospho-JNK1/2 or rabbit anti–phospho-MKK4 (1:1000 dilution) overnight at 4°C, then with HRP-conjugated secondary anti–mouse IgG or anti–rabbit IgG (1:4000 dilution). The blots were developed with use of ECL detection reagent, then stripped and re-blotted with antibodies to native proteins for normalization.
Statistical Analysis
The Kolmogorov–Smirnov test was used for confirming normal distribution, and data are presented as means ± SE. Comparison among groups involved ANOVA with post hoc least significant differences t test. P < 0.05 was considered statistically significant. Analyses involved use of SPSS 16.0 (SPSS Inc., Chicago, IL, USA).
RESULTS
OxLDL Treatment Generated PMVs
Flow cytometry detected the formation of microvesicles by labeling with PEcy5-conjugated anti-CD41a antibody (M gates in Figures 1A–D). The release of microvesicles increased after stimulation with oxLDL (50 μg/mL) or ADP (10 μmol/L), as quantified by mean fluorescence intensity (MFI) of CD41a (Figure 1E). For further confirmation, the supernatant of platelets was tested by ELISA. The amount of total PMVs (Figure 1F) and annexin V–positive PMVs (Figure 1G) increased significantly after treatment with oxLDL. The effect of oxLDL was comparable to that of ADP.
Figure 1.

Detection of PMVs by flow cytometry and ELISA. A–E, Flow cytometry detection of platelets and PMVs. Platelets were incubated with buffer control (A), 50 μg/mL native low-density lipoprotein (nLDL) (B), 50 μg/mL oxLDL (C) or 10 μmol/L ADP (D) for 15 min at 37°C. P and M indicate the gates for platelets and PMVs, respectively (E). (F–G) MFI of CD41a in M gates, ELISA quantification of total PMVs (F) and annexin V–positive PMVs (G). Platelets were incubated with 50 μg/mL oxLDL or nLDL, or 10 μmol/L ADP. Data are means ± SE from three independent experiments. *P < 0.05 compared with control.
PMVs Induced Platelet Activation
Next, we tested whether the PMVs collected from oxLDL-treated platelets could enhance platelet activation. We incubated resting platelets (1 × 106/mL) with PMVs (30 μg/mL) for 30 min at 22°C. Platelet activation is characterized by a conformation change in integrin αIIbβ3. As expected, the MFI and percentage of PAC-1 (recognizing the activated platelet integrin αIIbβ3) were enhanced significantly compared with the control. PMVs had almost the same effect as ADP (Figures 2A–B). Furthermore, PMVs increased platelet aggregation in response to ADP (Figure 2C).
Figure 2.

CD36-dependent platelet activation in response to PMVs. (A–C) The MFI and percentage of PAC-1, as well as platelet aggregation, were enhanced significantly after incubation of CD36-positive platelets with PMVs (30 μg/mL) for 30 min at 22°C. Data are means ± SE (n = 3). *P < 0.05 compared with control. (D–F) PAC-1 expression or platelet aggregation with CD36-deficient platelet incubation in response to PMVs. Data are means ± SE (n = 4). *P < 0.05 compared with control.
PMVs Were Ineffective in CD36-Deficient Platelets
We screened 2 CD36-deficient male donors in our laboratory. The CD36-deficient platelets were unable to bind PE-conjugated anti-CD36 antibody (Figure 3), and PMV-enhanced integrin αIIbβ3 expression and platelet aggregation were absent (Figures 2D–F).
Figure 3.
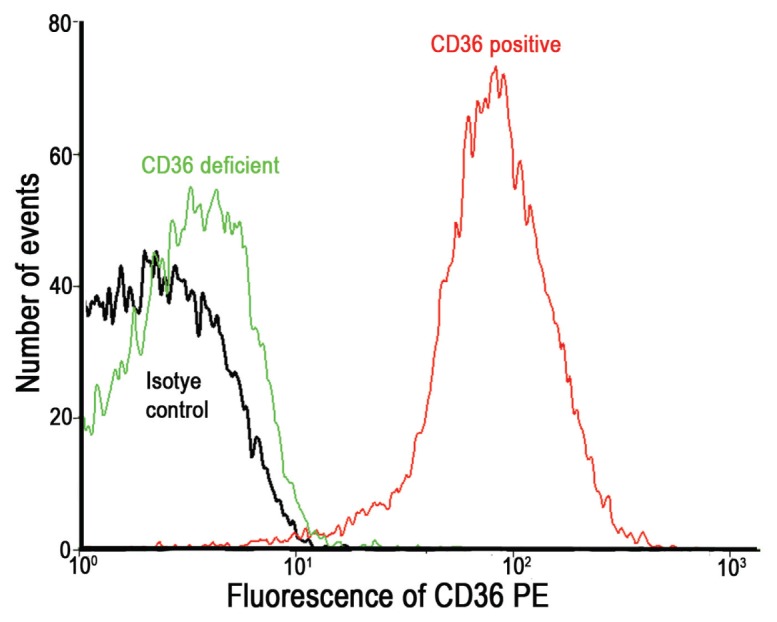
Identification of CD36-positive and CD36-deficient platelets by flow cytometry. MFI of binding of CD36-positive and CD36-deficient platelets with PE-conjugated anti-CD36 antibody or isotype controls.
PMVs Activated Platelets in a CD36-and PS-Dependent Manner
To define the influence of CD36 and PS on PMV–platelet interaction, we determined the expression of integrin αIIbβ3 (Figures 4A, B and D) and the secretion of P-selection (Figure 5C). PMVs substantially increased the expression of integrin αIIbβ3 and secretion of P-selectin. In all cases, the blocking of CD36 (by FA6, a CD36 antibody) or PS (by annexin V) could diminish the enhancement by PMVs, but IgG, an isotype-matched control of FA6, had no effect. As well, FA6 decreased the binding of PE-conjugated anti-CD36 antibody to platelets, with the expression of CD36 for other groups not changed significantly (Figure 5A).
Figure 4.

CD36-, PS- and JNK-dependent platelet activation by PMVs. Platelets were incubated with 2 μg/mL FA6 (A,D), 2 μg/mL mouse IgG or 20 μmol/L SP600125 (C,E) before incubation with PMVs (30 μg/mL) and analyzed by flow cytometry with FITC-labeled PAC-1. In some studies, PMVs were first incubated with 20 μg/mL annexin V to mask PS (B,D). (A–C) Histograms represent MFI and graphs; (D–E) show percentage of PAC-1–positive platelets. Data are means ± SE from at least three separate experiments. *P < 0.05 compared with control; #P < 0.05 compared with PMV treatment.
Figure 5.

(A–B) Expression of CD36 detected by flow cytometry. Preincubation of platelets with FA6, IgG, annexin V or SP600125 to examine binding of PE-conjugated CD36 antibody to platelets. (C–D) P-selection secretion determined by ELISA. The secretion of P-selection with blocking of CD36 or PS (C); the effect of JNK inhibitor SP600125 (final concentration of 20 μmol/L) on PMVs (D). *P < 0.05 compared with control, #P < 0.05 compared with PMV treatment. Data are means ± SE from at least three separate experiments.
Platelet Activation Induced by PMVs Was Mediated by JNK Signals
To elucidate the potential effect of JNK signaling in PMV-induced platelet activation, platelets were treated with the JNK inhibitor SP600125 before incubation with PMVs. Pharmacological inhibition of JNK reduced platelet activation by PMVs, including expression of integrin αIIbβ3 (Figure 4C, E) and secretion of P-selectin (Figure 5D). Thus, JNK signaling may contribute to PMV-induced platelet activation. Nevertheless, inhibition of JNK had no effect on expression of CD36 (Figure 5B).
Phosphorylation of Platelet JNK2 and its Upstream Activator MKK4 Induced by PMVs
To confirm the role of the JNK pathway in PMV-induced activation of platelets, we compared the phosphorylation of JNK2 in CD36-deficient and -positive platelets by immunoblotting. CD36-positive but not CD36-deficient platelets exposed to PMVs for 30 min showed a significant increase in phosphorylation of JNK2 and its upstream activator MKK4 (Figure 6A). The JNK2 phosphorylation was time and dose dependent, peaking at 15 min to approximately 1.3-fold that of baseline with 30 μg/mL PMVs (Figure 6B) and with increased concentrations of PMVs inducing stronger phosphorylation of JNK2 (Figure 6C).
Figure 6.

PMVs induced phosphorylation of JNK2 and its upstream activator MKK4 in CD36-positive platelets. CD36-positive and CD36-deficient platelets were incubated with PMVs and then lysed. Phosphorylated forms of JNK1/2 and MKK4 were detected by Western blot analysis for phospho-MKK4 (p-MKK4) and phospho-JNK (p-JNK1/2) and total MKK4 and JNK. (A) PMVs induced activation of JNK2 in CD36-positive but not CD36-deficient platelets. CD36-positive platelets were incubated with PMVs (30 μg/mL) for various times (B) and various concentrations of PMVs (C) for detection of phosphorylation of JNK2. Data are means ± SE of three independent experiments from different donors; *P < 0.05. □, Control; ■, PMVs.
PMVs Induced Oxidative Production of Platelets by CD36 and PS
Resting platelets showed a pronounced formation of 8-iso-PG-F2α on incubation with PMVs, which was significantly reversed by blocking CD36 or PS (Figure 7A). The change was similar to that of platelet activation. These data indicate that CD36 participates in a signaling pathway that regulates oxidative stress in platelets. The level of 8-iso-PG-F2α was not decreased with the inhibition of JNK by SP600125 (Figure 7B).
Figure 7.
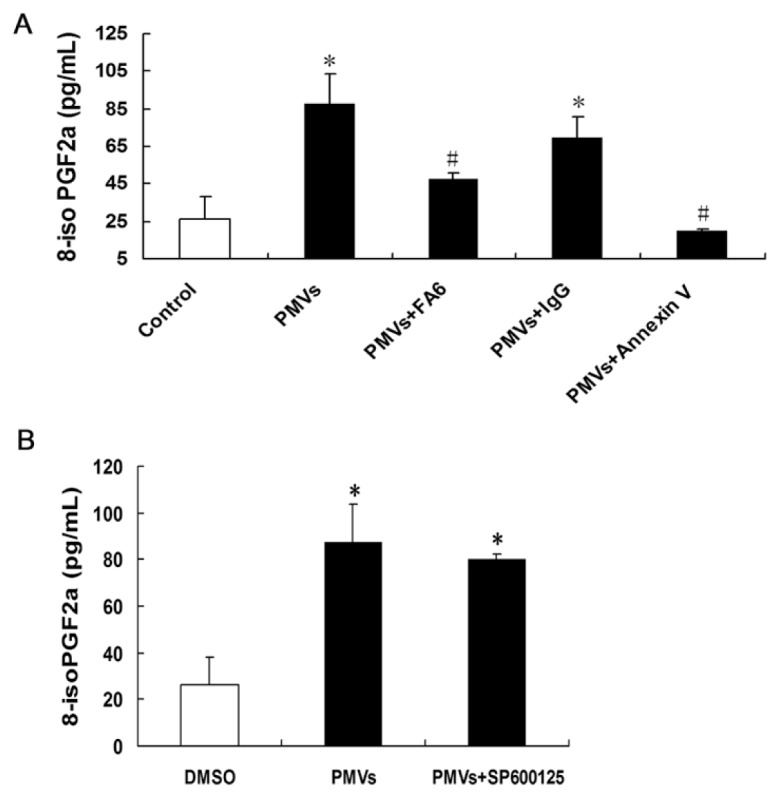
Eight (8)-iso-PG-F2α, a specific marker of oxidative stress, detected by enzyme immunoassay. (A) The level of 8-iso-PG-F2α after treatment of PMVs, and inhibition with FA6 and annexin V. (B) After blocking JNK with JNK inhibitor SP600125, the level of 8-iso-PG-F2α compared with PMV treatment. *P < 0.05 compared with control. #P < 0.05 compared with PMV treatment. Data are means ± SE of three experiments.
DISCUSSION
We explored the potential process of oxidative stress contributing to platelet activation and its mechanism. PMVs could act as a central mediator of oxidative damage, MAPK signaling and platelet activation. Briefly, oxLDL significantly promoted the generation of PMVs. Engagement of PMVs with platelet CD36 triggered MKK4/JNK2 signaling and activated platelets. In addition, PMVs enhanced platelet oxidative production in a CD36- and PS-dependent manner.
Oxidative stress is a central feature of the AS process. OxLDL, the active consequence of oxidative stress, favors AS effectively. Here, we reveal a powerful pathway whereby oxLDL results in platelet activation by PMVs. PMVs are small membrane-enclosed vesicles with strong procoagulant power discharged during platelet activation. They are leading players in thrombosis. PMVs and other microvesicles were found accumulated in the lipid core of plaques and thrombi (17). Our work demonstrated that oxLDL significantly promoted the generation of PMVs.
We tested the thromboplastic activity of PMVs derived from oxLDL-treated platelets. As expected, PMVs induced platelet activation, as assessed by the expression of integrin αIIbβ3, secretion of soluble P-selectin and platelet aggregation.
Nevertheless, PMVs were ineffective for CD36-deficient platelets. CD36 deficiency (Naka− phenotype) is common among Chinese (18). We screened 2 male volunteers with CD36 deficiency in our laboratory, neither with a history of bleeding diathesis. PMVs could not enhance integrin αIIbβ3 expression in CD36-deficient platelets. The occurrence of cardiovascular events in East Asia is lower than that in the Western world, and CD36 deficiency may be one of the reasons in addition to genotype and life style.
We used an inhibitory antibody to block platelet CD36 and annexin V to mask surface PS exposed on PMVs and found the activation function of PMVs to depend on CD36 and PS to some extent. Our results and those of others indicate that PS on the surface of PMVs binds to platelet CD36 and triggers platelet activation signals (11,19).
Earlier studies demonstrated that engaging CD36 with oxLDL initiated signal responses that result in foam-cell formation for macrophages and hyperreactivity for platelets (12,20). However, the signaling pathway of the PMV–CD36 complex triggering platelets was unknown. Most studies involving the signaling function of CD36 in other cells, such as macrophages and microvascular endothelial cells, have been focused on MAPKs. In addition, in a recent study (21) investigators found that MAPK/JNK1 plays an important role in platelet activation and thrombus formation. JNK1−/− mice showed longer bleeding times on tail-bleeding assays and platelet aggregation. We found JNK2 and its upstream activator MKK4 were activated on the interaction of CD36-positive but not CD36-deficient platelets with PMVS. Moreover, all platelet-stimulatory events induced by PMVs, including PAC-1 binding and P-selectin secretion, could be blocked with the JNK inhibitor SP600125. Therefore, MKK4/JNK2 signaling may contribute to the platelet hyperreactivity associated with PMVs. Our finding is consistent with those of recent studies showing MAPKs, including JNK, activated after oxLDL and thrombin exposure playing significant roles in platelet biology (12,22).
In addition, we found that the PMV–platelet interaction promoted the production of 8-iso-PG-F2α, a critical marker of oxidative stress. This function depended on CD36 and PS and implied that CD36 transduces signaling responses, not only activating platelets but also enhancing oxidative stress. For further insight into whether JNK was involved in this mechanism, we inhibited the JNK pathway and detected the level of oxidative stress. Surprisingly, the level of 8-iso-PG-F2α did not decrease, so the pro-oxidant signals are triggered by the PMV–CD36 complex but not JNK. In macrophages, CD36 activates src-family kinases, Vav family guanine nucleotide exchange factors and MAPKs (23,24). Whether the other two kinases mediated this signal pathway remains to be determined; it is the limitation of our study. We found that PMVs derived from oxLDL stimulation could promote platelet activation and oxidative stress, and enhanced oxidative stress further increases the generation of PMVs. Of course, atherothrombosis involves many other causative factors such as inflammation and various cytokines besides oxidative damage. We examined only oxidative stress, which represents key features of the AS process.
CONCLUSION
In summary, our data further confirm the potential roles of CD36 in platelet activation. One class of CD36 ligands, PMVs, is generated during system oxidant insult. The PMV–CD36 complex activates MKK4/JNK2 signals and contributes to platelet activation. In addition, PMVs feed-forward oxidative stress in a CD36- and PS-dependent manner. PMVs bearing various biologically active factors could be helpful in exploring the detailed mechanism of the prothrombotic state. Furthermore, PMVs and CD36 are potential targets for preventing thrombotic cardiovascular diseases.
ACKNOWLEDGMENTS
This work was supported by research grants from the Key Technologies Research and Development Program of Shandong Province (2006GG2202020 and 2010G0020262), the Natural Science Foundation of Shandong Province (Y2005C11, ZR2009CM022, ZR2009CM025 and BS2009YY026), the National Natural Science Foundation of China (30670874, 30871038, 30971215, 81070192 and 81070141) and the National Basic Research Program of China (973 Program, Grant no. 2009CB521904).
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
REFERENCES
- 1.Cocucci E, Racchetti G, Meldolesi J. Shedding microvesicles: artefacts no more. Trends Cell Biol. 2009;19:43–51. doi: 10.1016/j.tcb.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 2.Del Conde I, Shrimpton CN, Thiagarajan P, Lopez JA. Tissue-factor-bearing microvesicles arise from lipid rafts and fuse with activated platelets to initiate coagulation. Blood. 2005;106:1604–11. doi: 10.1182/blood-2004-03-1095. [DOI] [PubMed] [Google Scholar]
- 3.Bastida E, Ordinas A, Escolar G, Jamieson GA. Tissue factor in microvesicles shed from U87MG human glioblastoma cells induces coagulation, platelet aggregation, and thrombogenesis. Blood. 1984;64:177–84. [PubMed] [Google Scholar]
- 4.Ratajczak J, Wysoczynski M, Hayek F, Janowska-Wieczorek A, Ratajczak MZ. Membrane-derived microvesicles: important and underappreciated mediators of cell-to-cell communication. Leukemia. 2006;20:1487–95. doi: 10.1038/sj.leu.2404296. [DOI] [PubMed] [Google Scholar]
- 5.Gambim MH, do Carmo AO, Marti L, Verissimo-Filho S, Lopes LR, Janiszewski M. Platelet-derived exosomes induce endothelial cell apoptosis through peroxynitrite generation: experimental evidence for a novel mechanism of septic vascular dysfunction. Crit Care. 2007;11:R107. doi: 10.1186/cc6133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Siljander PR. Platelet-derived microparticles: an updated perspective. Thromb. Res. 2011;127(Suppl 2):S30–3. doi: 10.1016/S0049-3848(10)70152-3. [DOI] [PubMed] [Google Scholar]
- 7.Hunter MP, et al. Detection of microRNA expression in human peripheral blood microvesicles. PloS. One. 2008;3:e3694. doi: 10.1371/journal.pone.0003694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shantsila E, Kamphuisen PW, Lip GY. Circulating microparticles in cardiovascular disease: implications for atherogenesis and atherothrombosis. J Thromb Haemost. 2010;8:2358–68. doi: 10.1111/j.1538-7836.2010.04007.x. [DOI] [PubMed] [Google Scholar]
- 9.Matsumoto N, Nomura S, Kamihata H, Kimura Y, Iwasaka T. Increased level of oxidized LDL-dependent monocyte-derived microparticles in acute coronary syndrome. Thromb Haemost. 2004;91:146–54. doi: 10.1160/TH03-04-0247. [DOI] [PubMed] [Google Scholar]
- 10.Podrez EA, et al. Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nat Med. 2007;13:1086–95. doi: 10.1038/nm1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ghosh A, et al. Platelet CD36 mediates interactions with endothelial cell-derived microparticles and contributes to thrombosis in mice. J Clin Invest. 2008;118:1934–43. doi: 10.1172/JCI34904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen K, Febbraio M, Li W, Silverstein RL. A specific CD36-dependent signaling pathway is required for platelet activation by oxidized low-density lipoprotein. Circ Res. 2008;102:1512–9. doi: 10.1161/CIRCRESAHA.108.172064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang H, et al. Increased serum levels of microvesicles in nonvalvular atrial fibrillation determinated by ELISA using a specific monoclonal antibody AD-1. Clin Chim Acta. 2010;411:1700–4. doi: 10.1016/j.cca.2010.07.005. [DOI] [PubMed] [Google Scholar]
- 14.Deng JT, Parsons PG. Solid phase immunoassay for high molecular weight alkaline phosphatase in human sera using a specific monoclonal antibody. Clin Chim Acta. 1988;176:291–301. doi: 10.1016/0009-8981(88)90188-x. [DOI] [PubMed] [Google Scholar]
- 15.Miyazaki Y, et al. High shear stress can initiate both platelet aggregation and shedding of procoagulant containing microparticles. Blood. 1996;88:3456–64. [PubMed] [Google Scholar]
- 16.Heijnen HF, Schiel AE, Fijnheer R, Geuze HJ, Sixma JJ. Activated platelets release two types of membrane vesicles: microvesicles by surface shedding and exosomes derived from exocytosis of multivesicular bodies and alpha-granules. Blood. 1999;94:3791–9. [PubMed] [Google Scholar]
- 17.Leroyer AS, Tedgui A, Boulanger CM. Role of microparticles in atherothrombosis. J Intern Med. 2008;263:528–37. doi: 10.1111/j.1365-2796.2008.01957.x. [DOI] [PubMed] [Google Scholar]
- 18.Lin M, Shieh SH, Yang TF. Frequency of platelet-specific antigens among Chinese in Taiwan. Transfusion. 1993;33:155–7. doi: 10.1046/j.1537-2995.1993.33293158049.x. [DOI] [PubMed] [Google Scholar]
- 19.Greenberg ME, Sun M, Zhang R, Febbraio M, Silverstein R, Hazen SL. Oxidized phosphatidylserine-CD36 interactions play an essential role in macrophage-dependent phagocytosis of apoptotic cells. J Exp Med. 2006;203:2613–25. doi: 10.1084/jem.20060370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rahaman SO, Lennon DJ, Febbraio M, Podrez EA, Hazen SL, Silverstein RL. A CD36- dependent signaling cascade is necessary for macrophage foam cell formation. Cell Metab. 2006;4:211–21. doi: 10.1016/j.cmet.2006.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adam F, et al. Platelet JNK1 is involved in secretion and thrombus formation. Blood. 2010;115:4083–92. doi: 10.1182/blood-2009-07-233932. [DOI] [PubMed] [Google Scholar]
- 22.Bugaud F, Nadal-Wollbold F, Levy-Toledano S, Rosa JP, Bryckaert M. Regulation of c-jun-NH2 terminal kinase and extracellular-signal regulated kinase in human platelets. Blood. 1999;94:3800–5. [PubMed] [Google Scholar]
- 23.Rahaman SO, Swat W, Febbraio M, Silverstein RL. Vav family Rho guanine nucleotide exchange factors regulate CD36-mediated macrophage foam cell formation. J Biol Chem. 2010;286:7010–7. doi: 10.1074/jbc.M110.192450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baranova IN, et al. CD36 is a novel serum amyloid A (SAA) receptor mediating SAA binding and SAA-induced signaling in human and rodent cells. J Biol Chem. 2010;285:8492–506. doi: 10.1074/jbc.M109.007526. [DOI] [PMC free article] [PubMed] [Google Scholar]