

Abstract
Background
Colorectal (CRC) carcinogenesis through various morphological stages has been linked to several genetic and epigenetic changes. The Raf/MEK/ERK (MAPK) signal transduction cascade is an important mediator of a number of cellular fates.
Methods
In this study, we investigated the presence of B-raf and K-ras mutations in 94 consecutive cases of primary colon adenocarcinoma in correlation with the immunohistochemical expression of total and activated ERK and the expression of mismatch repair proteins (MMR) hMLH1 and hMSH2 as well as their correlations with standard clinicopathological parameters.
Results
The immunostaining pattern for total and activated ERK was nuclear and cytoplasmic. hMLH1 and hMSH2 proteins were preserved in 45/63 (71.43%) cases and 35/53 (66.04%) cases respectively. Total ERK nuclear expression, was positively correlated with tumor stage (p = 0.049), whereas nuclear pERK expression was positively correlated with histological grade (p = 0.0113) and tumor stage (p = 0.0952), although the latter relationship was of marginal significance. DNA sequencing showed that 12 samples (12.7%) had a mutation in B-RAF Exon 15 and none in Exon 11, whereas 22 (23.4%) had a K-ras mutation. Disruption of the MAP kinase pathway-either through K-ras or B-raf mutation-was detected in 37% of all the examined cases, although the overexpression of total and activated ERK1/2 was not correlated with the mutational status of K-ras or B-raf genes. Finally, the preservation of hMLH1 or hMSH2 immunoexpression was not correlated with the presence of B-raf and/or K-ras mutations.
Conclusions
In this study, we present evidence that ERK activation occurs in a K-ras or B-raf -independent manner in the majority of primary colon cancer cases. Moreover, B-raf mutations are not associated with mismatch-repair deficiency through loss of hMLH1 or hMSH2 expression. Activated ERK could possibly be implicated in tumor invasiveness as well as in the acquisition of a more aggressive phenotype.
Background
Colon cancer is the fourth most common malignancy in terms of both incidence and mortality worldwide [1]. The development of colon cancer is a complex multistep process dependent on both genetic and environmental factors, in which oncogenes, tumor suppressor genes as well as genes involved in DNA damage recognition and repair have been implicated. Gaining insight into the molecular pathways involved in the progression of colon cancer is imperative for the development of innovative individualised cancer treatment strategies.
Colorectal (CRC) carcinogenesis through various morphological stages has been linked to several genetic and epigenetic changes. Until recently, two main pathways of sporadic colorectal carcinogenesis were proposed: a) the chromosomal instability (CIN) pathway which affects proto-oncogenes and tumor suppressor genes and is characterized by alterations of chromosomal number and structure, and b) the microsatellite instability (MSI) pathway, responsible also for Lynch syndrome CRCs, which features size variations of repeated nucleotides (microsatellites) mainly in non coding sequences, due to defects in the mismatch repair system (hMLH1, hMSH2, mHMS6, PMS1, PMS2) [2,3]. In addition to CIN and MSI pathways, a third pathway, the epigenetic instability, which is thought to be largely driven by hypermethylation-induced silencing of tumor suppressor-like genes, has been implicated in the progression of colorectal carcinogenesis [4]. According to this notion, contemporary literature suggests that CRC in general develops through two independent pathways that involve sequences of genetic and epigenetic alterations associated with pathological and clinical features: the adenoma pathway in 70-80% and the newly recognized, the serrated pathway in the remaining 20-30% [5]. The somatic molecular features which characterize the newly introduced serrated pathway to CRC include activating mutations in B-raf [6] and widespread hypermethylation of gene promoters (CIMP) [7] with or without MSI [6].
The kinases of mitogen-activated protein (MAP) kinase superfamily participate in signaling cascades conserved through evolution, which transduce extracellular signals into intracellular responses. MAP kinases are major components of pathways controlling embryogenesis, cell differentiation, proliferation and death. The Ras/Raf/mitogen-extracellular signal-regulated kinase 1/2 (MEK1/2)/extracellular signal regulated kinase 1/2 (ERK1/2) cascade is activated by mitogenic factors, differentiation stimuli and cytokines [8,9]. The Raf family of protein kinases, which is one class of Ras effectors, phosphorylates the dual specific MAP kinases MEK1 and MEK2, which in turn phosphorylate and activate the effector MAP kinases ERK1 and ERK2 [9]. ERKs are multifunctional serine/threonine kinases that target a vast array of substrates localized in all cellular compartments, such as protein kinases, signaling effectors, receptors, cytoskeletal and nuclear proteins and transcription factors, that can influence cell fate [9-11].
Importantly, MAP kinases are capable of affecting gene expression via intermediary kinases by phosphorylating proteins in the cytoplasm, but also translocate to the nucleus, a critical step for the fulfilment of many cellular functions of ERK, such as gene transcription, cell proliferation and differentiation [12]. Through phosphorylation of these various substrates, constitutively activated ERKs are able to influence many of the hallmarks of carcinogenesis, as defined by Hanahan and Weinberg [13]. Constitutive activation of this pathway has been observed in several human malignancies and cell lines such as breast, colon, thyroid carcinomas and melanomas and provides a potent promitogenic force resulting in uncontrolled proliferation and differentiation [14,15].
The current study investigates the presence of mutations in K-ras and B-raf genes in colorectal carcinoma, in correlation with MAP kinase ERK expression and the expression of mismatch repair proteins (MMR) hMLH1 and hMSH2, attempting to elucidate the involvement of these MAP kinases in the development of colorectal cancer, as well as their correlations with standard clinicopathological parameters.
Methods
Patients
This is a retrospective study of 94 consecutive cases of primary colon adenocarcinoma (53 men and 41 women, median age of 60 years -range 35 to 82) for whom archival material from primary tumor resection surgical specimens was available. None of the patients had received chemotherapy or radiation before surgery. All cases were reviewed by two experienced pathologists (PK, GL) and assigned a histological grade according the standards laid down in the latest WHO classification of colon carcinoma [16]. There were 10 grade 1, 64 grade 2 and 20 grade 3 carcinomas. According to the latest TNM system of cancer staging adopted by the American Joint Committee on Cancer and the International Union Against Cancer (AJCC/IUCC), tumors were classified as stage I: 7 cases (T1N0M0, T2N0M0), stage II: 36 cases [stage IIA (T3N0M0), IIB (T4aN0M0) IIC (T4cN0M0), stage III: 47 cases [stage IIIA (T1-T2N1M0, T1N2aM0), IIIB (T3-T4aN1M0, T2-T3N2aM0, T1-T2N2bM0) and IIIC (T4aN2aM0, T3-T4aN2bM0, T4bN1-2M0)] and stage IV:4 (Any T Any N M1) cases.
Immunohistochemical analysis
Immunohistochemical analysis was performed in the same blocks that were used for molecular analysis. Accordingly, the tumor tissue that was available for immunohistochemistry was exhausted in some cases. Thus, the immunohistochemical expression of ERK protein was available in a subset of 55 specimens, the expression of pERK protein in 45 specimens, the expression of hMLH1 in 63 cases and the expression of hMSH2 in 53 cases. However, the distribution of mutations in K-ras and B-raf (exon 15 or 11) did not differ among the specimens for which immunohistochemical analysis was available and those that immunohistochemistry was not available, a fact that suggests that there was not any significant bias in this regard. ERK and pERK immunostaining was performed using a rabbit polyclonal (#9488, p44/42 MAP kinase, Cell Signaling, Beverly, MA) for ERK1/2 and a mouse monoclonal antibody (sc-7383, (E-4) Santa Cruz Biotechnology, Inc., California, USA) for pERK, whereas hMLH1 and hMSH2 immunostaining using mouse monoclonal antibodies (#554073, G168-15 for hMLH1, Pharmingen, San Diego, California and clone GB-12, for hMSH2, Oncogene Cambridge, MA). Sections (3 to 4 μm) were deparaffinized, rehydrated, immersed in 3% H2O2 for 30 min and microwaved at 750 W in 0.01 M citrate buffer (pH 6.0) for 15 min and left to cool down in TBS. Sections were incubated with ERK. pERK, hMLH1 and hMSH2 antibodies overnight at room temperature (37°C), at a dilution 1:100, 1:350, 1:180 and 1:180 respectively. Immunostaining was performed using the standard avidin-biotin complex (Ultra Vision Polymer, LabVision) and visualized with diaminobenzidine tetrahydrochloride solution.
Light microscopic evaluation of immunostained slides was performed independently by three (PK, NK, GL) experienced pathologists without previous knowledge of the clinical information. If a discrepancy occurred between the three assessments, the slides were reassessed jointly. In each case 1000-1500 neoplastic cells throughout the section were counted at high-power magnification. Nuclear and cytoplasmic pERK and ERK expression were evaluated separately. Nuclear or cytoplasmic pERK and ERK labeling index was defined as the percentage of neoplastic cells with nuclear or cytoplasmic immunoreactivity out of the total number of neoplastic cells counted. According to the percentage of positively stained neoplastic nuclei, each case was considered to display reduced (< 20%) or preserved (≥20%) expression of hMSH2 and hMLH. This is an arbitrary cut-off chosen for the purposes of this study, since no uniform threshold has been established in the respective literature. In this context studies evaluating the expression of MMR proteins in colorectal cancer have used cut-offs ranging from 5-50%, whereas in other tumors the more frequently cut-off value used is 20% [17-21].
Genomic DNA isolation
15 μm sections were used for DNA extraction. The samples were digested overnight at 55°C using 200 μl of digestion buffer consisting of 50 mM Tris, 1 mM EDTA, 0.5% SDS and 200 μg/ml Proteinase K. Genomic DNA was extracted with phenol-chloroform and precipitated in ice-cold ethanol. The DNA was redissolved in distilled water and quantified by spectrophotometry at 260 nm under UV light.
PCR
200 ng of total DNA were amplified in a 50 μl reaction mixture containing 25 pmoles of each primer, 25 mM each dNTP, 1.5 mM MgCl2, 1 mM KCl, 0.1% gelatin and 1.5 U Taq DNA polymerase (Advantage, Clontech, Takara, USA). The profile used in the Progene Techne thermal cycler was: 5 min at 95°C once; 30 sec at 95°C, 40 sec at 52-56°C, 1 min at 72°C for 40 cycles; 7 min at 72°C once. Sequences of the primers used are the following:
K-ras: F -actgaatataaacttgtggtagttggacct-
K-ras: R -tcaaagaatggtcctggacc-
B-raf exon 15: F- TCA TAA TGC TTG CTC TGA TAG GA-
B-raf exon 15: R- GGC CAA AAA TTT AAT CAG TGG A-
RFLP analysis
5-10 μl aliquots of the PCR product were digested with BstNI to reveal the presence of mutations in codon 12 of K-ras gene. Incubation was performed at 60°C for 3 hrs. The digestion products were then visualized by ethidium bromide staining under UV light after electrophoresis on a 4% (3:1 Nusieve agarose) gel (Lonza, Cologne, GmbH). The size of the PCR product for K-ras amplification is 157 bp and includes two restriction sites for BstNI restriction endonuclease. Thus the normal K-ras allele is indicated by the presence of a 114 bp band in the gel whereas the mutant K-ras allele by a 143 bp band. Heterozygous mutant cases display both bands of 143 bp and 114 bp
SSCP analysis
PCR products were screened for mutations in exons 11 and 15 of the B-raf gene. Firstly, PCR products were diluted in 10 μl formamide-dye solution (95% formamide, 20 mM EDTA, 0.05% bromophenol blue, 0.05% xylene cyanol), denatured for 7 min at 95°C and kept on ice until loaded onto a 0,5× MDE gel (BMA, USA). Electrophoresis was carried out at 3 W for 16-18 hours at 4°C. After electrophoresis, the gel was silver stained and examined for abnormal band patterns. The analysis of all cases displaying abnormal band patterns was repeated at least twice.
Sequencing
For B-raf mutation analysis the Big Dye terminator cycle sequencing kit was used. The aberrant SSCP cases were sequenced at least twice on an ABI prism 310 Genetic analyzer (Perkin-Elmer, California, USA). PCR primers were used for sequencing as well.
Statistical analysis
In the basic statistical analysis pERK and ERK nuclear or cytoplasmic expressions were treated as continuous variables, whereas hMLH1 and hMSH2 expression levels were treated as categorical variables. The relationships among ERK, pERK, hMLH1, hMSH2 expression and clinicopathological parameters, such as grade and stage were tested with non parametric tests (Spearman correlation coefficient, Kruskal-Wallis ANOVA and Mann Whitney U test), as appropriate. The respective correlations among mutation status (regarding K-ras or B-raf) and clinicopathological or immunohistochemical data were calculated using Fisher's exact test or Mann-Whitney U test. Statistical calculations were performed using the Statistical package STATA 9.0 for Windows. All results with a two-sided p ≤ 0.05 were considered statistically significant.
Results
Expression of ERK, pERK, hMLH1 and hMSH2 in colorectal carcinomas (Figure 1)
Figure 1.
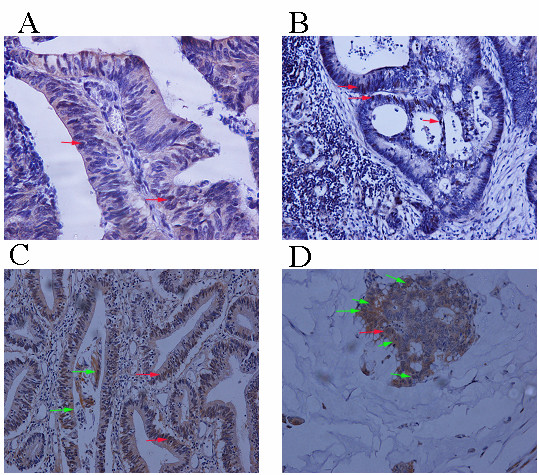
A. Grade 1, stage I colorectal carcinoma, displaying minimal nuclear pERK staining. B. Grade 3, stage III colorectal carcinoma, displaying increased nuclear pERK staining. C, D. ERK nuclear and cytoplasmic immunoreactivity in grade 1, stage II (C) and grade 3, stage III with production of extra-cellular mucin (D).
The detailed results of immunohistochemical analysis are shown in Additional file 1. The immunostaining pattern for ERK was nuclear and cytoplasmic. All cases (55/55) displayed either nuclear and/or cytoplasmic immunostaining. Nuclear immunoreactivity for ERK was detected in 85% (47/55) of cases, whereas cytoplasmic immunoreactivity in 92.7% of cases (51/55). In 43 cases (78.18%) concurrent cytoplasmic and nuclear positivity was recorded. pERK immunoreactivity was mainly nuclear found in 68.89% (31/45) of cases, while a small proportion of specimens displayed (11 cases, 24.4%) cytoplasmic positivity. In eight of these cases (17.78%) there was simultaneous nuclear and cytoplasmic pERK immunoexpression. Normal colorectal mucosa was present in 39 of the 55 examined slides and displayed membranous ERK immunoreactivity in the apical aspect of the superficial epithelium.
Nuclear immunoreactivity levels of ERK varied widely among tissue samples. We found that 13 samples had no expression (12.73%), 22 had a weak expression (40%, from 1 up to 15%), 13 had moderate expression (23.64%, from 15 up to 50%), whereas 13 had strong immunoexpression (23.64%, > 50%). Cytoplasmic immunoreactivity of ERK was in higher levels, since the majority of samples (40 in 55 cases, 72.73%) displayed strong positivity (> 50%). Most samples with strong cytoplasmic expression displayed increased nuclear expression, an observation which was confirmed with the calculation of the Spearman's correlation coefficient (rho = 0.3361, p = 0.0121).
pERK nuclear and cytoplasmic immunoexpression displayed a narrower range of variation, ranging from no expression up to 20% and 15% of tumor cells respectively. Also, there was no significant correlation between pERK nuclear and cytoplasmic expression (Spearman's correlation coefficient, p > 0.10). Normal colorectal mucosa showed minimal nuclear pERK expression whereas there was diffuse cytoplasmic pERK expression (results available in 36 cases in which there was normal mucosa adjacent to the tumor).
Additionally, there was no correlation between nuclear or cytoplasmic ERK labelling index and the presence of nuclear and/or cytoplasmic pERK immunoexpression (Mann Whitney U test, p > 0.10).
As far as hMLH1 and hMSH2 protein expression is concerned, it was preserved in 45/63 (71.43%) cases and 35/53 (66.04%) cases respectively. Adjacent normal colorectal mucosa preserved the expression of hMLH1 (available in 44 cases) and hMSH2 (available in 36 cases).
Associations between ERK, pERK, hMLH1 and hMSH2 with clinicopathological features
A significant association emerged between tumor's stage and ERK nuclear expression levels, suggesting lower expression levels (< 15%) in advanced tumors (Fisher's exact test, p = 0.049, Table 1). However, ERK nuclear immunoexpression was not correlated with histological grade, tumor location (Kruskal Wallis ANOVA, p > 0.10, Table 1) patients' gender or age (Spearman, p > 0.10, Table 1). Moreover, cytoplasmic ERK immunoreactivity was not correlated with any of the examined clinicopathological parameters (p > 0.10, Table 1).
Table 1.
Associations between nuclear ERK expression and clinicopathological parameters in 55 patients with colorectal carcinoma.
| Variables | ERK nuclear | ERK cytoplasmic expression | |||||
|---|---|---|---|---|---|---|---|
| N | Low expression group (< 15%) | High expression group (≥15%) | p-value | Low expression group (< 50%) | High expression group (≥50%) | p-value | |
| Gender | |||||||
| Male | 33 | 16 | 17 | 0.99 | 7 | 26 | 0.235 |
| Female | 22 | 10 | 12 | 8 | 14 | ||
| TNM stage | |||||||
| I | 4 | 0 | 4 | 0.049 | 2 | 2 | 0.298 |
| II-IV | 51 | 26 | 25 | 13 | 38 | ||
| Grade | |||||||
| I | 23 | 20 | 25 | > 0.10 | 10 | 33 | 0.404 |
| II/III | 8 | 4 | 4 | 3 | 5 | ||
| Tumor location | |||||||
| Right colon | 30 | 17 | 13 | 7 | 23 | 0.551 | |
| Left colon | 25 | 9 | 16 | 0.99 | 8 | 17 | |
| Median (range) | Median (range) | Median (range) | Median (range) | ||||
| Age (years) | 55 | 66.5 (51-77) | 61 (35-83) | 0.5562 | 59 (56-76) | 66.5 (35-83) | 0.5622 |
Results of Fisher's exact test.
pERK nuclear immunoreactivity was positively correlated with tumor grade (I vs II/III, Mann Whitney U test, p = 0.0113, Table 2, Figure 2). Thus, grade (II and III) tended to display higher pERK nuclear immunoreactivity levels. Accordingly, pERK nuclear immunoreactivity was mainly reported in advanced stage tumors (Fisher's exact test, p = 0.0952, Table 2), although this relationship was of marginal significance. The relationships between nuclear pERK expression levels and tumor location or patients' age, as well as pERK cytoplasmic immunoreactivity and all clinicopathological features failed to attain statistical significance (p > 0.10, Table 2). On the contrary the preservation of hMLH1 and hMSH2 expression was not correlated with tumor's stage, histological grade, tumor location or patients' age and gender (p > 0.10, table 3).
Table 2.
Associations between nuclear and cytoplasmic pERK expression and clinicopathological parameters in 45 patients with colorectal carcinoma.
| pERK nuclear LI | pERK cytoplasmic LI | ||||||
|---|---|---|---|---|---|---|---|
| N | Median | Range | P value | Median | Range | P value | |
| Gender | |||||||
| Male | 24 | 1 | 0-15 | 0 | 0-10 | ||
| Female | 21 | 1 | 0-20 | 0.6999 | 0 | 0-15 | 0.9518 |
| Histological grade | |||||||
| I | 4 | 0 | 0-0 | 0 | 0-0 | ||
| II/III | 41 | 1 | 0-20 | 0.0113 | 0 | 0-15 | 0.2504 |
| TNM stage | |||||||
| I | 5 | 0 | 0-10 | 0 | 0-0 | ||
| II-IV | 40 | 0-20 | 0.0952 | 0 | 0-15 | 0.1873 | |
| Tumor location | |||||||
| Right colon | 33 | 1 | 0-20 | 1 | 0-20 | ||
| Left colon | 12 | 1 | 0-10 | 0.3941 | 5 | 0-10 | 0.4452 |
| Age (years) | 45 | R = 0.3941, p = 0.5687 | R = 0.3153, p = 0.1529 | ||||
Results of Mann Whitney U test.
Figure 2.
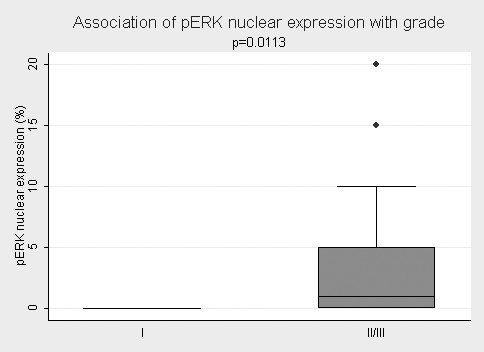
Schematical representation of the association between nuclear pERK expression and histological grade. Tumors with grade II/III tended to display higher pERK nuclear immunoreactivity compared to those with grade I (p = 0.0113, Mann- Whitney U test).
Table 3.
Associations between hMLH1 and hMSH2 expression and clinicopathological parameters in 53 and 63 patients with colorectal carcinoma respectively.
| Variables | hMLH1 expression | hMSH2 expression | ||||||
|---|---|---|---|---|---|---|---|---|
| N | Reduced (< 20%) | Preserved (≥20%) | p-value | N | Reduced (< 20%) | Preserved (≥20%) | p-value | |
| Gender | ||||||||
| Male | 34 | 10 | 24 | 0.99 | 30 | 10 | 20 | 0.99 |
| Female | 29 | 8 | 21 | 23 | 8 | 15 | ||
| TNM stage | ||||||||
| I | 5 | 1 | 4 | 0.99 | 6 | 3 | 3 | 0.397 |
| II-IV | 58 | 17 | 41 | 47 | 15 | 32 | ||
| Grade | ||||||||
| I | 8 | 1 | 7 | 0.421 | 6 | 2 | 4 | 0.99 |
| II/III | 55 | 17 | 33 | 47 | 16 | 31 | ||
| Tumor location | ||||||||
| Right colon | 36 | 11 | 25 | 33 | 11 | 22 | 0.99 | |
| Left colon | 27 | 7 | 20 | 0.782 | 20 | 7 | 13 | |
| Median (range) | Median (range) | Median (range) | Median (range) | |||||
| Age (years) | 63 | 58 (53-73) | 63(40-83) | 0.2558 | 60(45-83) | 60(40-81) | 0.9607 | |
Results of Fischer's exact test.
Associations between ERK, pERK, hMLH1 and hMSH2 expression levels with B-RAF and/or K-RAS mutational status
The results of molecular analysis are shown in Additional file 1. Of the 94 samples, 12 had a mutation in B-raf Exon 15 (Figure 3a,b) and none in Exon 11, whereas 22 had a K-ras mutation (Figure 4). In the subset of 55 specimens that were available for immunohistochemical evaluation of ERK protein tissue expression 4 had a B-raf and 14 a K-ras mutation (Table 4). Most of the cases with nuclear and cytoplasmic ERK expression did not have a K-RAS mutation (79.09%, 34/43), although this observation was not statistically significant (Table 4). The same applied to the cases with the presence of either nuclear (77.08%, 37/48) or cytoplasmic (76%, 38/50) expression. Moreover, three of the 4 cases with B-raf mutation had positive nuclear and cytoplasmic ERK expression (75%), whereas the fourth displayed only nuclear immunoreactivity. Importantly, the presence of either B-raf or K-ras mutation was marginally associated with the presence of cytoplasmic ERK expression (Fisher's exact test, p = 0.097), suggesting that the proportion of cytoplasmic ERK positivity is marginally higher in the mutated cases when compared to the unmutated ones.
Figure 3.

A. Representative SSCP* analysis of B-raf gene, exon 15 of seven colorectal cancer cells. Lanes 1, 3-7: tumor samples with normal band pattern (black arrows). Lane 2: tumor sample with the typical band pattern of a V600E mutation (arrow heads). *SSCP: Single strand conformation polymorphism. B. Sanger heterozygous analysis showing a heterozygous B-raf mutation (exon 15) at codon 600 (p.V600E, c.1799T > A).
Figure 4.
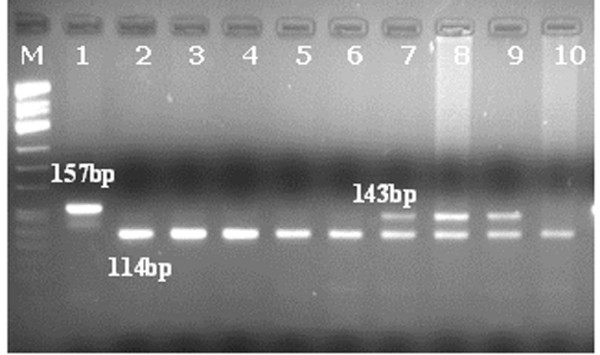
Mutational analysis of K-ras codon 12 by PCR-RFLP (digestion with BSTNI restriction endonuclease). PCR product size: 157 bp, Normal K-ras allele: 114 bp, Mutant K-ras allele: 143 bp. Heterozygous mutant case: 143 bp and 114 bp. Μ = molecular weight marker pUC Mix 8. Lane 1 = Undigested PCR product (157 bp). Lanes 7-9 = Mutant samples (143 bp and 114 bp). Lanes 2-6 & 10 = Normal samples (114 bp).
Table 4.
Correlations of the presence of K-ras and B-raf mutations (exon 15) with immunohistochemical results in patients for whom staining data were available.
| K-ras Mutation | B-raf Exon 15 mutation | |||||
|---|---|---|---|---|---|---|
| Absent | Present | p | Absent | Present | p | |
| ERK nuclear expression (n = 55) | ||||||
| Absent | 4 | 3 | 7 | 0 | ||
| Present | 37 | 11 | 0.354 | 44 | 4 | 0.99 |
| ERK cytoplasmic expression (n = 55) | ||||||
| Absent | 3 | 2 | 4 | 1 | ||
| Present | 38 | 12 | 0.592 | 47 | 3 | 0.325 |
| ERK nuclear and cytoplasmic expression (n = 55) | ||||||
| Absent | 7 | 5 | 11 | 1 | ||
| Present | 34 | 9 | 0.259 | 40 | 3 | 0.99 |
| pERK nuclear expression (n = 65) | ||||||
| Absent | 11 | 3 | 13 | 1 | ||
| Present | 24 | 7 | 0.99 | 27 | 4 | 0.99 |
| pERK cytoplasmic expression(n = 65) | ||||||
| Absent | 27 | 7 | 29 | 5 | ||
| Present | 8 | 3 | 0.687 | 11 | 0 | 0.313 |
| pERK nuclear and cytoplasmic expression (n = 65) | ||||||
| Absent | 29 | 8 | 32 | 5 | ||
| Present | 6 | 2 | 0.99 | 8 | 0 | 0.357 |
| hMLH1 expression (n = 63) | ||||||
| Reduced | 13 | 5 | 15 | 3 | ||
| Preserved | 33 | 12 | 0.99 | 41 | 4 | 0.397 |
| hMSH2 expression (n = 53) | ||||||
| Reduced | 12 | 6 | 13 | 5 | ||
| Preserved | 24 | 11 | 0.99 | 32 | 3 | 0.104 |
Results of Fischer's exact test.
In the subset of 45 specimens that were available for pERK immunohistochemical assessment, only 5 had a B-raf mutation whilst 10 had a K-ras mutation (Table 4). There was no difference in the distribution of K-ras mutations among the cases that showed nuclear and/or cytoplasmic pERK expression. Furthermore, all cases displayng cytoplasmic (11 cases) or both cytoplasmic and nuclear (8 cases) pERK expression had a wild-type B-raf. However, the presence of pERK immunoexpression, either nuclear or cytoplasmic was not correlated with the presence of either B-raf or K-ras mutations (Mann Whitney U test and Fisher's exact test, p > 0.10).
In the subset of the 63 patients for whom hMLH1 expression was available, 17 had a K-ras (12 of which preserved hMLH1 expression) and 7 a B-raf mutation (4 of which preserved hMLH1 expression) (Table 4). Accordingly, 64.71% of the cases (11/17) with K-ras mutation and 37.5% of the cases (3/8) with B-raf mutation preserved hMSH2 immunoreactivity (Table 4). However, the presence of hMLH1 or hMSH2 immunoexpression, was not correlated with the presence of B-raf and/or K-ras mutations (p > 0.10). Finally, the presence of K-ras and/or B-raf mutations could not be related to grade and stage (p > 0.10).
Discussion
Various molecular markers have been proposed for the classification of colon cancer with regard to clinical course and outcome, including cell cycle as well as apoptotic regulators (c-FLIP, Ki67, COX-2) [22,23]. Deregulation of the MAPK signalling pathway has often been associated with oncogenic transformation [24]. In this regard, there is accumulating evidence involving ERK activation in the tumorigenesis of various human cancers such as prostate, breast, colon and ovary [25-29]. Furthermore, activating B-raf mutations are frequently detected in different tumor types such as melanomas, thyroid and colon carcinomas implying their significance as potential targets for anticancer treatment [30,31]. In the present study we used immunohistochemistry to examine the expression of total and activated ERK1/2 in a panel of 94 colorectal carcinomas in parallel with the expression of two MMR proteins (hMLh1 and hMSH2), as well as K-ras and B-raf mutations, which may lead to constitutive activation of MAPK pathway.
To the best of our knowledge, this appears to be the first study examining ERK expression in mismatch repair deficient and mismatch repair proficient colorectal cancer simultaneously with upstream gene alterations such as K-ras and B-raf mutations, which contribute to MAPK activation. In our cohort, ERK immunoreactivity was observed in the cytoplasm as well as in the nucleus in the majority of the cases (93% and 91% respectively). More than 78% of the examined cases displayed concurrent ERK nuclear and cytoplasmic immunoreactivity, in concordance with previous observations [29,30]. Increased levels of nuclear ERK positivity correlated with strong cytoplasmic ERK expression. The observed correlation between nuclear and cytoplasmic expression as well as the concurrent nuclear and cytoplasmic immunolocalization of ERK in the present series is compatible with its multiple functions targeting proteins localized in the cytoplasm as well as the nucleus of the cell. Moreover, pERK immunoreactivity was predominantly nuclear (68%) whereas cytoplasmic positivity was found in a subset of tumors (24%). This staining pattern is in line with previous investigations in colon as well as other neoplasms, namely, endometrial, head and neck tumors and melanomas [27,29,32,33]. Additionally, total nuclear or cytoplasmic ERK staining was independent of nuclear or cytoplasmic pERK status, in our cohort. This observation is in agreement with previous studies in non small cell lung and endometrial cancer as well as in melanomas and could been explained by the hypothesis that pERK immunopositivity could arise due to ERK hyperactivation rather than overexpression [29,30,32].
Furthermore we show that high nuclear, but not cytoplasmic, ERK immunopositivity is correlated with tumor stage (Table 1). Interestingly, our results demonstrate that nuclear pERK expression parallels tumor grade and tumor stage (Table 2, Figure 2) in keeping with findings of previous studies in NSCLC and prostate cancer [30,34]. This observation is in contrast with a previous investigation [25] in which pERK expression was not correlated with tumor stage, whereas there are other studies in various other tumors (breast, pancreatic, endometrial and ovarian cancer) that have failed to substantiate a positive correlation between pERK expression and classical clinicopathological parameters [26,32,35-37]. Taking into account that in our study normal colorectal mucosa displays minimal nuclear pERK expression, our results speak in favor of the potential role of this molecule in tumor evolution as well as, in the acquisition of a more aggressive phenotype in colorectal carcinogenesis. Along this line, it has been suggested that constitutively active ERKs are capable of affecting gene expression, being able to influence many of the hallmarks of carcinogenesis [35]. It could be hypothesised that as colorectal cancer progresses to a more advanced disease, an increase in the activation of the MAP kinase signal transduction pathway occurs [34]. Unfortunately, the statistical power of the present investigation is reduced due to the small cohort of pERK positive cases. Further studies of larger cohorts are warranted to confirm our results. Along this line, pERK expression has been shown to be associated with poor prognosis in colorectal carcinomas [27]. It has been speculated that the activation of the MAPK pathway initiates cellular processes, which could result in either favorable or worse clinical outcome, a fact due to the complex signal transduction network ERKs are involved in. In fact, adverse effects can be observed, depending on the intensity and duration of the promitogenic signal [10].
The aforementioned findings are quite significant in view of the fact that protein kinases represent a group of molecular targets characterized by a cancer specific potential, allowing the development of new generation chemotherapeutic agents acting as kinase inhibitors. The first oral multi-kinase inhibitor that targets Raf kinases have already been approved for the treatment of renal cell cancer, whereas these factors display a broad spectrum antitumor activity in colon, breast and non-small-cell lung cancer in xenograft models and also hepatocellular carcinoma and sarcoma [38,39]. In this context, the presence of B-raf mutations has been suggested as a possible surrogate marker of sensitivity to those drugs which target the ERK pathway at the level of Raf kinase [38,39].
The frequency of K-ras mutations detected in this study (about 23% of the examined cases) was comparable to that found in previous reports (Table 4) [39-41]. Furthermore, the presence of B-raf mutations in colorectal cancer is estimated to be about 10% of unselected colorectal cancers [42-44]. In particular, B-raf T1799A mutation (V600E) has been reported in 4% of microsatellite stable (MSS) tumors whereas in microsatellite unstable tumors the percentage rises up to 27-52% [42,44-46]. By analogy to those investigations we detected V600E B-raf mutations in about 7% of MSS tumors and in 21% of MSI unstable tumors. Interestingly, our cases exhibited only either B-raf or K-ras mutations in accordance with previous observations suggesting that these are mutually exclusive defects that probably exert equivalent effects in tumorigenesis [39,46].
Moreover, in our series the expression levels of total and activated ERK1/2 were independent of the mutation status of B-raf and K-ras genes. These results are in favor of the view that constitutive pERK activation occurs in a K-ras or B-raf -independent manner in a large subset of primary colon cancer cases. Recently, several negative regulators of the MAPK signalling pathway upstream of ERK at the level of Raf were identified, including Sprouty and Spred. Activation of these negative regulators inhibits phosphorylation of ERK1/2, even in the presence of mutation in K-ras gene [32,47]. This finding has also previously been observed in ulcerative colitis-related carcinomas [48]. The genetic nature of constitutive activation of the RAS/RAF/MEK/ERK pathway in colorectal tumors with no B-raf or K-ras mutation remains unknown, although it may in part due to increased activity of growth factor receptor induced cell proliferation pathways. It could be speculated that in cancer constitutive activation of MAP kinase could be triggered by upstream oncogenic regulators due to the presence of paracrine/autocrine growth factor stimulation, rather than Ras or B-raf mutations or components of the various other signal transduction pathways that interact with MAPK, since the mutation of K-ras and B-raf obviously constitutes one of multiple ways to activate this pathway.
In sporadic colorectal carcinogenesis B-raf mutations like K-ras mutations appear to occur early at the transition from small to medium size adenoma and are extremely frequent in so-called serrated adenomas [49]. According to the MSI colorectal pathway, MSI in sporadic tumors has been suggested to be mostly due to hypermethylation of the promoters of MMR genes and is correlated with B-raf mutations (44). However, in our study B-raf mutations were not correlated with loss of hMLH1 or hMSH2 protein, suggesting that the B-raf mutated cases of our cohort may belong to more than one colorectal carcinogenesis pathways.
Conclusions
In this study, we have shown that mutations of the B-raf gene are not associated with mismatch-repair deficiency through loss of hMLH1 or hMSH2 expression. Disruption of the MAP kinase pathway-either through K-ras or B-raf mutation-was detected in 37% of all the examined cases, although the overexpression of total and activated ERK1/2 was not correlated with the mutational status of K-ras or B-Rraf genes. Interestingly, we present evidence that the expression of activated ERK expression parallels histological grade and stage in colon carcinomas, thus being implicated in tumor invasiveness as well as in the acquisition of a more aggressive phenotype. Our findings encourage prospective investigations to further elucidate and validate the potential role of pERK as a prognostic factor and therapeutic target in colon carcinogenesis.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
The manuscript was edited by GL, AAS Immunohistochemical staining was performed by PP and evaluated by GL, PK and NK. Molecular analysis was organized by AAS and performed by FG, MK and AS. Genomic DNA isolation was performed by MK. Statistical analysis was done by GL (MSc in Biostatistics). Clinical data of the patients were collected by NVM. The respective literature was collected by GA. The supervision and organization of the research program was performed by EP and PK. All the authors have read and approved the final manuscript.
Supplementary Material
Supplementary table presenting all samples with their clinical information and the staining results. This a comprehensive table in which all samples of the present study are listed along with their clinical information (i.e. age, gender) and the staining results of the examined proteins.
Contributor Information
Georgia Levidou, Email: glevidou@yahoo.gr.
Angelica A Saetta, Email: asaetta@med.uoa.gr.
Fanie Gigelou, Email: fgigelou@yahoo.gr.
Maria Karlou , Email: mary_karlou@yahoo.gr.
Polyanthi Papanastasiou, Email: pollyipapa@yahoo.gr.
Angeliki Stamatelli, Email: agstam@med.uoa.gr.
Nikolaos Kavantzas, Email: nkavan@med.uoa.gr.
Nikolaos V Michalopoulos, Email: nmichal@med.uoa.gr.
George Agrogiannis, Email: agrojohn@med.uoa.gr.
Efstratios Patsouris, Email: epatsour@med.uoa.gr.
Penelope Korkolopoulou, Email: pkorkol@med.uoa.gr.
References
- World Health Organization Fact Sheet No 297, October 2011 (Accessed December 2011)
- Narayan S, Roy D. Role of APC and DNA mismatch repair genes in the development of colorectal cancers. Mol Cancer. 2003;12(2):41. doi: 10.1186/1476-4598-2-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Hong SW, Shin SJ, Kim YM, Rhee Y, Jeon BI, Moon WC, Oh MR, Lim SK. Papillary thyroid carcinoma associated with familial adenomatous polyposis: molecular analysis of pathogenesis in a family and review of the literature. Endocr J. 2004;51:317–23. doi: 10.1507/endocrj.51.317. [DOI] [PubMed] [Google Scholar]
- Ahnen DJ. The American College of Gastroenterology Emily Couric Lecture-the adenoma-carcinoma sequence revisited: has the era of genetic tailoring finally arrived? Am J Gastroenterol. 2011;106(2):190–8. doi: 10.1038/ajg.2010.423. [DOI] [PubMed] [Google Scholar]
- Rosty C, Parry S, Young JP. Serrated polyposis: an enigmatic model of colorectal cancer predisposition. Patholog Res Int. 2011;2011:157073S. doi: 10.4061/2011/157073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Farraye A, Mack C, Posnik O, O'Brien MJ. "B-RAF and K-RAS mutations in hyperplastic polyps and serrated adenomas of the colorectum: relationship to histology and CpG island methylation status,". American Journal of Surgical Pathology. 2004;28(11):1452–1459. doi: 10.1097/01.pas.0000141404.56839.6a. [DOI] [PubMed] [Google Scholar]
- Chan AOO, Issa JPJ, Morris JS, Hamilton SR, Rashid A. "Concordant CpG islandmethylation in hyperplastic polyposis,". American Journal of Pathology. 2002;160(2):529–536. doi: 10.1016/S0002-9440(10)64872-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCubrey JA, Steelman LS, Abrams SL, Lee JT, Chang F, Bertrand FE, Navolanic PM, Terrian DM, Franklin RA, D'Assoro AB, Salisbury JL, Mazzarino MC, Stivala F, Libra M. mRoles of the RAF/MEK/ERK and PI3K/PTEN/AKT pathways in malignant transformation and drug resistance. nAdv Enzyme Regul. 2006;46:249–79. doi: 10.1016/j.advenzreg.2006.01.004. [DOI] [PubMed] [Google Scholar]
- Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev. 2001;22:53–83. doi: 10.1210/er.22.1.53. [DOI] [PubMed] [Google Scholar]
- Meloche S, Pouysségur J. The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene. 2007;26:3227–39. doi: 10.1038/sj.onc.1210414. [DOI] [PubMed] [Google Scholar]
- Mercer K, Chiloeches A, Huser M, Kiernan M, Marais R, Pritchard C. ERK signalling and oncogene transformation are not impaired in cells lacking A-Raf. Oncogene. 2002;21:347–55. doi: 10.1038/sj.onc.1205101. [DOI] [PubMed] [Google Scholar]
- Chen CH, Wang WJ, Kuo JC, Tsai HC, Lin JR, Chang ZF, Chen RH. Bidirectional signals transduced by DAPK-ERK interaction promote the apoptotic effect of DAPK. EMBO J. 2005;24:294–304. doi: 10.1038/sj.emboj.7600510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. doi: 10.1016/S0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J, Hargrave D, Pritchard-Jones K, Maitland N, Chenevix-Trench G, Riggins GJ, Bigner DD, Palmieri G, Cossu A, Flanagan A, Nicholson A, Ho JW, Leung SY, Yuen ST, Weber BL, Seigler HF, Darrow TL, Paterson H, Marais R, Marshall CJ, Wooster R, Stratton MR, Futreal PA. Mutations of the B-RAF gene in human cancer. Nature. 2002;417:949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- Hoshino R, Chatani Y, Yamori T, Tsuruo T, Oka H, Yoshida O, Shimada Y, Ari-i S, Wada H, Fujimoto J, Kohno M. Constitutive activation of the 41-/43-kDa mitogen-activated protein kinase signaling pathway in human tumors. Oncogene. 1999;18:813–22. doi: 10.1038/sj.onc.1202367. [DOI] [PubMed] [Google Scholar]
- Bosman FT, Carneiro F, Hruban RH, Theise ND. WHO Classification of Tumors of the digestive system. 4. 2009. pp. 131–181. [Google Scholar]
- Saetta AA, Korkolopoulou P, Karlou M, Levidou G, Goudopoulou A, Thymara I, Stamatelli A, Tzivras M, Michalopoulos NV, Thomas-Tsagli E, Patsouris E. TGF-betaRII, BAX, IGFIIR, caspase-5, hMSH3 and hMSH6 alterations are not associated with microsatellite instability or p53 mutations in invasive urothelial carcinoma of the urinary bladder. Pathology. 2007;39(4):425–32. doi: 10.1080/00313020701444457. [DOI] [PubMed] [Google Scholar]
- Jin TX, Furihata M, Yamasaki I, Kamada M, Liang SB, Ohtsuki Y, Shuin T. Human mismatch repair gene (hMSH2) product expression in relation to recurrence of transitional cell carcinoma of the urinary bladder. Cancer. 1999;85(2):478–84. doi: 10.1002/(SICI)1097-0142(19990115)85:2<478::AID-CNCR28>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Hameed F, Goldberg PA, Hall P, Algar U, van Wijk R, Ramesar R. Immunohistochemistry detects mismatch repair gene defects in colorectal cancer. Colorectal Dis. 2006;8(5):411–7. doi: 10.1111/j.1463-1318.2006.00956.x. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Taguchi K, Baba H, Endo K, Kohnoe S, Okamura T, Maehara Y. Loss of protein expression of hMLH1 and hMSH2 with double primary carcinomas of the stomach and colorectum. Oncol Rep. 2006;16(1):41–7. [PubMed] [Google Scholar]
- Ashktorab H, Brim H, Al-Riyami M, Date A, Al-Mawaly K, Kashoub M, Al-Mjeni R, Smoot DT, Al-Moundhri M, Al-Hashemi S, Ganguly SS, Raeburn S. Sporadic colon cancer: mismatch repair immunohistochemistry and microsatellite instability in Omani subjects. Dig Dis Sci. 2008;53(10):2723–31. doi: 10.1007/s10620-007-0189-3. [DOI] [PubMed] [Google Scholar]
- Kumar R. Commentary: targeting colorectal cancer through molecular biology. Semin Oncol. 2005;32:S37–9. doi: 10.1053/j.seminoncol.2005.06.012. [DOI] [PubMed] [Google Scholar]
- Korkolopoulou P, Saetta AA, Levidou G, Gigelou F, Lazaris A, Thymara I, Scliri M, Bousboukea K, Michalopoulos NV, Apostolikas N, Konstantinidou A, Tzivras M, Patsouris E. c-FLIP expression in colorectal carcinomas: association with Fas/FasL expression and prognostic implications. Histopathology. 2007;51:150–6. doi: 10.1111/j.1365-2559.2007.02723.x. [DOI] [PubMed] [Google Scholar]
- Seger R, Krebs EG. The MAPK signaling cascade. FASEB J. 1995;9:726–35. [PubMed] [Google Scholar]
- McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M, Tafuri A, Stivala F, Libra M, Basecke J, Evangelisti C, Martelli AM, Franklin RA. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta. 2007;1773:1263–84. doi: 10.1016/j.bbamcr.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Givant-Horwitz V, Davidson B, Lazarovici P, Schaefer E, Nesland JM, Trope CG, Reich R. Mitogen-activated protein kinases (MAPK) as predictors of clinical outcome in serous ovarian carcinoma in effusions. Gynecol Oncol. 2003;91:160–72. doi: 10.1016/S0090-8258(03)00434-7. [DOI] [PubMed] [Google Scholar]
- Schmitz KJ, Wohlschlaeger J, Alakus H, Bohr J, Stauder MA, Worm K, Winde G, Schmid KW, Baba HA. Activation of extracellular regulated kinases (ERK1/2) but not AKT predicts poor prognosis in colorectal carcinoma and is associated with K-ras mutations. Virchows Arch. 2007;450:151–9. doi: 10.1007/s00428-006-0342-y. [DOI] [PubMed] [Google Scholar]
- Ji Z, Flaherty KT, Tsao H. Targeting the RAS pathway in melanoma. Trends Mol Med. 2011. [DOI] [PMC free article] [PubMed]
- Jorgensen K, Holm R, Maelandsmo GM, Florenes VA. Expression of activated extracellular signal-regulated kinases 1/2 in malignant melanomas: relationship with clinical outcome. Clin Cancer Res. 2003;9:5325–31. [PubMed] [Google Scholar]
- Vicent S, Lopez-Picazo JM, Toledo G, Lozano MD, Torre W, Garcia-Corchon C, Quero C, Soria JC, Martin-Algarra S, Manzano RG, Montuenga LM. ERK1/2 is activated in non-small-cell lung cancer and associated with advanced tumors. Br J Cancer. 2004;90:1047–52. doi: 10.1038/sj.bjc.6601644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karhoff D, Sauer S, Schrader J, Arnold R, Fendrich V, Bartsch DK, Hörsch D. Rap1/B-raf signaling is activated in neuroendocrine tumors of the digestive tract and Raf kinase inhibition constitutes a putative therapeutic target. Neuroendocrinology. 2007;85:45–53. doi: 10.1159/000100508. [DOI] [PubMed] [Google Scholar]
- Mizumoto Y, Kyo S, Mori N, Sakaguchi J, Ohno S, Maida Y, Hashimoto M, Takakura M, Inoue M. Activation of ERK1/2 occurs independently of K-RAS or B-RAF status in endometrial cancer and is associated with favorable prognosis. Cancer Sci. 2007. pp. 652–8. [DOI] [PMC free article] [PubMed]
- Albanell J, Codony-Servat J, Rojo F, Del Campo JM, Sauleda S, Anido J, Raspall G, Giralt J, Rosello J, Nicholson RI, Mendelsohn J, Baselga J. Activated extracellular signal-regulated kinases: association with epidermal growth factor receptor/transforming growth factor alpha expression in head and neck squamous carcinoma and inhibition by anti-epidermal growth factor receptor treatments. Cancer Res. 2001;61:6500–10. [PubMed] [Google Scholar]
- Gioeli D, Mandell JW, Petroni GR, Frierson HF Jr, Weber MJ. Activation of mitogen-activated protein kinase associated with prostate cancer progression. Cancer Res. 1999;59:279–84. [PubMed] [Google Scholar]
- Lugli A, Zlobec I, Minoo P, Baker K, Tornillo L, Terracciano L, Jass JR. Role of the mitogen-activated protein kinase and phosphoinositide 3-kinase/AKT pathways downstream molecules, phosphorylated extracellular signal-regulated kinase, and phosphorylated AKT in colorectal cancer-a tissue microarray-based approach. Hum Pathol. 2006;37:1022–31. doi: 10.1016/j.humpath.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Adeyinka A, Nui Y, Cherlet T, Snell L, Watson PH, Murphy LC. Activated mitogen-activated protein kinase expression during human breast tumorigenesis and breast cancer progression. Clin Cancer Res. 2002;8:1747–53. [PubMed] [Google Scholar]
- Chadha KS, Khoury T, Yu J, Black JD, Gibbs JF, Kuvshinoff BW, Tan D, Brattain MG, Javle MM. Activated Akt and Erk expression and survival after surgery in pancreatic carcinoma. Ann Surg Oncol. 2006;13:933–9. doi: 10.1245/ASO.2006.07.011. [DOI] [PubMed] [Google Scholar]
- Gollob JA, Wilhelm S, Carter C, Kelley SL. Role of Raf kinase in cancer: therapeutic potential of targeting the Raf/MEK/ERK signal transduction pathway. Semin Oncol. 2006;33:392–406. doi: 10.1053/j.seminoncol.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Pratilas CA, Solit DB. Targeting the mitogen-activated protein kinase pathway: physiological feedback and drug response. Clin Cancer Res. 2010;16(13):3329–34. doi: 10.1158/1078-0432.CCR-09-3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikehara N, Semba S, Sakashita M, Aoyama N, Kasuga M, Yokozaki H. B-RAF mutation associated with dysregulation of apoptosis in human colorectal neoplasms. Int J Cancer. 2005;115:943–50. doi: 10.1002/ijc.20957. [DOI] [PubMed] [Google Scholar]
- Yuen ST, Davies H, Chan TL, Ho JW, Bignell GR, Cox C, Stephens P, Edkins S, Tsui WW, Chan AS, Futreal PA, Stratton MR, Wooster R, Leung SY. Similarity of the phenotypic patterns associated with B-RAF and K-RAS mutations in colorectal neoplasia. Cancer Res. 2002;62:6451–5. [PubMed] [Google Scholar]
- Samowitz WS, Sweeney C, Herrick J, Albertsen H, Levin TR, Murtaugh MA, Wolff RK, Slattery ML. Poor survival associated with the B-RAF V600E mutation in microsatellite-stable colon cancers. Cancer Res. 2005;65:6063–9. doi: 10.1158/0008-5472.CAN-05-0404. [DOI] [PubMed] [Google Scholar]
- Li WQ, Kawakami K, Ruszkiewicz A, Bennett G, Moore J, Iacopetta B. B-RAF mutations are associated with distinctive clinical, pathological and molecular features of colorectal cancer independently of microsatellite instability status. Mol Cancer. 2006;5:2. doi: 10.1186/1476-4598-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubomierski N, Plotz G, Wormek M, Engels K, Kriener S, Trojan J, Jungling B, Zeuzem S, Raedle J. B-RAF mutations in colorectal carcinoma suggest two entities of microsatellite-unstable tumors. Cancer. 2005;104:952–61. doi: 10.1002/cncr.21266. [DOI] [PubMed] [Google Scholar]
- Oliveira C, Pinto M, Duval A, Brennetot C, Domingo E, Espín E, Armengol M, Yamamoto H, Hamelin R, Seruca R, Schwartz S Jr. B-RAF mutations characterize colon but not gastric cancer with mismatch repair deficiency. Oncogene. 2003;22:9192–6. doi: 10.1038/sj.onc.1207061. [DOI] [PubMed] [Google Scholar]
- Koinuma K, Shitoh K, Miyakura Y, Furukawa T, Yamashita Y, Ota J, Ohki R, Choi YL, Wada T, Konishi F, Nagai H, Mano H. Mutations of B-RAF are associated with extensive hMLH1 promoter methylation in sporadic colorectal carcinomas. Int J Cancer. 2004;108:237–42. doi: 10.1002/ijc.11523. [DOI] [PubMed] [Google Scholar]
- Rajagopalan H, Bardelli A, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature. 2002;418:934. doi: 10.1038/418934a. [DOI] [PubMed] [Google Scholar]
- Aust DE, Terdiman JP, Willenbucher RF, Chang CG, Molinaro-Clark A, Baretto GB, Loehrs U, Waldman FM. The APC/beta-catenin pathway in ulcerative colitis-related colorectal carcinomas: a mutational analysis. Cancer. 2002;94(5):1421–7. doi: 10.1002/cncr.10334. [DOI] [PubMed] [Google Scholar]
- Yuen ST, Davies H, Chan TL, Ho JW, Bignell GR, Cox C, Stephens P, Edkins S, Tsui WW, Chan AS, Futreal PA, Stratton MR, Wooster R, Leung SY. Similarity of the phenotypic patterns associated with B-RAF and K-RAS mutations in colorectal neoplasia. Cancer Res. 2002;62(22):6451–5. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary table presenting all samples with their clinical information and the staining results. This a comprehensive table in which all samples of the present study are listed along with their clinical information (i.e. age, gender) and the staining results of the examined proteins.