

Abstract
Primary aldosteronism is the most common cause of secondary hypertension, most frequently due to an aldosterone-producing adenoma or idiopathic hyperaldosteronism. Somatic mutations of the potassium channel KCNJ5 in the region of the selectivity filter have been found in a significant number of aldosterone-producing adenomas. There are also familial forms of primary aldosteronism, one of which, familial hyperaldosteronism type 3 which to date has been found in one family who presented with a severe abnormality in aldosterone and 18-oxocortisol production and hypertrophy and hyperplasia of the transitional zone of the adrenal cortex. In familial hyperaldosteronism type 3, there is a genomic mutation causing a T158A change of amino acids within the selectivity filter region of the KCNJ5 gene.
We are reporting our studies demonstrating that lentiviral-mediated expression of a gene carrying the T158A mutation of the KCNJ5 in the HAC15 adrenal cortical carcinoma cell line causes a 5.3-fold increase in aldosterone secretion in unstimulated HAC15-KCNJ5 cells and that forskolin-stimulated aldosterone secretion was greater than that of angiotensin II. Expression of the mutated KCNJ5 gene decreases plasma membrane polarization, allowing sodium and calcium influx into the cells. The calcium channel antagonist nifedipine and the calmodulin inhibitor W-7 variably inhibited the effect. Overexpression of the mutated KCNJ5 channel resulted in a modest decrease in HAC15 cell proliferation.
These studies demonstrate that the T158A mutation of the KCNJ5 gene produces a marked stimulation in aldosterone biosynthesis that is dependent on membrane depolarization and sodium and calcium influx into the HAC15 adrenal cortical carcinoma cells.
Primary aldosteronism (PA) is characterized by the autonomous excessive production of aldosterone from the adrenal zona glomerulosa (1). Patients with PA are hypertensive and have a high prevalence of cardiovascular and cerebrovascular complications (1, 2). PA is the most frequent cause of the secondary hypertension with a frequency of 5–10% among hypertensives (1). The two most common forms of PA are aldosterone-producing adenomas (APA) and idiopathic hyperaldosteronism, also called bilateral adrenal zona glomerulosa hyperplasia (1, 3). Some forms of PA are familial, including familial primary aldosteronism type I or glucocorticoid-suppressible aldosteronism, due to a gene duplication produced by the uneven recombination between the 5′-regulatory segments of the cytochrome P450 (CYP)11B1 gene (exons 2–4) and the last exons of the CYP11B2 gene, resulting in a hybrid aldosterone synthase gene that is regulated by ACTH (4, 5). These patients excrete large quantities of the hybrid steroids 18-hydroxycortisol and 18-oxocortisol in addition to aldosterone (6). Familial hyperaldosteronism type 2 is the most common familial form, but it is of unknown etiology with a linkage to chromosome 7p22 in some families (7). In familial hyperaldosteronism type 3 (FH3), of which only one family has been reported to date, patients have severe hypertension and the highest recorded excretion of the hybrid steroids 18-hydroxycortisol and 18-oxocortisol (8).
The resting membrane potential of the zona glomerulosa cell is regulated by potassium (K+) channel activity (9). Voltage-gated calcium (Ca2+) channels are activated by membrane depolarization by hyperkalemia and by angiotensin II (A-II). The resulting increase in intracellular Ca2+ initiates the signaling events that increase aldosterone biosynthesis (9). The etiology of APA or idiopathic hyperaldosteronism is unknown. Recently, Choi et al. (10) reported the presence either of two somatic mutations of the KCNJ5 gene coding for the potassium channel Kir3.4 in eight of 22 aldosterone-producing adenomas, as well as in the FH3 family. The mechanisms by which a KCNJ5 mutation causes increased aldosterone production in adrenal zona glomerulosa cells have not been fully elucidated, although the KCNJ5 mutations, G151R or L168R, found in the APA tumors were in or near the selectivity filter in the glycine-tyrosine-glycine (GYG) motif of the Kir3.4 protein (10). The family with FH3 has an inherited mutation, T158A, within the same region associated with severe hyperaldosteronism and massive bilateral adrenal cortical hyperplasia with transitional zone characteristics (8, 10).
K+ selectivity of KCNJ potassium channel is conferred by a GYG motif at the narrowest part of the pore. Inflow of K+ through the channel hyperpolarizes the cell membrane (11). The mutation around the GYG motif in the APA and FH3 patients was shown to alter selectivity for cations including Na+, thereby depolarizing the cell membrane (12), triggering the opening of the voltage-gated Ca2+ channel (13), resulting in an influx of Ca2+ into the cell that activates sequential cascades including calmodulin and calmodulin kinase, and leading to increased steroidogenesis in adrenal cortical cells (14).
In this study, we hypothesized that expression in the adrenocortical carcinoma cell line HAC15 (15) using a lentivirus carrying a Kir 3.4 mutation (T158A) would increase aldosterone secretion and provide a model with which to address the mechanism of action of KCNJ5 in adrenal zona glomerulosa cells.
Materials and Methods
Cell culture and materials
The HAC15 human adrenocortical carcinoma cell line, a subclone of the H295R, a human adrenocortical carcinoma cell (15, 16), was provided by W. E. Rainey (Georgia Health Care University). The HAC15 cells were cultured in DMEM-F12 (1:1) supplemented with 10% Cosmic Calf serum (HyClone Laboratories, Logan, UT) at 37C under an atmosphere of 5% CO2.
A-II and nifedipine were purchased from Sigma Aldrich Co. Ltd. (St. Louis, MO). Forskolin was from LC Laboratories (Woburn, MA), and W-7, an inhibitor of calmodulin, was from Merck KGaA (Darmstadt, Germany). G418 and Blasticidin used to select lentiviral infected cells were purchased from InvivoGen (San Diego, CA). The dye to detect membrane voltage, DiSBAC2 (3), was purchased from AnaSpec (Fremont, CA); those to detect Na+ inflow, CoroNa Green, and intracellular Ca2+ concentration, Fluo-4 AM, were purchased from Invitrogen (Carlsbad, CA).
Plasmids
The full-length cDNA of KCNJ5 (pCR4-TOPO) was purchased from Open Biosystems (Huntsville, AL). The KCNJ5 mutation (T158A) plasmid was made using the QuikChange II XL Site Directed Mutagenesis kit from Stratagene (Santa Clara, CA) and the following primers: forward, GGCTTCCGAGTCATCGCAGAGAAGTGTCCAG; reverse, CTGGACACTTCTCTGCGATGACTCGGAAGCC. The cDNA was then amplified by PCR using the 5′-primer CACCGCTATGGCTGGCGATTCTAGGAA and 3′-primer TTATCACACCGAGCCCCTGGCC and inserted into pENTR/d-TOPO for subsequent use in LR reactions with the Gateway-compatible lentivector pLX303 plasmid (Addgene plasmid 25897; Addgene, Cambridge MA) (17) by LR reaction method (Invitrogen), resulting in pLX303-KCNJ5 T158A. Control plasmid was prepared as pLX303 without KCNJ5. The sequence of the mutation was confirmed by a PCR-based direct sequencing method.
The lentiviral transfer plasmid pBM14 was obtained from Dr. Fusseneger (18). Its promoter was eliminated by amplifying the plasmid using primers that excluded the EF1 promoter and introducing a restriction site. The 5′-flanking DNA of CYP11B2 (−1521 bp) from a pGL3 plasmid (a kind gift from Dr W. Rainey) was fused immediately upstream of the gaussia luciferase cDNA resulting in pBM14-CYP11B2 promoter/gaussia. The psPAX2 (Addgene plasmid 12260 from Didier Trono's Laboratory), and pCMV-VSV-G (Addgene plasmid 8454 from the Stewart laboratory (19) for virus production were purchased from Addgene, Inc.
Lentiviral production and infection
The 293TN cell line (System Biosciences, Mountain View, CA) was cultured in branched PEI25-coated plates (Sigma Aldrich) with DMEM supplemented with FetalClone II serum (HyClone Laboratories, Logan, UT) until 80–90% confluent, and then transfected with lentiviral transfer vector, psPAX2, and pCMV-VSV-G (0.41 μg/cm2; 12:14:8 molar ratio) using PEI22 (20). Medium was replaced after an overnight incubation, after which cells were cultured for an additional 48 h. Supernatant collected from this cell culture was centrifuged at 3000 × g for 30 min at 4C and stored at −80C.
Transduction with control or KCNJ5-T158A lentivirus was performed 24 h after seeding HAC15 (21, 22). To assess steroid production, membrane voltage, mRNA expression, and intracellular Ca2+ concentration 72 h after infection, cells were serum deprived using DMEM-F12 supplemented with 0.1% cosmic calf serum for 24 h. The medium was replaced with fresh DMEM-F12 containing 0.1% cosmic calf serum with or without 10 nm A-II, 10 μm forskolin, 3 or 10 μm nifedipine, or 10 or 30 μm W-7. Cells were incubated for 18 h, after which the supernatants were collected for steroid measurement and the cells were harvested for analysis of mRNA and protein expression.
To investigate the effect of KCNJ5-T158A on cell proliferation and apoptosis assay, HAC15 cells were stably infected with control or KCNJ5-T158A lentivirus as previously described (23). Briefly, 72 h after infection, the cells were selected with 5 μg/ml of blasticidin. The cells were cultured for at least 4 wk in the absence of the selecting agent before performing the experiments to avoid any confounding effect due to the selecting antibiotic.
Luciferase assay
To assess the CYP11B2 activity, we transduced HAC15 cells with lentivirus carrying the pBM14-CYP11B2 promoter/gaussia luciferase as above, using G418 (0.5 mg/ml) for antibiotic selection. The stably transduced cells were plated in 48-well plates and then transduced with control or KCNJ5-T158A lentivirus. The supernatants were collected and placed in 96-well plates for luminescence detection using the gaussia luciferase substrate coelenterazine (Gold Biotechnology, St. Louis, MO), and read with a BMG FLUOstar Omega Reader (BMG Labtech, Ortenberg, Germany) (24).
RNA extraction and RT-PCR
Total RNA was extracted with the RNAzol-RT Reagent (Molecular Research Center, Inc., Cincinnati, OH). For reverse transcription, 1.5 μg of total RNA was incubated with SUPERase-In (Applied Biosystems/Ambion, Austin, TX) or SuperScript III (Invitrogen) following the manufacturer's protocol. Table 1 shows real-time PCR primers designed to generate approximately 100-bp amplicons. CYP11B2, CYP11B1, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA expression were determined by the Taqman Gene expression assay as previously reported (21). mRNA expression of steroidogenic acute regulatory protein (STAR), cytochrome P450, family 11, subfamily A, polypeptide 1 (CYP11A1), 3β-hydroxysteroid dehydrogenase, cytochrome P450, family 21, subfamily A, polypeptide 2 (CYP21A2), cytochrome P450, family 17, subfamily A, polypeptide 1 (CYP17A1) and GAPDH were quantified with 1 μl reverse transcription product, 1 μl Titanium Taq DNA polymerase (CLONTECH Laboratories, Inc., Mountain View, CA), 1:20000 dilution SYBR Green I (Molecular Probes, Carlsbad, CA), 10 nm (Bio-Rad Laboratories, Hercules, CA), 0.2 mm deoxynucleotide triphosphates, and 0.1 μm each primer. Real-time data were obtained during the extension phase, and critical threshold cycle values were calculated at the log phase of each gene amplification curve. Gene expression levels were analyzed as arbitrary units normalized against GAPDH mRNA expression.
Table 1.
Real-time PCR primers
| Gene symbol | Forward primer | Reverse primer |
|---|---|---|
| STAR | CATACTCTAAACACGAACCCCACC | GTCCCACCCTGCCTCTGAAG |
| CYP11A1 | CTTCTTCGACCCGGAAAATTT | CCGGAAGTAGGTGATGTTCTTGT |
| HSD3B2 | AGAAGAGCCTCTGGAAAACACATG | TAACGCACAAGTGTACAAGGTATCACCA |
| CYP21A2 | TCCCAGCACTCAACCAACCT | CAGCTCAGAATTAAGCCTCAATCC |
| CYP17A1 | AGCCGCACACCAACTATCAG | TCACCGATGCTGGAGTCAAC |
| CYP11B1 | GGCAGAGGCAGAGATGCTG | TCTTGGGTTAGTGTCTCCACCTG |
| CYP11B2 | GGCAGAGGCAGAGATGCTG | CTTGAGTTAGTGTCTCCACCAGGA |
| GAPDH | CCCCTTCATTGACCTCAACTAC | GATGACAAGCTTCCCGTTCTC |
Cell proliferation and apoptosis assay
The 2,3-Bis(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide inner salt (XTT) cell proliferation assay kit (American Type Culture Collection, Manassas, VA) was used to evaluate the effect of KCNJ5-T158A on cell proliferation (25). Briefly, the stably transduced control and KCNJ5-T158A cells were seeded on 96-well plates at a concentration of 5 × 104 cells per well and incubated with 50 μl of activated-XTT solution for 4 h, following the manufacture's protocol. The absorbance of the wells was measured at a wavelength of 475 nm and 660 nm.
For crystal violet staining, the stably transduced cells were plated at 1 × 105 cells per well in 48-well plates, the supernatant was aspirated at the indicated times, and the cells were fixed for 20 min in 10% paraformaldehyde. They were then stained for 30 min with a 0.05% solution of crystal violet, washed with water, then solubilized in 1% sodium dodecyl sulfate solution for 1 h, and absorbance was read at 590-nm wavelength.
The commercial Caspase-3/CPP32 detection kit (InvivoGen) was used to investigate the effect of KCNJ5-T158A on the apoptotic state. The stably transduced cells were plated at 1 × 106 cells per well in 12-well plates, and cells were harvested and incubated following the manufacturer's instruction.
Analysis of inflow of Na+ into cytoplasm, plasma membrane voltage, and intracellular Ca2+ concentration
After transduction with control or KCNJ5-T158A lentiviruses and serum starving as above, the medium was replaced with fresh DMEM-F12 containing 0.1% cosmic calf serum with 10 μm CoroNa Green dye to assess Na+ flow into cytoplasm, 3 μm DiSBAC2 for membrane voltage, or 3 μm Fluo-4 AM for intracellular Ca2+ concentration. After incubation for the indicated times, the cells were washed three times with HEPES buffer, and fluorescence was detected with a BMG FLUOstar Omega Reader.
Steroid and protein assays
Aldosterone, cortisol, and 18-oxocortisol levels were measured in cell culture supernatants by time-resolved fluorescence. The primary antibodies were developed in our laboratory as previously described (26, 27). The protein levels were detected by ELISA using a Micro BCA Protein Assay kit (Thermo Scientific, Rockford, IL).
Statistical analysis
All results were expressed as mean ± se of at least three separate experiments in which each sample was assayed in triplicate or quadruplicate. Differences between two groups were analyzed for statistical significance by t test and multiple groups were analyzed by one-way ANOVA followed by Bonferroni comparisons. The differences were considered to be significant at P < 0.05. Analyses were performed using SPSS for Windows (release 12.0; SPSS Inc., Chicago, IL).
Results
The effects of KCNJ5-T158A on aldosterone production
The sequence of the KCNJ5-T158A plasmid was confirmed by a direct sequence method. Transduction of HAC15 cells with the lentivirus carrying the KCNJ5-T158A mutation potentiated basal aldosterone production 5.3-fold over empty lentivirus-infected HAC15 control cells (Fig. 1A). Cells transduced with the wild KCNJ5 lentivirus decreased aldosterone production and were not used as controls in these experiments (data not shown). To compare the effects of KCNJ5-T158A on aldosterone production induced by A-II and forskolin, we measured aldosterone levels in the supernatants of control and KCNJ5-T158A cells stimulated by A-II and forskolin. There was no difference in the level of aldosterone stimulation between A-II and forskolin in control cells, but aldosterone secretion stimulated by forskolin was significantly greater than by A-II in KCNJ5-T158A cells (Fig. 1A). Cortisol production was only slightly, but significantly, stimulated by KCNJ5-T158A (Fig. 1B). KCNJ5-T158A-infected cells also showed enhanced 18-oxocortisol production compared with control HAC15 cells (Fig 1C).
Fig. 1.
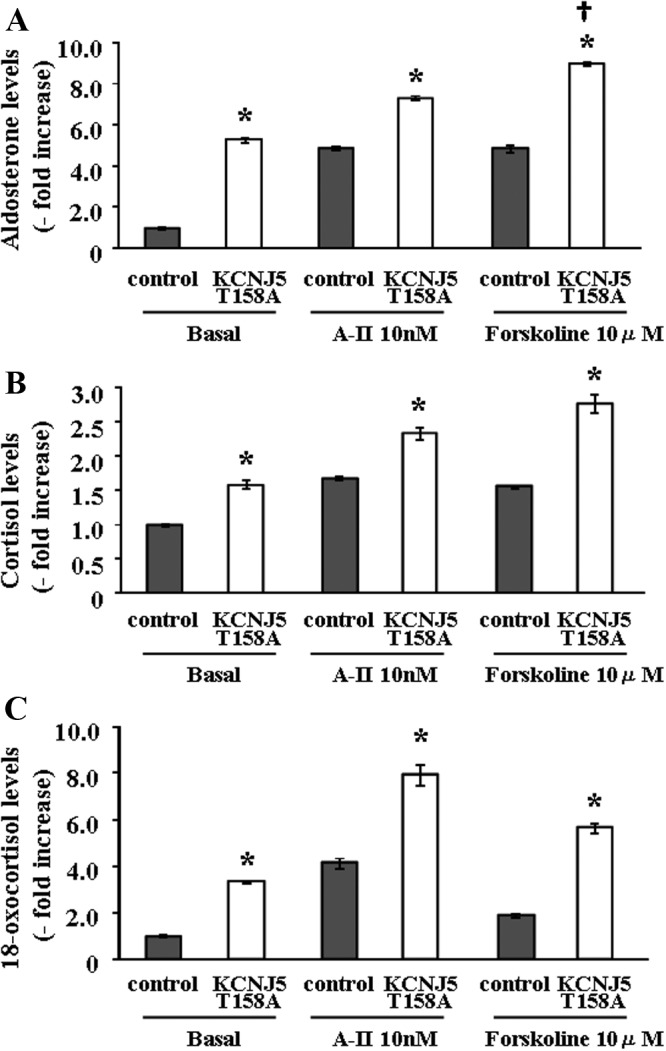
Basal and stimulated aldosterone (A), cortisol (B), and 18-oxocortisol (C) production by HAC15 infected with control or KCNJ5 T158A lentiviruses. Cells were incubated for 24 h with no secretagogue or 10 nm of either A-II or 10 μm forskolin. *, P < 0.01 vs. each control, n = 4. †, P < 0.01 vs. aldosterone levels after A-II stimulation in KCNJ5–T158A cells (n = 4).
The alteration of extracellular Na+ inflow into cytoplasm, membrane voltage, and intracellular Ca2+ concentration by the KCNJ5-T158A mutation in HAC15 cells
To elucidate the mechanism of the effect of the KCNJ5-T158A mutation on aldosterone production, we first investigated its effect on Na+ influx in the HAC15 cells using a cell-impermeant dye, CoroNa Green. KCNJ5-T158A cells had a 1.2-fold higher fluorescence for Na+ than control cells, indicating that Na+ inflow was increased by the mutation (Fig. 2A). KCNJ5-T158A cells showed a 2.3-fold increased accumulation of DiSBAC2 (3), an indicator of higher plasma membrane voltage than control cells (Fig. 2B). As shown in Fig. 2C, KCNJ5-T158A caused a 1.6-fold increase in Fluo-4 AM, an indicator of intracellular Ca2+ concentration, compared with control. These results suggested that the KCNJ5-T158A mutation led to a sequential increase in Na influx, membrane voltage, and intracellular Ca2+ accumulation.
Fig. 2.
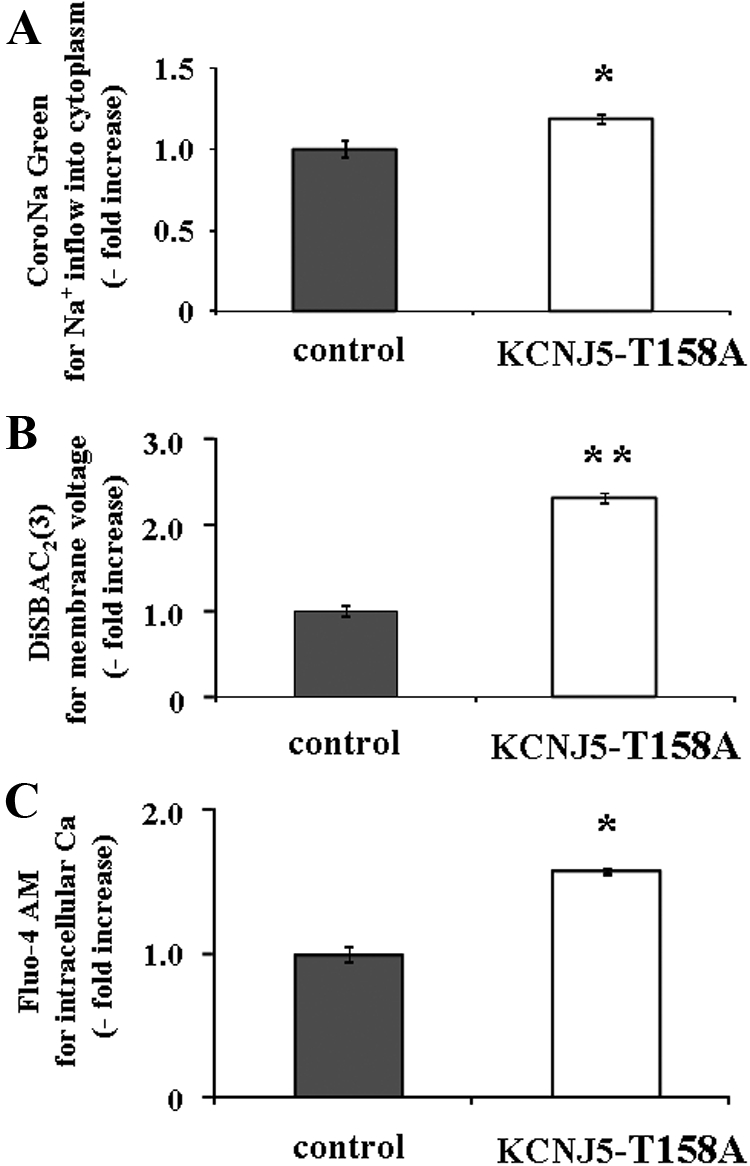
Effect of the KCNJ5–T158A mutation on Na+ influx, membrane voltage, and intracellular Ca2+ concentration in HAC15 cells transduced with control or KCNJ5–T158A lentiviruses. Seventy two hours after infection, cells were serum deprived in DMEM-F12 containing 0.1% cosmic calf serum for 24 h, and then incubated with fresh media with 0.1% serum and 10 μm CoroNa Green for 2 h (A), 3 μm DiSBAC2(3) for 5 min (B), or 3 μm Fluo-4 AM for 10 min (C). Fluorescence was detected by a plate reader. Results were expressed as fold increase vs. control cells. *, P < 0.05; and **, P < 0.01 vs. control (n = 4).
mRNA expression and reporter assay of steroid biosynthetic enzymes
The expression of CYP11B1 and CYP11B2 mRNA in KCNJ5-T158A cells was significantly elevated by 5.8-fold and 17.7-fold in comparison to control cells, respectively (Fig. 3). However, no differences of other steroidogenic enzymes or StAR were observed between control and KCNJ5-T158A cells (Fig. 3), except for CYP17A1, which was significantly decreased.
Fig. 3.
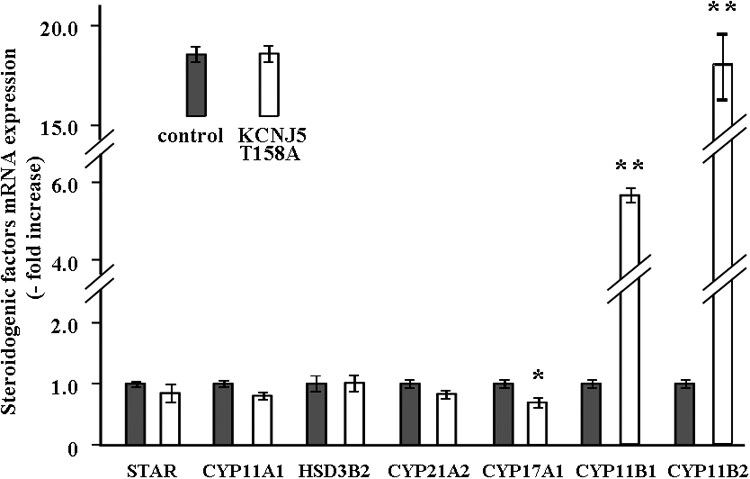
Effect of the KCNJ5–T158A mutation on mRNA expression of the key proteins for adrenal steroid synthesis: STAR, CYP11A1, 3β-hydroxysteroid dehydrogenase, CYP21A2, CYP17A1, CYP11B1, and CYP11B2. Seventy two hours after infection, cells were serum deprived in DMEM-F12 containing 0.1% cosmic calf serum for 24 h, and then incubated with fresh media with 0.1% serum for 3 h. After aspiration of media, the cells were collected for RNA extraction, and real time RT-PCR was performed with the primers shown in Table 1. Results were normalized by GAPDH mRNA expression and expressed as fold increase vs. control. *, P < 0.05; and **, P < 0.01 vs. control (n = 3).
We investigated the transcriptional regulation of CYP11B2 analyzed by luciferase assays using HAC15 cells stably infected with each reporter construct. As shown in Fig. 4, CYP11B2 transcriptional activity in KCNJ5-T158A cells showed 2.8-fold increase compared with that in control cells.
Fig. 4.
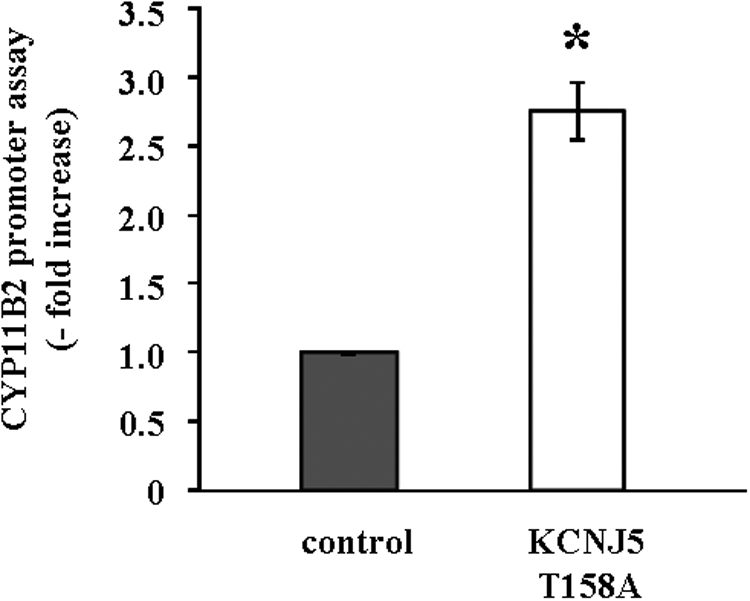
Effect of the KCNJ5-T158A mutation on CYP11B2 reporter gene expression. HAC15 cells stably infected with pBM14-CYP11B2 promoter/gaussia were infected with control or KCNJ5-T158A lentiviruses. After 72 h incubation, cells were serum deprived in DMEM-F12 containing 0.1% cosmic calf serum for 24 h, incubated in fresh media with 0.1% serum for 24 h, and supernatants collected to assess luminescence. Results shown as fold increase vs. control cells. *, P < 0.01 vs. control (n = 3).
Calcium channels and calmodulin mediate the aldosterone production in KCNJ5-T158A cells
To determine the role of increased intracellular Ca2+ in aldosterone production in KCNJ5-T158A cells, aldosterone levels in the supernatant of KCNJ5-T158A cells treated with or without 3 or 10 μm nifedipine or 10 or 30 μm W-7 was measured. Nifedipine (10 μm) and W-7 (30 μm) showed 75% and 18% inhibition of aldosterone production, respectively (Fig. 5A). Nifedipine (10 μm) and W-7 (30 μm) also inhibited CYP11B2 reporter activity by 46% and 20%, respectively (Fig. 5B). These results indicated that increased intracellular Ca2+ has a role in the regulation of aldosterone production in KCNJ5-T158A cells.
Fig. 5.
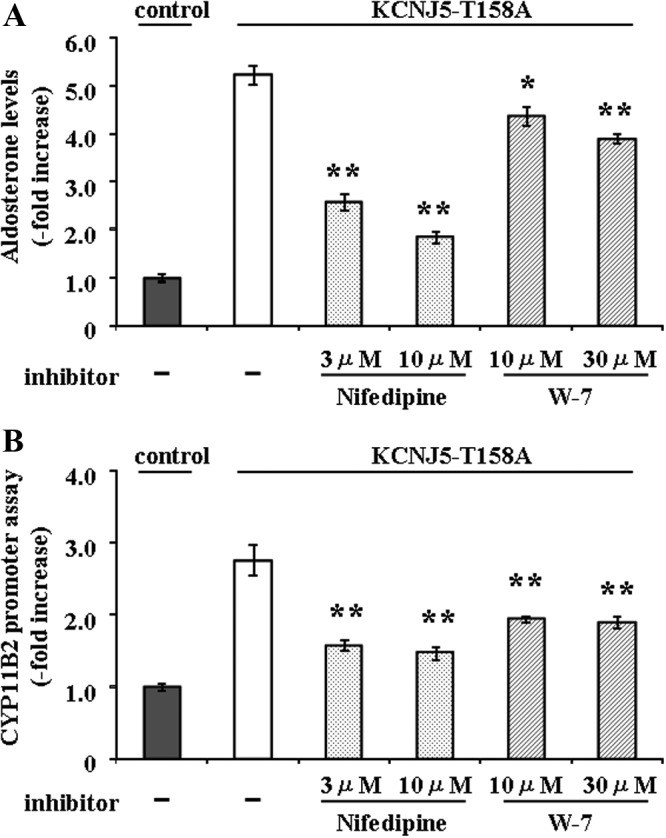
Effect of nifedipine and W-7 on aldosterone production (A) and CYP11B2 reporter gene activation (B) on control and KCNJ5-T158A mutant HAC15 cells stably transduced with pBM14-CYP11B2 promoter/gaussia reporter gene. Seventy two hours after infection with control or KCNJ5-T158A lentivirus, HAC15 cells were serum deprived in DMEM-F12 containing 0.1% cosmic calf serum for 24 h and then incubated in the fresh media with 0.1% serum and with or without 3 μm or 10 μm nifedipine or 10 μm or 30 μm W-7 for 24 h. Results were expressed as fold increase vs. control cells. *, P < 0.05; and **, P < 0.01 vs. control (n = 4) (A); **, P < 0.01 vs. control (n = 3) (B).
Effects of KCNJ5-T158A on adrenal cell proliferation
Proliferation of cells transduced with KCNJ5-T158A lentivirus was significantly less than that of control cells as detected by crystal violet staining (25.2% reduction) and XTT assay (31.8% reduction) after 72 h incubation (Fig. 6, A and B). Cells transduced with wild KCNJ5 lentivirus had no effect on proliferation (data not shown). We performed an apoptosis assay using Caspase-3 protein measurement, because the protein is reported to be regulated in human adrenal cells for apoptosis (28). No difference of Caspase-3 protein levels between control and KCNJ5-T158A cells (1.07 ± 0.07-fold increase) was found. Next, to know whether the negative effect of KCNJ5-T158A on cell proliferation was mediated by intracellular Ca2+ concentration, we investigated the effects of nifedipine on cell proliferation in control and KCNJ5-T158A cells. Nifedipine caused a 22.7% and 33.0% reduction of cell proliferation by crystal violet staining and XTT assay in control cells, respectively (Fig. 6, A and B). KCNJ5-T158A cells were further negatively regulated with nifedipine treatment as detected by crystal violet staining (24.2% reduction) and XTT assay (21.0% reduction), compared with control cells with nifedipine (Fig. 6, A and B). Therefore, nifedipine does not influence the difference of cell proliferation between control and KCNJ5-T158A cells, indicating that the effect of cell proliferation is independent of intracellular calcium concentration.
Fig. 6.
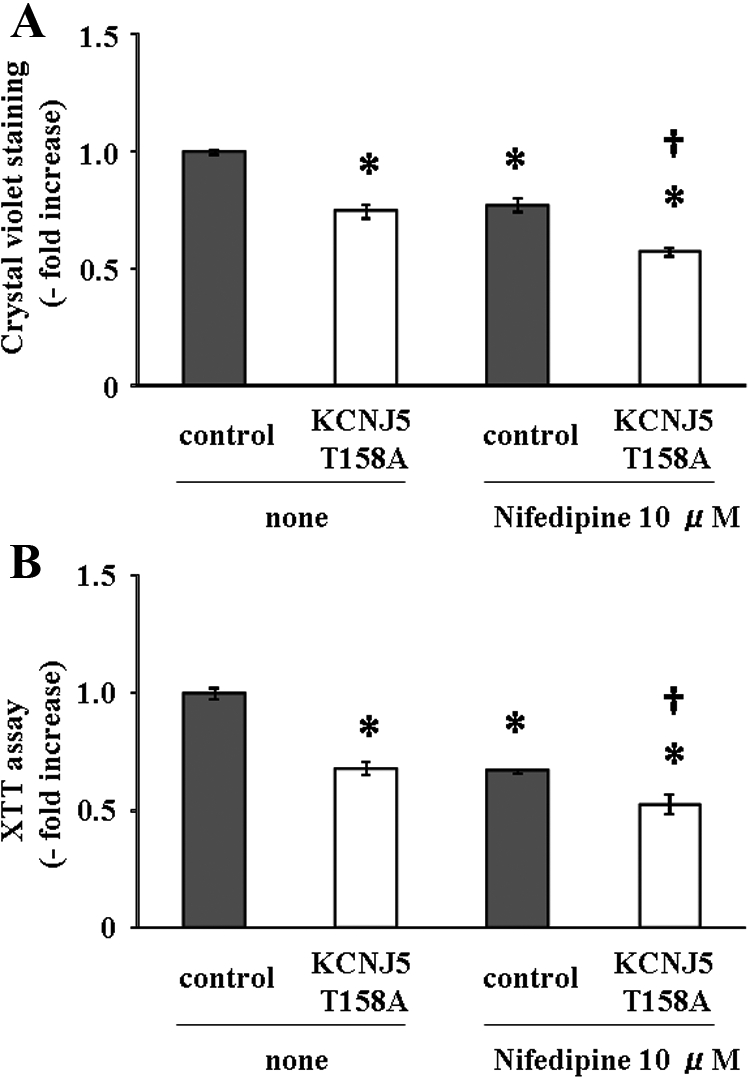
Effect of the KCNJ5 T158A mutation on HAC15 cell proliferation with or without nifedipine (10 μm). The identical numbers of control and KCNJ5-T158A cells were plated. After 72 h, crystal violet staining (A) and XTT assays (B) were performed. The results of crystal violet staining and XTT assay were expressed as the KCNJ5 T158A mutation cells compared with that of the control cells for each time point. *, P < 0.01 vs. control (n = 4). †, P < 0.01 vs. control with nifedipine (n = 4).
Discussion
Certain mutations around the GYG motif in KCNJ5 cause a loss in K+ selectivity and an increase in Na+ influx into cytoplasm (12), resulting in the depolarization of the plasma membrane, thereby activating the voltage-gated Ca2+ channel, and accumulation of intracellular Ca2+. We showed that KCNJ5-T158A-transduced adrenocortical cells had a greater influx of Na+ into cytoplasm, increased intracellular Ca2+ concentration, and higher membrane voltage. Increased intracellular Ca2+ activates calmodulin-dependent kinase (CaMK) I and subsequent regulation of gene transcription, resulting in the synthesis of steroidogenic enzymes and increased aldosterone production (14). mRNA expression and transcriptional activity of CYP11B2 were up-regulated in KCNJ5-T158A cells, resulting in the increase in aldosterone production. Because the Ca2+ channel blocker and calmodulin antagonist suppressed the CYP11B2 transcriptional activity and aldosterone production in KCNJ5-T158A cells, the increased CYP11B2 expression in these cells appears to be mediated by the Ca2+/calmodulin cascade. Whereas transduction of HAC15 cells with KCNJ5-T158A lentiviruses stimulated aldosterone production, cell proliferation was inhibited, consistent with published results showing that increased intracellular Ca2+ negatively regulates cell proliferation (29, 30).
The KCNJ5-T158 corresponds to the chicken KCNJ12-T152, which is highly conserved among species and identical to other inward-rectifier K+ channels (10, 31). The crystal structure of chicken KCNJ12 (Kir2.2), the only one available for the inward-rectifier K+ channels, shows that the pore helix and selectivity filter is 89% identical to the human KCNJ5 (31). The KCNJ5-T158 is near the GYG motif in the pore and makes hydrogen bonds with conserved positions P128 and C129 to maintain pore function (10). Mutations near GYG motif change the plasma membrane voltage by altering K+ selectivity (12). Therefore, disruption of hydrogen binding by the KCNJ5-T158A mutation could alter pore morphology, resulting in loss of K+ selectivity (10). Our results show that there is an increase in Na+ inflow in cells transduced with the KCNJ5-T158A lentiviruses and support this explanation.
Other potassium channels participate in aldosterone regulation. TWIK-related acid-sensitive K TASK-1 and TASK-3 subunits, of the KCNK family of two-pore domain/four-transmembrane K channels, regulate the rectification properties in physiological asymmetric K+ solutions. TASK 1 subunit-knockout mice and TASK 1 + 3 double-knockout mice exhibited depolarized membrane voltage in the adrenal cortex and activation of voltage-dependent Ca2+ channel, producing aldosterone hypersecretion (32, 33). Aldosterone synthase expression is normally restricted to the adrenal zona glomerulosa cell. TASK 1-knockout mice exhibited a gender-specific alteration in adrenal aldosterone synthesis characterized by expression of the aldosterone synthase in the zona fasciculata of female mice, which was glucocorticoid suppressible (32). The large Ca2+-activated K+ (BK) channel mediates steady hyperpolarization as a result of transient outward currents carried by BK channels, which were spontaneously activated by the local release of Ca2+ from intracellular stores (34). Recent studies show that aldosterone concentration in blood is elevated in both BK-α- and BK-β1-knockout mice (35).
Calmodulin is highly expressed in the adrenal cortex (36), and the increase in intracellular Ca2+ by voltage-gated receptors activates calmodulin, which activates CaMK I and stimulates aldosterone production (14). We observed that the increase of membrane voltage by the KCNJ5 mutation enhanced the intracellular Ca2+ concentration and increased in CYP11B2 mRNA expression and aldosterone production. Calcium channel and calmodulin antagonists suppressed these increases in the mutant cells.
The steroid production profile of HAC15 transduced with KCNJ5-T158A lentiviruses was comparable to that of patients with aldosterone-producing adenomas and FH3 (8, 10). H295R, the parent cell line of the HAC15 cell, and the HAC15 cell do not respond or respond poorly to ACTH but respond well to the PKA activator forskolin (15, 37). As shown in Fig 1A, the response to forskolin was more prominent than that to A-II. Aldosterone-producing adenomas respond better to ACTH than to A-II (38–40). Our in vitro findings regarding enhancement of the Ca2+/calmodulin-related cascade are consistent with recent evidence showing that APA have activated Ca2+/CaMK pathways (41), and a microarray study indicated that the CaMK-dependent pathways are enhanced in a subset of the APA (42). Patients with APA synthesize significant quantities of the hybrid steroid 18-oxocortisol (43, 44), and patients with FH3 produce massive amounts of 18-oxocortisol (8). Consistent with the in vivo findings in patients with FH3, compared with control cells, the HAC15-KCNJ5-T158A cells produced significantly greater amounts of 18-oxocortisol, as well as aldosterone, under basal conditions, and synthesis was further enhanced by stimulation. In the family with FH3, the very high production of aldosterone and 18-oxocortisol in FH3 was not suppressible with dexamethasone, probably reflecting the marked increase in cells with transitional zone characteristics (8). The KCNJ5-T158A mutation has only been found in FH3, although the KCNJ5-G151R and G168R mutations found in some APA adenomas are in the same selectivity region of the KCNJ5 potassium channel, thus would be expected to show similar characteristics with respect to basal and stimulated alterations in aldosterone secretion (10).
Aldosterone-producing adenomas are generally small, but in the initial report of patients with the somatic mutations in KCNJ5, the tumors were relatively large (8). Activation of Ca2+ signaling inhibits growth in the G2 phase of the cell cycle in mammalian cell lines and budding yeast (29). Expression of a constitutive form of calcium/CaMK II leads to the arrest of the cell cycle in G2 (30). Similarly, colon cancer appears to be inhibited by the increase in intracellular Ca2+ concentration in clinical and experimental studies (45, 46). In FH3, characterized by the genomic KCNJ5-T158A mutation, there is atrophy or suppression of zona glomerulosa and marked hyperplasia and cellular hypertrophy of an area characteristic of the transitional zone and/or zona fasciculata (8). The discrepancy between the cell-specific hyperplasia and hypertrophy in FH3 adrenals and the decreased proliferation of HAC15-KCNJ5-T158A cells might be due to the fact that the HAC15 cells were derived from an adrenal carcinoma, which might differ from in vivo development of an adenoma (47).
In conclusion, we have demonstrated the mechanism of increased aldosterone synthesis produced by a mutation near the GYG motif in the selectivity filter of the potassium channel that mediates inward rectifying currents. HAC15 infected with a KCNJ5-T158A lentivirus carrying a mutation near the GYG motif in the selectivity filter results in decreased ion selectivity of the channel, membrane depolarization, activation of voltage-gated Ca2+ channels, increased intracellular Ca2+ concentration, CYP11B2 overexpression, and increased aldosterone and 18-oxocortisol production.
Acknowledgments
This work was supported by National Institutes of Health grants HL27255 and HL105383 and from medical research funds from the Department of Veterans Affairs.
Disclosure Summary: K.O., M.W.P., M.L.L., E.P.G.-S., and C.E.G.-S. have nothing to declare.
For editorial see page 1575
- A-II
- Angiotensin II
- APA
- aldosterone-producing adenoma
- CaMK
- calmodulin-dependent protein kinase
- CYP
- cytochrome P450
- FH3
- familial hyperaldosteronism type 3
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- GYG
- glycine-tyrosine-glycine
- PA
- primary aldosteronism
- STAR
- steroidogenic acute regulatory protein
- XTT
- 2,3-Bis(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide inner salt.
References
- 1. Funder JW, Carey RM, Fardella C, Gomez-Sanchez CE, Mantero F, Stowasser M, Young WF, Jr, Montori VM. 2008. Case detection, diagnosis, and treatment of patients with primary aldosteronism: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 93:3266–3281 [DOI] [PubMed] [Google Scholar]
- 2. Milliez P, Girerd X, Plouin PF, Blacher J, Safar ME, Mourad JJ. 2005. Evidence for an increased rate of cardiovascular events in patients with primary aldosteronism. J Am Coll Cardiol 45:1243–1248 [DOI] [PubMed] [Google Scholar]
- 3. Mulatero P, Bertello C, Rossato D, Mengozzi G, Milan A, Garrone C, Giraudo G, Passarino G, Garabello D, Verhovez A, Rabbia F, Veglio F. 2008. Roles of clinical criteria, computed tomography scan, and adrenal vein sampling in differential diagnosis of primary aldosteronism subtypes. J Clin Endocrinol Metab 93:1366–1371 [DOI] [PubMed] [Google Scholar]
- 4. Lifton RP, Dluhy RG, Powers M, Rich GM, Gutkin M, Fallo F, Gill JR, Jr, Feld L, Ganguly A, Laidlaw JC, Murnaghan DJ, Kauffman C, Stockigt J, Ulick l, Lalouel J-M. 1992. Hereditary hypertension caused by chimeric gene duplication and ectopic expression of aldosterone synthase. Nat Genet 2:66–74 [DOI] [PubMed] [Google Scholar]
- 5. Pascoe L, Curnow KM, Slutsker L, Connell JM, Speiser PW, New MI, White PC. 1992. Glucocorticoid-suppressible hyperaldosteronism results from hybrid genes created by unequal crossovers between CYP11B1 and CYP11B2. Proc Natl Acad Sci USA 89:8327–8331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gomez-Sanchez CE, Gill JR, Jr, Ganguly A, Gordon RD. 1988. Glucocorticoid-suppressible aldosteronism: a disorder of the adrenal transitional zone. J Clin Endocrinol Metab 67:444–448 [DOI] [PubMed] [Google Scholar]
- 7. Sukor N, Mulatero P, Gordon RD, So A, Duffy D, Bertello C, Kelemen L, Jeske Y, Veglio F, Stowasser M. 2008. Further evidence for linkage of familial hyperaldosteronism type II at chromosome 7p22 in Italian as well as Australian and South American families. J Hypertens 26:1577–1582 [DOI] [PubMed] [Google Scholar]
- 8. Geller DS, Zhang J, Wisgerhof MV, Shackleton C, Kashgarian M, Lifton RP. 2008. A novel form of human mendelian hypertension featuring nonglucocorticoid-remediable aldosteronism. J Clin Endocrinol Metab 93:3117–3123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Spät A, Hunyady L. 2004. Control of aldosterone secretion: a model for convergence in cellular signaling pathways. Physiol Rev 84:489–539 [DOI] [PubMed] [Google Scholar]
- 10. Choi M, Scholl UI, Yue P, Björklund P, Zhao B, Nelson-Williams C, Ji W, Cho Y, Patel A, Men CJ, Lolis E, Wisgerhof MV, Geller DS, Mane S, Hellman P, Westin G, Åkerström G, Wang W, Carling T, Lifton RP. 2011. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science 331:768–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Heginbotham L, Abramson T, MacKinnon R. 1992. A functional connection between the pores of distantly related ion channels as revealed by mutant K+ channels. Science 258:1152–1155 [DOI] [PubMed] [Google Scholar]
- 12. Dibb KM, Rose T, Makary SY, Claydon TW, Enkvetchakul D, Leach R, Nichols CG, Boyett MR. 2003. Molecular basis of ion selectivity, block, and rectification of the inward rectifier Kir3.1/Kir3.4 K(+) channel. J Biol Chem 278:49537–49548 [DOI] [PubMed] [Google Scholar]
- 13. Yao J, Davies LA, Howard JD, Adney SK, Welsby PJ, Howell N, Carey RM, Colbran RJ, Barrett PQ. 2006. Molecular basis for the modulation of native T-type Ca2+ channels in vivo by Ca2+/calmodulin-dependent protein kinase II. J Clin Invest 116:2403–2412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Condon JC, Pezzi V, Drummond BM, Yin S, Rainey WE. 2002. Calmodulin-dependent kinase I regulates adrenal cell expression of aldosterone synthase. Endocrinology 143:3651–3657 [DOI] [PubMed] [Google Scholar]
- 15. Parmar J, Key RE, Rainey WE. 2008. Development of an adrenocorticotropin-responsive human adrenocortical carcinoma cell line. J Clin Endocrinol Metab 93:4542–4546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang T, Rainey WE. 5 September 2011. Human adrenocortical carcinoma cell lines. Mol Cell Endocrinol 10.1016/j.mce.2011.08.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yang X, Boehm JS, Yang X, Salehi-Ashtiani K, Hao T, Shen Y, Lubonja R, Thomas SR, Alkan O, Bhimdi T, Green TM, Johannessen CM, Silver SJ, Nguyen C, Murray RR, Hieronymus H, Balcha D, Fan C, Lin C, Ghamsari L, Vidal M, Hahn WC, Hill DE, Root DE. 2011. A public genome-scale lentiviral expression library of human ORFs. Nat Methods 8:659–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mitta B, Rimann M, Ehrengruber MU, Ehrbar M, Djonov V, Kelm J, Fussenegger M. 2002. Advanced modular self-inactivating lentiviral expression vectors for multigene interventions in mammalian cells and in vivo transduction. Nucleic Acids Res 30:e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, Sabatini DM, Chen IS, Hahn WC, Sharp PA, Weinberg RA, Novina CD. 2003. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 9:493–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Thomas M, Lu JJ, Ge Q, Zhang C, Chen J, Klibanov AM. 2005. Full deacylation of polyethylenimine dramatically boosts its gene delivery efficiency and specificity to mouse lung. Proc Natl Acad Sci USA 102:5679–5684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Romero DG, Gomez-Sanchez EP, Gomez-Sanchez CE. 2010. Angiotensin II-regulated transcription regulatory genes in adrenal steroidogenesis. Physiol Genomics 42A:259–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Romero DG, Plonczynski MW, Welsh BL, Gomez-Sanchez CE, Zhou MY, Gomez-Sanchez EP. 2007. Gene expression profile in rat adrenal zona glomerulosa cells stimulated with aldosterone secretagogues. Physiol Genomics 32:117–127 [DOI] [PubMed] [Google Scholar]
- 23. Romero DG, Zhou MY, Yanes LL, Plonczynski MW, Washington TR, Gomez-Sanchez CE, Gomez-Sanchez EP. 2007. Regulators of G-protein signaling 4 in adrenal gland: localization, regulation, and role in aldosterone secretion. J Endocrinol 194:429–440 [DOI] [PubMed] [Google Scholar]
- 24. Tannous BA. 2009. Gaussia luciferase reporter assay for monitoring biological processes in culture and in vivo. Nat Protoc 4:582–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Berridge MV, Herst PM, Tan AS. 2005. Tetrazolium dyes as tools in cell biology: new insights into their cellular reduction. Biotechnol Annu Rev 11:127–152 [DOI] [PubMed] [Google Scholar]
- 26. Gomez-Sanchez CE, Foecking MF, Ferris MW, Chavarri MR, Uribe L, Gomez-Sanchez EP. 1987. The production of monoclonal antibodies against aldosterone. Steroids 49:581–587 [DOI] [PubMed] [Google Scholar]
- 27. Morra di Cella S, Veglio F, Mulatero P, Christensen V, Aycock K, Zhu Z, Gomez-Sanchez EP, Gomez-Sanchez CE. 2002. A time-resolved fluoroimmunoassay for 18-oxocortisol and 18-hydroxycortisol. Development of a monoclonal antibody to 18-oxocortisol. J Steroid Biochem Mol Biol 82:83–88 [DOI] [PubMed] [Google Scholar]
- 28. Mikhaylova IV, Kuulasmaa T, Jääskeläinen J, Voutilainen R. 2007. Tumor necrosis factor-α regulates steroidogenesis, apoptosis, and cell viability in the human adrenocortical cell line NCI-H295R. Endocrinology 148:386–392 [DOI] [PubMed] [Google Scholar]
- 29. Planas-Silva MD, Means AR. 1992. Expression of a constitutive form of calcium/calmodulin dependent protein kinase II leads to arrest of the cell cycle in G2. EMBO J 11:507–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chanklan R, Aihara E, Koga S, Takahashi H, Mizunuma M, Miyakawa T. 2008. Inhibition of Ca2+-signal-dependent growth regulation by radicicol in budding yeast. Biosci Biotechnol Biochem 72:132–138 [DOI] [PubMed] [Google Scholar]
- 31. Tao X, Avalos JL, Chen J, MacKinnon R. 2009. Crystal structure of the eukaryotic strong inward-rectifier K+ channel Kir2.2 at 3.1 A resolution. Science 326:1668–1674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Heitzmann D, Derand R, Jungbauer S, Bandulik S, Sterner C, Schweda F, El Wakil A, Lalli E, Guy N, Mengual R, Reichold M, Tegtmeier I, Bendahhou S, Gomez-Sanchez CE, Aller MI, Wisden W, Weber A, Lesage F, Warth R, Barhanin J. 2008. Invalidation of TASK1 potassium channels disrupts adrenal gland zonation and mineralocorticoid homeostasis. EMBO J 27:179–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Davies LA, Hu C, Guagliardo NA, Sen N, Chen X, Talley EM, Carey RM, Bayliss DA, Barrett PQ. 2008. TASK channel deletion in mice causes primary hyperaldosteronism. Proc Natl Acad Sci USA 105:2203–2208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nelson MT, Cheng H, Rubart M, Santana LF, Bonev AD, Knot HJ, Lederer WJ. 1995. Relaxation of arterial smooth muscle by calcium sparks. Science 270:633–637 [DOI] [PubMed] [Google Scholar]
- 35. Grimm PR, Irsik DL, Settles DC, Holtzclaw JD, Sansom SC. 2009. Hypertension of Kcnmb1−/− is linked to deficient K secretion and aldosteronism. Proc Natl Acad Sci USA 106:11800–11805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Coyne MD, Cornelius P, Venditti N, Toscano DG, Gross MK, Toscano WA., Jr 1985. Purification and properties of calmodulin from adrenal cortex. Arch Biochem Biophys 236:629–637 [DOI] [PubMed] [Google Scholar]
- 37. Antonini SR, Baldacchino V, Tremblay J, Hamet P, Lacroix A. 2006. Expression of ACTH receptor pathway genes in glucose-dependent insulinotrophic peptide (GIP)-dependent Cushing's syndrome. Clin Endocrinol (Oxf) 64:29–36 [DOI] [PubMed] [Google Scholar]
- 38. Wisgerhof M, Brown RD, Hogan MJ, Carpenter PC, Edis AJ. 1981. The plasma aldosterone response to angiotensin II infusion in aldosterone-producing adenoma and idiopathic hyperaldosteronism. J Clin Endocrinol Metab 52:195–198 [DOI] [PubMed] [Google Scholar]
- 39. Kem DC, Weinberger MH, Higgins JR, Kramer NJ, Gomez-Sanchez C, Holland OB. 1978. Plasma aldosterone response to ACTH in primary aldosteronism and in patients with low renin hypertension. J Clin Endocrinol Metab 46:552–560 [DOI] [PubMed] [Google Scholar]
- 40. Seccia TM, Miotto D, De Toni R, Pitter G, Mantero F, Pessina AC, Rossi GP. 2009. Adrenocorticotropic hormone stimulation during adrenal vein sampling for identifying surgically curable subtypes of primary aldosteronism: comparison of 3 different protocols. Hypertension 53:761–766 [DOI] [PubMed] [Google Scholar]
- 41. Sackmann S, Lichtenauer U, Shapiro I, Reincke M, Beuschlein F. 2011. Aldosterone producing adrenal adenomas are characterized by activation of calcium/calmodulin-dependent protein kinase (CaMK) dependent pathways. Horm Metab Res 43:106–111 [DOI] [PubMed] [Google Scholar]
- 42. Lenzini L, Seccia TM, Aldighieri E, Belloni AS, Bernante P, Giuliani L, Nussdorfer GG, Pessina AC, Rossi GP. 2007. Heterogeneity of aldosterone-producing adenomas revealed by a whole transcriptome analysis. Hypertension 50:1106–1113 [DOI] [PubMed] [Google Scholar]
- 43. Hamlet SM, Gordon RD, Gomez-Sanchez CE, Tunny TJ, Klemm SA. 1988. Adrenal transitional zone steroids, 18-oxo and 18-hydroxycortisol, useful in the diagnosis of primary aldosteronism are ACTH-dependent. Clin Exp Pharmacol Physiol 15:317–322 [DOI] [PubMed] [Google Scholar]
- 44. Yamakita N, Mune T, Morita H, Yoshida H, Yasuda K, Miura K, Gomez-Sanchez CE. 1994. Plasma 18-oxocortisol levels in the patients with adrenocortical disorders. Clin Endocrinol (Oxf) 40:583–587 [DOI] [PubMed] [Google Scholar]
- 45. Rey O, Young SH, Jacamo R, Moyer MP, Rozengurt E. 2010. Extracellular calcium sensing receptor stimulation in human colonic epithelial cells induces intracellular calcium oscillations and proliferation inhibition. J Cell Physiol 225:73–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Baron JA, Beach M, Mandel JS, van Stolk RU, Haile RW, Sandler RS, Rothstein R, Summers RW, Snover DC, Beck GJ, Bond JH, Greenberg ER. 1999. Calcium supplements for the prevention of colorectal adenomas. Calcium Polyp Prevention Study Group. N Engl J Med 340:101–107 [DOI] [PubMed] [Google Scholar]
- 47. Roderick HL, Cook SJ. 2008. Ca2+ signalling checkpoints in cancer: remodelling Ca2+ for cancer cell proliferation and survival. Nat Rev Cancer 8:361–375 [DOI] [PubMed] [Google Scholar]