

Abstract
Trypanothione synthetase (TryS) is essential for the survival of the protozoan parasite Trypanosoma brucei, which causes human African trypanosomiasis. It is one of only a handful of chemically validated targets for T. brucei in vivo. To identify novel inhibitors of TbTryS we screened our in-house diverse compound library that contains 62 000 compounds. This resulted in the identification of six novel hit series of TbTryS inhibitors. Herein we describe the SAR exploration of these hit series, which gave rise to one common series with potency against the enzyme target. Cellular studies on these inhibitors confirmed on-target activity, and the compounds have proven to be very useful tools for further study of the trypanothione pathway in kinetoplastids.
Keywords: antiprotozoal agents, drug design, Trypanosoma brucei, trypanothione synthetase
Introduction
Human African trypanosomiasis (HAT), or sleeping sickness, is endemic in sub-Saharan Africa, claiming the lives of about 30 000 people every year and putting approximately 60 million people at risk of infection.1 HAT is a progressive and fatal disease caused by the protozoan parasites Trypanosoma brucei gambiense and T. b. rhodesiense, which are transmitted to the human host by the bite of the tsetse fly. If left untreated the disease progresses to the central nervous system and is ultimately fatal. There is a clinical need for more effective drug therapies. Current therapies are toxic and have inappropriate treatment regimens for a rural African setting. There are also problems with treatment failures.1–3
Differences in metabolic pathways have been discovered between parasite and host, which may be exploited for drug discovery programmes. An example of such a difference is found in thiol metabolism and the response of T. brucei to oxidative stress.4–8 Studies have shown that trypanosomatid parasites are uniquely dependent on trypanothione (N1,N8-bis(glutathionyl)spermidine) as their principal thiol, in contrast to most other organisms (including their mammalian hosts) that use glutathione (γ-l-glutamyl-l-cysteinylglycine, GSH).9 In T. brucei trypanothione is synthesised from GSH and spermidine (Spd) by an ATP-dependent C–N ligase, trypanothione synthetase (TryS; EC 6.3.1.9), with N1- and N8-glutathionylspermidine as intermediates.10, 11
Selective inhibition of the trypanothione pathway with chemical agents (targeting trypanothione reductase, tryparedoxin, and tryparedoxin peroxidise) or classical gene knockout studies have shown a clear trypanocidal effect.12, 13 TbTryS has also been genetically validated as a drug target, with RNAi and gene knockout studies confirming that TbTryS is essential for T. brucei growth in both bloodstream and procyclic forms, and that there is no alternative bypass mechanism available to the parasite.14, 15
Before commencing a drug discovery programme, TbTryS was assessed for its suitability as a drug target using the traffic light scoring system that we have developed in house.16 The assessment indicated TbTryS is an attractive target for drug development, especially as it is unlikely to have resistance or toxicity issues, as there is no obvious bypass metabolism or equivalent enzyme in humans.10 The main concern was the potential druggability of the target. Because the active site of TbTryS is large enough to accommodate trypanothione and precursors, this may be an issue if the active site is a large featureless pocket, as is observed in T. brucei trypanothione reductase (TbTryR).17 However, the structure of TryS from Leishmania major suggests this is not the case,18 and the potential to co-crystallise ligands with the protein to inform a chemistry programme was a distinct advantage. Importantly, TbTryS is a bifunctional enzyme, which catalyzes the biosynthesis and hydrolysis of the GSH–Spd adduct trypanothione. The two catalytic domains are separate in Leishmania. The N-terminal domain is a cysteine-containing amidohydrolase/peptidase amidase site, with the C-terminal ATP grasp domain responsible for the synthetase activity of the enzyme.18
Figure 1 shows the only previously disclosed inhibitor of TbTryS, compound 1.19 Whilst a valuable tool molecule, the optimisation and development of this phosphinate inhibitor into a potential clinical candidate is limited due to the peptidic nature of such a compound, with a high polar surface area (PSA), and charges at physiological pH (which are detrimental for cellular penetration, metabolic stability, bioavailability, and blood–brain barrier permeability).
Figure 1.
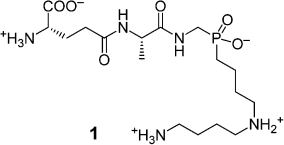
Previously published TbTryS inhibitor: compound 1, Kiapp: 500–1200 nm.
Herein we describe a medicinal chemistry programme to develop inhibitors of TbTryS, which gave rise to some potent compounds. We recently reported the biological experiments to chemically validate TbTryS using five of these compounds: 9, 20, 71, 84, and 89.15, 20
Results and Discussions
High-throughput screening
As previously described, a high-throughput screening assay for TbTryS was developed and validated.20 TbTryS was then screened against a 62 000 compound diversity set at single-point concentration (30 μm). This gave rise to >720 hits (compounds with TbTryS percentage inhibition >33 % at 30 μm). These hits were clustered and filtered down to 174 compounds that underwent potency testing (10-point, half-log dilution dose–response curves from which an accurate IC50 could be calculated). This gave rise to the six putative hit series, plus a number of singletons. Appropriate hits were then re-purchased to confirm identity and activity. A round of purchasing (where available) and synthesis of analogues was initiated to validate the series and to investigate the SAR around these potential hit series to assess optimisation towards lead series.
Table 1 shows the hit series identified from the HTS screen, and the ligand efficiencies of compounds from these series.21 Although there are six distinct chemical series shown in Table 1, the series can be clustered into two distinct groups that share common pharmacophoric features. Attempts were made to co-crystallise hit ligands in the protein to obtain an X-ray crystal structure showing ligands in the binding domain, but unfortunately these have been unsuccessful so far.
Table 1.
Hit series clusters identified from HTS.
| Series | Structure | LE[a] |
|---|---|---|
| 1 a | 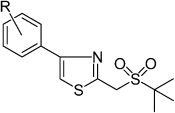 |
0.44 |
| 1 b | 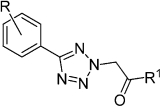 |
0.45 |
| 1 c | 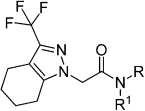 |
0.33 |
| 2 a | 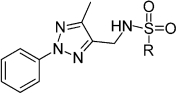 |
0.33 |
| 2 b | 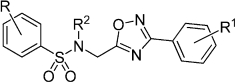 |
0.30 |
| 2 c | 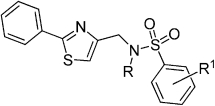 |
0.29 |
Ligand efficiency, calculated as −0.6×ln(IC50)/(heavy atom count).21
Initial hit exploration around series/group 1
Group 1 series were identified as having a common pharmacophore with two hydrogen bond acceptors (HBA) in 1,5 relationship, with one of the HBAs from a heterocyclic ring. The pharmacophore also includes regions of hydrophobicity, indicating a potential lipophilic pocket.
Hit series 1 a: thiazole methylene sulfone
The synthetic route to prepare this series involved the condensation of a thioamide with an appropriate α-bromoketone (Table 2), based on the methodology of Dunn et al.22 The corresponding α-bromoketones could be bought or synthesised from the corresponding acetyl compound (see Experimental Section below for more details). Activities of hit series 1 a compounds against TbTryS are listed in Table 2.
Table 2.
SAR around hit series 1 a.
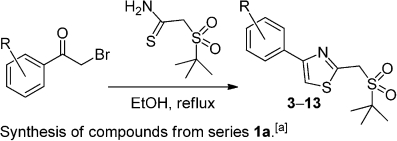 | |||||
|---|---|---|---|---|---|
| Compd | R | IC50 [μm][b] | Compd | R | IC50 [μm][b] |
| 3 | H | 1.2 | 9 | 3-F | 0.4 |
| 4 | 2-CH3 | 3.2 | 10 | 4-CH3 | >100 |
| 5 | 2-F | 1.1 | 11 | 4-CF3 | >100 |
| 6 | 3-OCH3 | 12.0 | 12 | 3,4-di-Cl | 2.5 |
| 7 | 3-CF3 | 1.1 | 13 | 3,4-di-F | 0.4 |
| 8 | 3-Cl | 0.4 | |||
[a] Yields: 60–87 %; the synthesis of compound 9 was previously described by Torrie et al.20 [b] IC50 values against TbTryS.
Exploration around the scaffold of 1 a (Table 2) shows that 2-substitution with a small substituent, methyl (compound 4) and fluorine (compound 5), is tolerated without loss of potency. An electron-donating substituent at the 3-position, as in compound 6, gave a 10-fold loss in potency relative to unsubstituted compound 3; however, all potency was regained if the 3-position group was changed to an electron-withdrawing group, such as trifluoromethyl (compound 7). In addition, a slight improvement in potency was observed for the 3-F (9) and 3-Cl (8) derivatives (IC50: 0.4 μm). These analogues showed sub-micromolar potency is achievable within this hit series. Substitution at the 4-position was not tolerated with either an electron-withdrawing or -donating group. Both methyl (10) and trifluoromethyl (11) were found to be inactive. However, 3,4-disubstitution with an electron-withdrawing substituent at the 3-position restored potency (compounds 12 and 13), although ligand efficiency was decreased.
Hit series 1 b: tetrazole methylene carbonyl
In this series, the thiazole subunit is replaced by a tetrazole, and the sulfone by a carbonyl group. The tetrazoles were alkylated with α-bromoketones (Table 3). Amide analogue 23 was synthesised via an amide coupling of the corresponding acid, which in turn was obtained from saponification of the ethyl ester.
Table 3.
SAR around hit series 1 b.
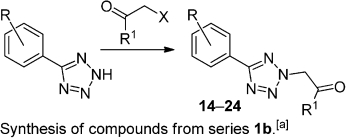 | |||
|---|---|---|---|
| Compd | R | R1 | TbTryS IC50 [μm] |
| 14 | H | tBu | 8.8 |
| 15 | 4-CF3 | tBu | >100 |
| 16 | 3-Cl, 4-CH3 | tBu | 7.2 |
| 17 | 3-F | tBu | 1.5 |
| 18 | 3-CF3 | tBu | 3.5 |
| 19 | 3,5-di-F | tBu | 1.9 |
| 20 | 3,5-di-Cl | tBu | 0.3 |
| 21 | 3,5-di-Cl | CH3 | 6.5 |
| 22 | 3,5-di-Cl | CH2CH3 | 2.7 |
| 23 | 3,5-di-Cl | N(CH3)2 | 0.6 |
| 24 | 3,5-di-F | Ph | 45 |
[a] Conditions: NaI, K2CO3, DMF, 90 °C; yields: 44–86 %; the synthesis of compound 20 was previously described by Torrie et al.20
Subtle trends in SAR similar to series 1 a were observed in series 1 b (Table 3). In particular, there was a 25-fold gain in potency for the 3,5-dichloro analogue 20, relative to the unsubstituted scaffold 14. This could be due to the greater lipophilicity of this compound causing an increase in nonspecific binding, although the ligand efficiency improved (LE from 0.39 to 0.45), suggesting a favourable specific interaction. It was possible to modify the ketone moiety to an amide without loss of potency (compare amide 23 (IC50=0.6 μm), with ketone 20 (IC50=0.3 μm)). In going from methyl (21) to ethyl (22) to tert-butyl ketone (20), an improvement in potency was observed (IC50: 8.8 to 6.5 to 0.3 μm, respectively), indicating lipophilic bulk in this area is required. In contrast, the phenyl ketone 24 shows a marked loss in potency, with an IC50 value of 45 μm.
Hit series 1 c: tetrahydroindazole methylene amide
The pyrazole core for series 1 c was made by condensation of ethyl hydrazinoacetate with the commercially available dione. The ester was then hydrolysed, allowing amide couplings using standard methodology (Table 4).23, 24
Table 4.
SAR around hit series 1 c.
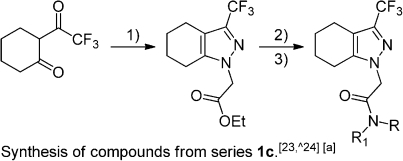 | |||
|---|---|---|---|
| Compd | R1 | R | TbTryS IC50 [μm] |
| 25 | H | (+/−)-CH(CH3)Ph | 5.6 |
| 26 | H | CH2-4-chlorophenyl | 3.0 |
| 27 | H | CH2-2-thiophene | 3.9 |
| 28 | H | CH2CH2Ph | 5.6 |
| 29 | H | (+/−)-CH(CH3)CH2CH3 | 12 |
| 30 | H | CH2CH2CH3 | 13 |
| 31 | H | CH2CH2OCH3 | 30 |
| 32 | H | Ph | >100 |
| 33 | H | cHex | >100 |
| 34 | CH2Ph | CH2Ph | >100 |
| 35 | CH2CH3 | CH2CH3 | 15 |
[a] Conditions: 1) ethyl 2-hydrazinylacetate, Et3N, EtOH, reflux, 61 % yield; 2) 2 m NaOH, THF/H2O (2:1), 89 % yield; 3) RR1NH, EDC⋅HCl, HOBt, DIPEA, DMF, 50 °C; yields: 79–80 %.
Small changes to optimise the amide group in this series generated a “flat” SAR plateau (25–28; Table 4). Aliphatic amides (29–31) were tolerated, albeit at weaker potency, but bulky rigid substitution with the phenyl 32 and cyclohexyl 33 moieties was not tolerated, with complete loss in potency observed. Tertiary amide 34 with two bulky benzyl groups also lost all potency, but the less bulky diethyl amide 35 retained some inhibitory activity.
Initial hit exploration around series/group 2
Following the HTS campaign we also identified three hit series based around a second pharmacophore. This second group of hit series described a slightly different pharmacophore, with the heteroatom of the heterocycle in a 1,6 relationship with the HBA motif of the sulfonamide. These hit series had higher molecular weight and generally lower potency, with lower ligand efficiency (see Table 1). To explore the SAR around these series, compounds were purchased and screened. Data for these are listed in Tables 5, 6, and 7.
Table 5.
SAR around hit series 2 a.
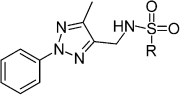 | ||
|---|---|---|
| Compd | R | TbTryS IC50 [μm] |
| 36 | 2-thiophene | 8.5 |
| 37 | 5-(2,4-dimethylthiazole) | 2.2 |
| 38 | 4-(1,3,5-trimethylpyrazole) | 2.6 |
| 39 | 1-naphthyl | >100 |
| 40 | 2,5-dimethoxyphenyl | >100 |
| 41 | 3-(2,5-dimethylthiophene) | 24 |
| 42 | tBu | 37 |
Table 6.
SAR around hit series 2 b.
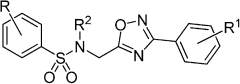 | ||||
|---|---|---|---|---|
| Compd | R2 | R1 | R | TbTryS IC50 [μm] |
| 43 | H | H | H | >50 |
| 44 | H | 2-CH3 | H | 15 |
| 45 | H | 2-Cl | H | 25 |
| 46 | H | H | 2,5-di-Cl | 16 |
| 47 | H | H | 4-NHCOCH3 | 5.9 |
| 48 | H | H | 4-OCH3 | 11 |
| 49 | H | 2-CH3 | 4-OCH3 | 4.0 |
| 50 | H | 2-CH3 | 4-NHCOCH3 | 1.7 |
| 51 | H | 2-Cl | 4-NHCOCH3 | 3.7 |
| 52 | CH3 | H | 4-NHCOCH3 | >50 |
Table 7.
SAR around hit series 2 c.
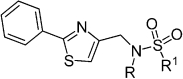 | |||
|---|---|---|---|
| Compd | R | R1 | TbTryS IC50 [μm] |
| 53 | H | 2,5-difluorophenyl | 6.1 |
| 54 | H | phenyl-4-COCH3 | 8.7 |
| 55 | H | 2,5-dichlorophenyl | 6.3 |
| 56 | H | 3,4-dichlorophenyl | 5.8 |
| 57 | H | 2-naphthyl | 2.4 |
| 58 | Ph | 4-methylphenyl | >100 |
None of the group 2 series offered any benefits in terms of potency over the compound series from group 1. In addition, the DMPK data indicated less favourable properties of group 2 series compounds, relative to data for compounds from group 1 (see Table 12 below). Therefore, further optimisation work was focussed on the group 1 series.
Table 12.
Trypanosome cell, human (MRC5) cell, and pooled human microsomal intrinsic clearance (Cli) data for key (representative) compounds from series 1–3.
| Compd | Series | TbTryS IC50 [μm] | Tb EC50 [μm] | MRC5 EC50 [μm] | Cli [mL min−1 g−1] |
|---|---|---|---|---|---|
| 8 | 1 a | 0.41 | 22 | >50 | 0.9 |
| 26 | 1 c | 14 | 14 | >50 | 3.0 |
| 36 | 2 a | 8.5 | 32 | >50 | 9.1 |
| 57 | 2 c | 2.4 | 19 | >50 | 17 |
| 71 | 3 | 0.09 | 7.9 | >50 | 1.6 |
| 84 | 3 | 0.045 | 5.4 | 20 | ND[a] |
| 89 | 3 | 0.14 | 5.1 | >50 | ND[a] |
[a] Not determined.
Hit to lead optimisation strategy
Hit series 1 a–c were successfully validated and shared an overlapping pharmacophore for TbTryS activity (Figure 2), demonstrating clear SAR and a visible potential for further optimisation. The pharmacophoric features of the three group 1 series scaffolds were hybridised into one new core scaffold, which was based on an indazole (Figure 3). One of the indazole nitrogen atoms becomes the HBA, and the other is used for attachment of the other HBA. This indazole series is also predicted to have reasonable physicochemical properties: low molecular weight, clogP<5, and low PSA. For example, compound 60 (Table 8) has Mr=292 Da, clogP=4.0, and PSA=35 Å2.
Figure 2.
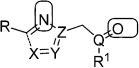
Group 1 common pharmacophore: R and R1 are hydrophobic binding domains; N and O (boxed) are the hydrogen bond acceptor motifs (1,5 relationship); X, Y, and Z are C, N, O, or S; Q is C or SO (sulfone).
Figure 3.
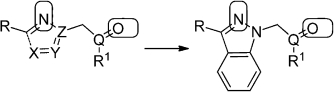
Generation of the new scaffold.
Table 8.
SAR around lead series 3.
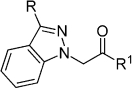 | ||||
|---|---|---|---|---|
| Compd | R | R1 | TbTryS IC50 [μm] | LE[a] |
| 59 | Ph | OCH3 | 20 | 0.32 |
| 60 | Ph | tBu | 0.15 | 0.43 |
| 61 | 4-chlorophenyl | tBu | >100 | |
| 62 | 4-indole | tBu | 3.7 | 0.30 |
| 63 | 4-(N-methyl)indole | tBu | >100 | |
| 64 | 2-naphthyl | tBu | >100 | |
| 65 | 4-pyridyl | tBu | 0.3 | 0.41 |
| 66 | 2-furanyl | tBu | 1.9 | 0.38 |
| 67 | 3-pyridyl | tBu | 3.0 | 0.35 |
| 68 | 3-thiophenyl | tBu | 0.4 | 0.42 |
| 69 | 4-(N-isobutyl)pyrazole | tBu | >100 | |
| 70 | 3-chlorophenyl | tBu | 0.35 | 0.39 |
| 71[b] | 3-fluorophenyl | tBu | 0.09 | 0.42 |
Validation of the indazole series
The indazole scaffold was prepared as shown in Scheme 1. Indazole was first iodinated (at the 3-position) using standard conditions.25 A Suzuki reaction afforded the 3-aryl intermediate. Alkylation of the N1 nitrogen atom with the required α-chloroketone (or arylmethylchloride) gave final products 60, 71, and 75–79. Alternatively for compounds 61–70, the alkylation of the 3-iodoindazole was completed first and the Suzuki reaction, to add the aryl substituent, was employed as a final step.
Scheme 1.
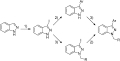
General synthesis of indazole analogues. Conditions: 1) KOH, I2, DMF, as in Edwards et al.,25 89 % yield; 2) ArB(OH)2, Na2CO3, DME, EtOH, H2O, yields: 50–78 %; 3) NaH, DMF, α-haloketone (or other), yields: 37–62 %. Synthesis of compounds 71 and 89 were described previously.20
3-Indazole substitution
Table 8 shows the data for key compounds from the initial SAR study, describing changes to the aromatic group at position 3 of the indazole (depicted R in the structure). The initial compound 60 (R=phenyl), had an IC50 value of 150 nm in the TbTryS enzyme assay (LE calculated as 0.43) and was the most potent compound to date. Para substituents led to loss in activity (e.g. 61, 63, 64, and 69). This is possible evidence for the presence of a tight binding pocket into which the aromatic subunit sits. A variety of heterocycles were tolerated, although some gave a 10-fold loss in activity, such as the 2-furanyl compound 66 and 3-pyridyl compound 67. The 3-fluorophenyl motif (71, IC50=90 nm, LE=0.42) was more potent than 3-chlorophenyl (70, IC50=354 nm), and was used as the standard template for exploration of substituent SAR around the R1 position (see Tables 9–11 below).
Table 9.
SAR around indazole sulfone analogues.
 | |||
|---|---|---|---|
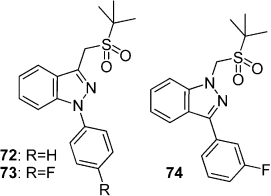
| Compd | R | TbTryS IC50 [μm] | LE[a] |
| 72 | H | 0.22 | 0.40 |
| 73 | 4-fluoro | 0.08 | 0.41 |
| 74 | – | 0.12 | 0.40 |
[a] Ligand efficiency, calculated as −0.6×ln(IC50)/(heavy atom count).21
Table 11.
Amide side chain indazoles.
 | ||||
|---|---|---|---|---|
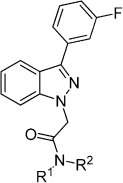
| Compd | R1 | R2 | TbTryS IC50 [μm] | LE[a] |
| 80 | (CH2)3-N-methylpiperazine | H | 0.83 | 0.28 |
| 81 | (CH2)3-N1-imidazole | H | 0.35 | 0.32 |
| 82 | (CH2)2-N-Boc-piperazine | H | 2.9 | 0.22 |
| 83 | (CH2)2N(CH3)2 | H | 0.145 | 0.38 |
| 84[b] | (CH2)3N(CH3)2 | H | 0.045 | 0.39 |
| 85 | (CH2)3N(CH3)2 | CH3 | 0.17 | 0.35 |
| 86 | CH2-4-chlorophenyl | H | >100 | |
| 87 | CH2CH3 | CH2CH3 | 0.20 | 0.39 |
| 88 | CH3 | CH3 | 0.11 | 0.44 |
| 89[b] | fused into piperidine ring | 0.14 | 0.38 | |
| 90 | (CH2)2-N-methylpiperazine | H | 0.86 | 0.29 |
Having established the indazole as a potent heterocyclic core with good ligand efficiency, a number of SAR studies were carried out to explore chemical optimisation around this scaffold.
Investigation of alternative HBA moieties
We were concerned that the ketone group could react with nucleophiles, so a number of alternative HBAs were investigated. The initial sulfone analogue 74 (TbTryS IC50=120 nm) was found to be equipotent to the ketone 60 (Table 9). The position from which the pendant HBA is attached to the core scaffold was also investigated. In compounds 72 and 73 the sulfone HBA is appended from the 3-position of the indazole core (Table 9). This modification had little effect on activity, which is probably due to the symmetry of the molecule. For ease of synthesis, further HBA modifications were investigated at the N1 position of the indazole.
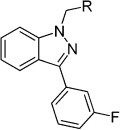
Approaches were made to use a heterocycle as the second HBA motif (Table 10). Pyridine and oxazole were investigated, but showed lower activity than the ketone or sulfone groups.
Table 10.
SAR around pendant heterocyclic side chain indazoles.
 | |||
|---|---|---|---|
| Compd | R | TbTryS IC50 [μm] | LE[a] |
| 74 | SO2tBu | 0.12 | 0.40 |
| 75 | 2-pyridyl | 2.9 | 0.33 |
| 76 | 3-pyridyl | 4.5 | 0.32 |
| 77 | 2,5-dimethyl-4-oxazole | 28 | 0.26 |
| 78 | 5-phenyl-3-isoxazole | >100 | |
| 79 | 5-methyl-3-isoxazole | >100 | |
[a] Ligand efficiency, calculated as −0.6×ln(IC50)/(heavy atom count).21
Amide HBA analogues
Alkylation of the 3-arylindazole (synthesis shown in Scheme 2) with ethylbromoacetate, followed by saponification, yielded the indazole-N-acetic acid intermediate. This was coupled under standard amide coupling conditions to the appropriate amine to give access to amide analogues.
Scheme 2.
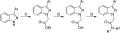
Synthesis of amide analogues. Conditions: 1) NaH, DMF, ethyl bromoacetate, as in Fujimura et al.,29 yields: 37–62 %; 2) NaOH, THF/H2O, 95 % yield; 3) HOBt, EDC, DMF, DIPEA, R1R2NH; yields: 40–48 %.
Encouragingly, the ketone subunit could be replaced by a simple amide without loss of potency or ligand efficiency (Table 11). Thus the diethyl and dimethyl amides (87 and 88) had IC50 values similar to that of the ketone 71, with compound 88 having a ligand efficiency of 0.44. Potency was retained when fusing the dialkyl amide into an N-piperidine amide (89 TbTryS IC50=135 nm), but activity was completely abolished with a larger alkyl aromatic substituent (e.g. 86 IC50>100 μm).
Addition of an appended basic amine, to potentially pick up the TbTryS endogenous substrate Spd binding domain interactions and improve ligand potency, was investigated. This basic group could also improve the aqueous solubility of our compounds and lower the lipophilicity (logP). Although the piperazine-containing compounds 90 and 80 lost potency (ninefold relative to 71), and were less efficient binders (ligand efficiencies of 0.29 and 0.28 respectively), they were still sub-micromolar inhibitors of the TbTryS enzyme, with TbTryS IC50 values of 0.86 and 0.83 μm, respectively. If the second basic amine centre was removed and the compounds were truncated to make the C2- and C3-linked dimethyl amine compounds 83 and 84, a significant improvement in potency (six- and 18-fold, respectively) over the analogous piperazines was observed. These compounds were also significantly more efficient binders than compounds 80 and 90, with respective ligand efficiencies of 0.38 and 0.39. The C3-linked dimethylamine compound 84 was observed to be the most potent compound to date, with a TbTryS IC50 value of 45 nm. Compound 84 shows improved physicochemical properties over compound 60, especially in decreased lipophilicity, with a clogP value of 2.8 (1.3 units lower than that of 60), and Mr=354 Da and PSA=50 Å2.
As compound 84 shows similar potency to an analogue not containing an appended amine (71, IC50 90 nm) it is unlikely that compound 84 has picked up the Spd binding domain. This conclusion is supported by competition binding studies which revealed that the compounds displayed mixed inhibition with respect to Spd, and did not show classical competitive binding kinetics.20 Finally, capping the NH group of the amide of 84 with a methyl group (compound 85) resulted in a fourfold decrease in potency, and the C3-linked imidazole 81 was eightfold less potent than the dimethylamine.
Cell potency and DMPK parameters for key compounds
Following the discovery of promising novel TryS inhibitors, compounds were progressed into a trypanosomal cell proliferation assay and a human cell counter-screen. Selected compounds were also screened in an in vitro metabolic clearance assay (Table 12), to assess suitability for series progression. Metabolic stability studies using pooled human liver microsomes indicate a range of stabilities. Compounds from the group 2 series are highly metabolically unstable. However, compounds 8, 26, and 71 are reasonably metabolically stable, suggesting nothing fundamentally problematic with these scaffolds in terms of cytochrome P450-driven metabolism.
Table 12 also shows cell data for key compounds from several of the various series of TbTryS inhibitors discovered. Although these compounds were not toxic to the MRC5 mammalian cell line, there was up to a 100-fold decrease in going from enzyme to trypanosomal cell efficacy, even with the lead compounds 71 and 84 (TbTryS IC50: 90 and 45 nm, respectively). While these cell potencies are equivalent to the drugs eflornithine (22 μm) and nifurtimox (2 μm) currently in clinical use for late-stage human African trypanosomiasis, they are much less potent than the alternative arsenic-containing drug melarsoprol (8 nm), although arsenic-based compounds do show significant toxicity in the clinic.26
As this large potency shift between the enzyme IC50 values and parasite EC50 values was unexpected, further experiments were carried out to confirm whether hit compounds were entering the cell and acting on-target. As described fully elsewhere, exposing T. brucei parasites to the model TbTryS inhibitors 89 and 84 (2×EC50 for 72 h) resulted in trypanothione levels dropping to <10 % of wild-type levels.15, 20 In addition, there was a corresponding increase in the TbTryS substrate GSH, providing strong evidence that these compounds were acting on-target.
As previously reported, the on-target effects of these hit compounds were further confirmed by generating TbTryS single knockout (SKO) and TbTryS overexpressing (OE) cell lines. Western blot analysis and densitometry demonstrated that TbTryS protein levels were decreased in the SKO cells and elevated in the OE cell line, relative to wild-type cells, and these cell lines showed the expected changes in potency to 89 (EC50 values: 20.4, 6.9, and 44.5 μm for wild-type, SKO, and OE cell lines, respectively) and 84 (EC50 values: 7.1, 1.2, and 23.3 μm for wild-type, SKO, and OE cell lines, respectively), confirming that TbTryS is the specific target of these compounds.15, 20
Conclusions
In this work we successfully took HTS hits, clustered them into putative hit series, and rationalised their activities based on common pharmacophores. Initial investigation of SAR around the hit series confirmed an overlapping pharmacophore, and the optimisation potential of group 1 hit series in particular. Following the SAR on group 1 series, a hybridisation strategy and scaffold-hopping approach led us to discover the indazole lead series. Optimisation of this series for potency and improved DMPK properties led to compounds 71 and 84, which displayed in vitro enzyme potencies >10-fold improved over the best HTS hits. Attempts so far to co-crystallise our inhibitors with the TbTryS enzyme have failed to produce robust data.
Although these indazoles inhibit TbTryS with IC50 values of <100 nm, they failed to show sub-micromolar potency in a trypanosome proliferation assay. This can be rationalised by the observation that parasites can survive with low levels of trypanothione beyond the timeframe of the standard whole-parasite proliferation assay. The extension of the time-course in screening assay format is prohibited by the need for repeated dilutions of samples to remain in log-phase growth, leading to unacceptable variability. The lead compounds do, however, show a robust biochemical effect in T. brucei, and are proven to act on-target, inhibiting TbTryS in cells.15, 20 The current lead compounds could also prove very useful in combination therapy with known trypanocides (such as melarsoprol), as studies have revealed TryS-depleted T. brucei procyclics are significantly more susceptible to trypanocides.27 Our compounds are the most advanced, potent, and drug-like (as predicted by physicochemical and in vitro DMPK properties) inhibitors of TbTryS reported to date, and are extremely useful leads to further explore the trypanothione pathway in kinetoplastids.
Experimental section
Chemistry
1H NMR spectra were recorded on either Bruker Avance DPX 500 or Bruker Avance 300 spectrometers. Chemical shifts (δ) are expressed in ppm. Signal splitting patterns are described as singlet (s), broad singlet (bs), doublet (d), triplet (t), quartet (q), multiplet (m) or combination thereof. LC–MS analyses were performed with either an Agilent HPLC 1100 series instrument connected to a Bruker Daltonics MicrOTOF, or an Agilent Technologies 1200 series HPLC connected to an Agilent Technologies 6130 quadrupole LC–MS; both instruments were connected to an Agilent diode-array detector. LC–MS chromatographic separations were conducted with a Phenomenex Gemini C18 column, 50×3.0 mm, 5 μm particle size; mobile phase, H2O/CH3CN +0.1 % HCOOH 80:20→5:95 over 3.5 min, and then held for 1.5 min; flow rate: 0.5 mL min−1. High-resolution electrospray MS measurements were performed on a Bruker Daltonics MicrOTOF mass spectrometer. Thin-layer chromatography (TLC) was carried out on Merck silica gel 60 F254 plates using UV light and/or KMnO4 for visualisation. TLC data are given as the Rf value with the corresponding eluent system specified in brackets. Column chromatography was performed using RediSep® 4 or 12 g silica pre-packed columns. LCMS chromatographic separations were conducted with a Waters Xbridge C18 column, 50 mm × 2.1 mm, 3.5 μm particle size; Method A: mobile phase, H2O/CH3CN + 0.1 % NH3; linear gradient 80:20→5:95 over 3.5 min, and then held for 1.5 min; flow rate 0.5 mL min−1. All reactions were carried out under dry and inert conditions, unless otherwise stated.
Compounds in series 1 a:
2-(tert-Butylsulfonylmethyl)-4-(3-fluorophenyl)thiazole (9): Synthesis previously described.20 To a stirred solution of 3-fluoroacetophenone (250 mg, 1.81 mmol) in THF (6 mL), was added trimethylphenylammonium tribromide (681 mg, 1.81 mmol) solution in THF (4 mL). The reaction was stirred at room temperature for 18 h; the resulting white precipitate was filtered off, and the filtrate was added to petroleum ether (PE; 20 mL). The PE solution containing the product was washed with H2O (30 mL) and then dried (MgSO4). The solvent was then removed in vacuo to give intermediate 2-bromo-1-(3-fluorophenyl)ethanone (390 mg, 99 %) as a pale-yellow oil; [M+H]+=217/219. To a stirred solution of 2-bromo-1-(3-fluorophenyl)ethanone (326 mg, 1.8 mmol) in EtOH (20 mL) was added 2-(tert-butylsulfonyl)ethanethioamide (386 mg, 1.98 mmol), and the reaction was heated at reflux and stirred for 2 h. The solvent was then removed in vacuo to give a crude residue which was purified by column chromatography (CH2Cl2→10 % MeOH/CH2Cl2 eluent) to give the title compound 9 (380 mg, 81 %) as a white solid: 1H NMR ([D6]DMSO, 500 MHz): δ=8.40 (1 H, bs), 8.31 (2 H, m), 7.70 (2 H, m), 5.15 (2 H, bs), 1.4 (9 H, bs); [M+H]+=314; HRMS C14H16FNO2S2 calcd: 314.0679, obsd: 314.0690.
Compounds 3–8 were prepared according to the same procedures as described above for the preparation of compound 9, using the corresponding acetophenone.
2-((tert-Butylsulfonyl)methyl)-4-(phenyl)thiazole (3): 1H NMR ([D6]DMSO, 500 MHz): δ=8.23 (1 H, s), 7.98 (2 H, dd, J=1.4, 8.5 Hz), 7.46 (2 H, t, J=7.5 Hz), 7.37 (1 H, t, J=12.5 Hz), 5.07 (2 H, bs), 1.39 ppm (9 H, bs); [M+H]+=296; HRMS C14H17NO2S2 calcd: 296.0767, obsd: 296.0773.
2-((tert-Butylsulfonyl)methyl)-4-(2-methylphenyl)thiazole (4): 1H NMR ([D6]DMSO, 500 MHz): δ=7.88 (1 H, m), 7.59 (1 H, m), 7.31 (3 H, m), 5.06 (2 H, bs), 2.42 (3 H, s), 1.38 ppm (3 H, s); [M+H]+=310; HRMS C15H19NO2S2 calcd: 310.0930, obsd: 310.0943.
2-((tert-Butylsulfonyl)methyl)-4-(2-fluorophenyl)thiazole (5): 1H NMR ([D6]DMSO, 500 MHz): δ=8.10 (2 H, m), 7.44 (1 H, m), 7.35 (2 H, m), 5.10 (2 H, m), 1.39 ppm (9 H, s); [M+H]+=314; HRMS C14H16FNO2S2 calcd: 314.0679, obsd: 314.0690.
2-((tert-Butylsulfonyl)methyl)-4-(3-methoxyphenyl)thiazole (6): 1H NMR ([D6]DMSO, 500 MHz): δ=8.26 (1 H, s), 7.55 (1 H, m), 7.52 (1 H, m), 7.38 (1 H, t, J=8 Hz), 6.95 (1 H, m), 5.08 (2 H, s), 3.82 (3 H, s), 1.39 ppm (9 H, s); [M+H]+=326; HRMS C15H19NO3S2 calcd: 326.0879, obsd: 326.0888.
2-(tert-Butylsulfonylmethyl)-4-(3-trifluoromethylphenyl)thiazole (7): 1H NMR ([D6]DMSO, 500 MHz): δ=8.49 (1 H, bs), 8.30 (2 H, m), 7.74 (2 H, m), 5.12 (2 H, bs), 1.4 ppm (9 H, bs); [M+H]+=364; HRMS C15H16F3NO2S2 calcd: 364.0647, obsd: 364.0639.
2-((tert-Butylsulfonyl)methyl)-4-(3-chlorophenyl)thiazole (8): 1H NMR ([D6]DMSO, 500 MHz): δ=8.38 (1 H, bs), 8.03 (1 H, m), 7.95 (1 H, m), 7.51 (1 H, m), 7.44 (1 H, m), 5.09 (2 H, bs), 1.39 (9 H, s); 13C NMR (125 MHz, CDCl3): δ=23.8, 52.3, 61.6, 116.9, 124.4, 126.6, 128.4, 130.1, 134.9, 135.6, 154.0, 156.9 ppm; [M+H]+=330/332; HRMS C14H16ClNO2S2 calcd: 330.0384, obsd: 330.0384.
Compounds 9–13 were obtained from Maybridge.
Compounds in series 1 b:
1-(5-(3,5-Dichlorophenyl)-2H-tetrazol-2-yl)-3,3-dimethylbutan-2-one (20): Synthesis previously described.20 A mixture of commercially available 5-(3,5-dichlorophenyl)tetrazole (0.215 g, 1.0 mmol), 1-bromopinacolone (0.14 mL, 1.0 mmol) and diisopropylethylamine (0.19 mL, 1.1 mmol) in CH3CN (5 mL) was heated at 75 °C for 3 days. The mixture was cooled, diluted with EtOAc (15 mL), and filtered through a plug of silica. The filtrate was concentrated and purified by chromatography on silica (eluent: EtOAc/hexane 0→50 %) to give the title compound 20 (0.270 g, 86 %) as a white solid: 1H NMR (CDCl3, 300 K): δ=8.08 (2 H, d, J=1.9 Hz), 7.48 (1 H, t, J=1.9 Hz), 5.71 (2 H, s), 1.36 ppm (9 H, s); 13C NMR (125 MHz, CDCl3): δ=26.2, 43.7, 56.6, 125.3, 130.0, 130.3, 135.7, 163.4, 204.2 ppm; [M+H]+=313; HRMS C13H14Cl2N4O calcd: 313.0586, obsd: 313.0601.
2-(5-(3,5-Dichlorophenyl)-2H-tetrazol-2-yl)-N,N-dimethylacetamide (23): A mixture of 5-(3,5-dichlorophenyl)tetrazole (0.215 g, 1.0 mmol), NaI (0.150 g, 1.0 mmol) and K2CO3 (0.210 g, 1.5 mmol) in DMF (5 mL) was heated at 90 °C until effervescence was nearly complete. 2-Chloro-N,N-dimethylacetamide (0.11 mL, 1.1 mmol) was added, and the mixture was heated at 90 °C overnight. The mixture was diluted with H2O (15 mL) and allowed to stand for several hours. The precipitate was collected by filtration and washed with H2O and Et2O to give the title compound 23 (0.150 g, 50 %) as a white solid: 1H NMR (CDCl3, 300 K): δ=8.09 (2 H, d, J=1.9 Hz), 7.47 (1 H, t, J=1.9 Hz), 5.58 (2 H, s), 3.17 (3 H, s), 3.07 ppm (3 H, s); [M+H]+=300; HRMS C11H11Cl2N5O calcd: 300.0413, obsd: 300.0411.
Compounds below were prepared according to the same procedures as described above for the preparation of compound 23, using the corresponding aryl tetrazole and α-haloketone.
3,3-Dimethyl-1-(5-phenyl-2H-tetrazol-2-yl)butan-2-one (14): 1H NMR (CDCl3, 300 K): δ=8.17 (2 H, m), 7.51 (3 H, m), 5.70 (2 H, s), 1.35 ppm (9 H, s); [M+H]+=245; HRMS C13H16N4O calcd: 245.1397, obsd: 245.1387.
1-(5-(3-Fluorophenyl)-2H-tetrazol-2-yl)-3,3-dimethylbutan-2-one (17): 1H NMR (CDCl3, 300 K): δ=7.98 (1 H, d, J=7.7 Hz), 7.88 (1 H, d, J=9.3 Hz), 7.48 (1 H, m), 7.19 (1 H, td, J=8.3 and 2.1 Hz), 5.71 (2 H, s), 1.32 ppm (9 H, s); [M+H]+=263; HRMS C13H15FN4O calcd: 263.1303, obsd: 263.1291.
1-(5-(3,5-Difluorophenyl)-2H-tetrazol-2-yl)-3,3-dimethylbutan-2-one (19): 1H NMR (CDCl3, 300 K): δ=7.72 (2 H, m), 6.94 (1 H, tt, J=8.8 and 2.4 Hz), 5.71 (2 H, s), 1.36 ppm (9 H, s); [M+H]+=281; HRMS C13H14F2N4O calcd: 281.1208, obsd: 281.1210.
1-(5-(3,5-Dichlorophenyl)-2H-tetrazol-2-yl)propan-2-one (21): 1H NMR (CDCl3, 300 K): δ=8.08 (2 H, d, 1.9 Hz), 7.48 (1 H, t, 1.9 Hz), 5.51 (2 H, s), 2.30 ppm (3 H, s); [M+H]+=271/273; HRMS C10H8Cl2N4O calcd: 271.0148, obsd: 271.0141.
1-(5-(3,5-Dichlorophenyl)-2H-tetrazol-2-yl)butan-2-one (22): 1H NMR (CDCl3, 300 K): δ=8.09 (2 H, d, 1.9 Hz), 7.50 (1 H, t, 1.9 Hz), 5.52 (2 H, s), 2.57 (2 H, q, J=7.2 Hz), 1.19 ppm (3 H, t, J=7.2 Hz); [M+H]+=285/287; HRMS C11H10Cl2N4O calcd: 285.0304, obsd: 285.0292.
2-(5-(3,5-Difluorophenyl)-2H-tetrazol-2-yl)-1-phenylethanone (24): 1H NMR (CDCl3, 300 K): δ=8.02 (2 H, m), 7.72 (3 H, m), 7.58 (2 H, t, J=7.8 Hz), 6.93 (1 H, tt, J=8.8 and 2.4 Hz), 6.16 ppm (2 H, s); [M+H]+=301; HRMS C11H6F2N4O calcd: 301.0895, obsd: 301.0901.
Compounds 15, 16 and 18 were obtained from Maybridge.
Compounds in series 1 c:
N-(4-Chlorobenzyl)-2-(3-(trifluoromethyl)-4,5,6,7-tetrahydro-1H-indazol-1-yl)acetamide (26): To a stirred suspension of 2-(trifluoroacetyl)cyclohexanone (1 g, 5.151 mmol) and ethylhydrazinoacetate hydrochloride (2.39 g, 15.5 mmol) in EtOH (30 mL) was added Et3N (2.17 mL, 15.5 mmol) and the reaction heated at reflux for 24 h. The reaction mixture was cooled to room temperature and the solvent removed in vacuo. The resultant crude residue was taken up in CH2Cl2, washed (H2O, brine), dried (MgSO4) and concentrated in vacuo. The resultant crude residue was purified by column chromatography, eluting with 0–50 % EtOAc/hexane, to give ethyl 2-(3-(trifluoromethyl)-4,5,6,7-tetrahydro-1H-indazol-1-yl)acetate (864 mg, 61 %) as a white solid; [M+H]+=277.
To a stirred solution of ethyl 2-(3-(trifluoromethyl)-4,5,6,7-tetrahydro-1H-indazol-1-yl)acetate (691 mg, 2.5 mmol) in 1:1 THF/H2O (30 mL) was added 2 m (aq) NaOH (5 mL, 10 mmol), and the reaction stirred for 3 h. The solvent was then removed in vacuo to give a crude residue which was suspended in H2O, acidified (1 m aq. HCl), stirred for 5 min, filtered and the precipitate dried in vacuo to give 2-(3-(trifluoromethyl)-4,5,6,7-tetrahydro-1H-indazol-1-yl)acetic acid (550 mg, 89 %) as a white solid; [M+H]+=249.
To a stirred solution of 2-(3-(trifluoromethyl)-4,5,6,7-tetrahydro-1H-indazol-1-yl)acetic acid (124 mg, 0.5 mmol), 4-chlorobenzylamine (0.061 mL, 0.5 mmol) and 1-hydroxybenzotriazole hydrate (68 mg, 0.5 mmol) in N,N-dimethylformamide (DMF; 3 mL) at 50 °C was added N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (96 mg, 0.5 mmol) and N,N′-diisopropylethylamine (0.18 mL, 1 mmol) and the reaction stirred for 16 h. The reaction mixture was cooled to room temperature, taken up in EtOAc, washed (H2O, brine), dried (MgSO4) and concentrated in vacuo. The resultant crude residue was purified by reversed-phase HPLC (method A), to give title compound 26 (120 mg, 79 %) as a white solid: 1H NMR ([D6]DMSO, 500 MHz): δ=8.78 (1 H, t, J=5.9 Hz), 7.41 (2 H, m), 7.30 (2 H, m), 4.84 (2 H, s), 4.30 (2 H, d, J=3.0 Hz), 2.56 (2 H, m), 2.51 (2 H, m), 1.74 (2 H, m), 1.67 ppm (2 H, m); 13C NMR (125 MHz, CDCl3): δ=19.9, 21.2, 21.9, 22.2, 42.8, 52.3, 116.1, 120.6, 122.7, 128.8, 128.9, 133.5, 136.0, 140.1, 166.3 ppm; [M+H]+=372; HRMS C17H17ClF3N3O calcd: 372.1085, obsd: 372.1103.
N,N-Diethyl-2-(3-(trifluoromethyl)-4,5,6,7-tetrahydro-1H-indazol-1-yl)acetamide (35): By proceeding in a similar manner to compound 26, using 2-(3-(trifluoromethyl)-4,5,6,7-tetrahydro-1H-indazol-1-yl)acetic acid and diethylamine the title compound 35 (100 mg, 80 %) was obtained as a white solid. 1H NMR ([D6]DMSO, 500 MHz): δ=5.10 (2 H, s), 3.37 (2 H, q, J=7.2, 7.5 Hz), 3.27 (2 H, q, J=7.1 Hz), 1.71 (4 H, m), 1.17 (3 H, t, J=7.1 Hz), 1.03 ppm (3 H, t, J=7.1 Hz); [M+H]+=304; HRMS C14H20F3N3O calcd: 304.1631, obsd: 304.1628.
Compounds 25, 27, 28, 31, 32, 33 and 34 were obtained from Asinex. Compounds 29 and 30 were obtained from ChemDiv.
Compounds in series 2 a:
2-Methyl-N-((5-methyl-2-phenyl-2H-1,2,3-triazol-4-yl)methyl)propane-2-sulfonamide (42): tert-Butylsulfinyl chloride (0.18 mL, 1.5 mmol) was added to a solution of (5-methyl-2-phenyl-2H-1,2,3-triazol-4-yl)methanamine (0.188 g, 1.0 mmol) and Et3N (1.4 mL, 10 mmol) in CH2Cl2 (10 mL) at 0 °C. The mixture was stirred at 0 °C for 2 h and then quenched with aqueous NaHCO3. The phases were separated and the aqueous phase was extracted with CH2Cl2. The combined organic phases were dried (Na2SO4) and concentrated. Chromatography on silica (50–100 % EtOAc/PE 40:60) gave 2-methyl-N-((5-methyl-2-phenyl-2H-1,2,3-triazol-4-yl)methyl)propane-2-sulfinamide as an orange solid (0.240 g, 82 %): 1H NMR (CDCl3, 300 K): δ=8.01 (2 H, d, J=8.4 Hz), 7.47 (2 H, t, J=7.9 Hz), 7.33 (1 H, t, J=7.3 Hz), 4.50 (1 H, dd, J=14.1 and 4.7 Hz), 4.36 (1 H, dd, J=14.1 and 7.5 Hz), 3.60 (1 H, m), 2.42 (3 H, s), 1.27 ppm (9 H, s); [M+H]+=293.
A solution of 2-methyl-N-((5-methyl-2-phenyl-2H-1,2,3-triazol-4-yl)methyl)propane-2-sulfinamide (0.146 g, 0.5 mmol) in CH2Cl2 (7.5 mL) was treated with mCPBA (Aldrich, ≤77 %; 0.225 g, ≤1.0 mmol) and stirred at room temperature for 45 min. The reaction was quenched with 2 m aqueous NaHSO3 (10 mL) and saturated aqueous NaHCO3 (10 mL). The organic phase was applied to a plug of silica and eluted with CH2Cl2/EtOAc (0→100 %) to give title compound 42 as a white solid (0.107 g, 69 %): 1H NMR (CDCl3, 300 K): δ=8.01 (2 H, m), 7.49 (2 H, m), 7.34 (1 H, tt, J=7.4 and 1.1 Hz), 4.51 (2 H, d, J=5.7 Hz), 4.43 (1 H, m), 2.44 (3 H, s), 1.47 ppm (9 H, s); [M+H]+=309; HRMS C14H20N4O2S calcd: 309.1307, obsd: 309.1312.
Compounds in series 2 a, 2 b and 2 c:
Compounds 36–41 were obtained from Maybridge. Compounds 43, 44, 45, 53, and 54 were obtained from ChemDiv. Compound 46 was obtained from ChemBridge. Compounds 47–52 were obtained from Asinex. Compounds 55–57 were obtained from Life Chemicals. Compound 58 was obtained from Enamine.
Compounds in series 3:
1-(3-(3-Fluorophenyl)-1H-indazol-1-yl)-3,3-dimethylbutan-2-one (71): The synthesis was previously described elsewhere.20
3-Iodo-1H-indazole was prepared as reported by Edwards et al;25 [M+H]+=245.
A capped process vial containing 3-iodo-1H-indazole (366 mg, 1.5 mmol), 3-fluorophenylboronic acid (252 mg, 1.8 mmol), 2 m (aq) Na2CO3 (1.12 mL, 2.25 mmol) and tetrakis-(triphenylphosphine)palladium(0) in 7:3:2 DME/H2O/EtOH (3 mL) was degassed, flooded with argon and irradiated (Biotage Initiator) at 150 °C for 20 min. 3-Fluorophenylboronic acid (126 mg, 0.9 mmol) and 2 m (aq) Na2CO3 (0.56 mL, 1.12 mmol) were added, the system degassed, flooded with argon and irradiated (Biotage Initiator) at 150 °C for 20 min. The reaction mixture was taken up in EtOAc, washed (H2O, 1 m (aq) NaOH, brine), dried (MgSO4) and concentrated in vacuo. The resultant crude residue was purified by column chromatography, eluting with 0–50 % EtOAc/hexane, to give 3-(3-fluorophenyl)-1H-indazole (196 mg, 62 %) as a white solid.
To a stirred suspension of 60 % NaH (72 mg, 1.79 mmol) in DMF (4 mL) was added 3-(3-fluorophenyl)-1H-indazole (95 mg, 0.448 mmol) and the reaction stirred for 10 min. To the reaction mixture 1-bromopinacolone (0.24 mL, 1.79 mmol) was added, the reaction heated at 75 °C and stirring continued for 66 h. The reaction mixture was cooled to room temperature, diluted with EtOAc, washed (H2O, brine), dried (MgSO4) and concentrated in vacuo. The crude residue was purified by column chromatography, eluting with 0–50 % EtOAc/hexane to give title compound 71 (80 mg, 58 %) was obtained as a white solid: 1H NMR ([D6]DMSO, 500 MHz): δ=8.12 (1 H, d, J=4.1 Hz), 7.84 (1 H, m), 7.71 (1 H, m), 7.59 (1 H, m), 7.55 (1 H, m), 7.45 (1 H, m), 7.27 (2 H, m), 5.75 (2 H, s), 1.38 ppm (9 H, s); [M+H]+=311; HRMS C19H19FN2O calcd: 311.1554, obsd: 311.1555.
By proceeding in a similar manner to compound 71, using 3-(3-fluorophenyl)-1H-indazole (or 3-phenyl-1H-indazole) and the corresponding alkylating agent, the following compounds were obtained.
1-(3-Phenyl-1H-indazol-1-yl)-3,3-dimethylbutan-2-one (60): 1H NMR ([D6]DMSO, 500 MHz): δ=8.09 (1 H, d, J=8.2 Hz), 7.97 (2 H, m), 7.54 (3 H, m), 7.43 (2 H, m), 7.25 (1 H, m), 5.73 (2 H, s), 1.28 ppm (9 H, s); 13C NMR (125 MHz, CDCl3): δ=26.4, 43.5, 53.4, 109.0, 121.2, 121.6, 122.1, 126.7, 127.6, 128.0, 128.8, 133.5, 141.8, 144.9, 207.9 ppm; [M+H]+=293; HRMS C19H20N2O calcd: 293.1648, obsd: 293.1658.
3-(3-Fluorophenyl)-1-(pyridin-2-ylmethyl)-1H-indazole (75): 1H NMR ([D6]DMSO, 500 MHz): δ=8.63 (1 H, d, J=4.5 Hz), 8.06 (1 H, d, J=8.5 Hz), 7.83 (1 H, m), 7.75 (1 H, m), 7.58 (1 H, m), 7.47 (2 H, m), 7.42 (1 H, m), 7.27 (1 H, m), 7.21 (1 H, m), 7.12 (1 H, m), 6.95 (1 H, d, J=8.0 Hz), 5.83 ppm (2 H, s); [M+H]+=304; HRMS C19H14FN3 calcd: 304.1245, obsd: 304.1258.
3-(3-Fluorophenyl)-1-(pyridin-3-ylmethyl)-1H-indazole (76): 1H NMR ([D6]DMSO, 500 MHz): δ=8.63 (1 H, d, J=2.0 Hz), 8.49 (1H dd, J=1.5, 5.0 Hz), 8.13 (1 H, d, J=8.5 Hz), 7.87 (2 H, t, J=8.5 Hz), 7.72 (1 H, dt, J=2.0, 9.5 Hz), 7.69 (1 H, dt, J=2.0, 9.5 Hz), 7.58 (1 H, m), 7.50 (1 H, m), 7.35 (1 H, dd, J=4.5, 8.0 Hz), 7.28 (2 H, m), 5.81 ppm (2 H, s); [M+H]+=304; HRMS C19H14FN3 calcd: 304.1245, obsd: 304.1235.
4-((3-(3-Fluorophenyl)-1H-indazol-1-yl)methyl)-2,5-dimethyloxazole (77): 1H NMR ([D6]DMSO, 500 MHz): δ=8.10 (1 H, d, J=8.5 Hz), 7.83 (1 H, m), 7.77 (1 H, m), 7.71 (1 H, m), 7.58 (1 H, m), 7.48 (1 H, m), 7.27 (2 H, m), 5.52 (2 H, bs), 3.35 (3 H, bs), 2.36 ppm (3 H, bs); [M+H]+=322; HRMS C19H16FN3O calcd: 322.1350, obsd: 322.1345.
3-((3-(3-Fluorophenyl)-1H-indazol-1-yl)methyl)-5-phenylisoxazole (78): 1H NMR ([D6]DMSO, 500 MHz): δ=8.15 (1 H, d, J=8.0 Hz), 7.86 (4 H, m), 7.77 (1 H, m), 7.59 (1 H, m), 7.51 (5 H, m), 7.30 (2 H, m), 6.95 ppm (2 H, s); [M+H]+=370; HRMS C23H16FN3O calcd: 370.1350, obsd: 370.1345.
3-((3-(3-Fluorophenyl)-1H-indazol-1-yl)methyl)-5-methylisoxazole (79): 1H NMR ([D6]DMSO, 500 MHz): δ=8.13 (1 H, d, J=8.50), 7.86 (1 H, m), 7.80 (1 H, m), 7.74 (1 H, m), 7.59 (1 H, m), 7.51 (1 H, m), 7.28 (2 H, m), 6.08 (1 H, s), 5.80 (2 H, s), 2.32 ppm (3 H, s); [M+H]+=308; HRMS C18H14FN3O calcd: 308.1194, obsd: 308.1179.
By proceeding in a similar manner to compound 71, using the corresponding boronic acid and 1-(3-iodo-1H-indazol-1-yl)-3,3-dimethylbutan-2-one, the following compounds were obtained:
1-(3-(4-Chlorophenyl)-1H-indazol-1-yl)-3,3-dimethylbutan-2-one (61): 1H NMR ([D6]DMSO, 500 MHz): δ=8.09 (1 H, d, J=8.3 Hz), 8.00 (2 H, m), 7.59 (2 H, m), 7.55 (1 H, d, J=8.5 Hz), 7.45 (1 H, m), 7.27 (1 H, m), 5.74 (2 H, s), 1.28 ppm (9 H, s); [M+H]+=327/329; HRMS C19H19ClN2O calcd: 327.1259, obsd: 327.1265.
1-(3-(1H-Indol-4-yl)-1H-indazol-1-yl)-3,3-dimethylbutan-2-one (62): 1H NMR ([D6]DMSO, 500 MHz): δ=11.21 (1 H, s), 8.00 (1 H, d, J=8.2 Hz), 7.55 (2 H, m), 7.44 (3 H, m), 7.24 (2 H, m), 6.88 (1 H, m), 5.75 (2 H, s), 1.30 ppm (9 H, s); [M+H]+=332; HRMS C21H21N3O calcd: 332.1757, obsd: 332.1754.
3,3-Dimethyl-1-(3-(1-methyl-1H-indol-4-yl)-1H-indazol-1-yl)butan-2-one (63): 1H NMR ([D6]DMSO, 500 MHz): δ=8.01 (1 H, d, J=8.4 Hz), 7.57 (3 H, m), 7.43 (1 H, t, J=7.5 Hz), 7.39 (1 H, d, J=3.2 Hz), 7.33 (1 H, t, J=7.8 Hz), 7.23 (1 H, t, J=7.5 Hz), 6.87 (1 H, d, J=3.0 Hz), 5.75 (2 H, s), 3.86 (3 H, s), 1.30 ppm (9 H, s); [M+H]+=346; HRMS C22H23N3O calcd: 346.1914, obsd: 346.1913.
3,3-Dimethyl-1-(3-(naphthalen-2-yl)-1H-indazol-1-yl)butan-2-one (64): 1H NMR ([D6]DMSO, 500 MHz): δ=8.56 (1 H, s), 8.30 (1 H, d, J=8.4 Hz), 8.14 (2 H, m), 8.06 (1 H, d, J=8.6 Hz), 7.97 (1 H, d, 7.7 Hz), 7.57 (3 H, m), 7.47 (1 H, m), 7.31 (1 H, t, J=7.5 Hz), 5.78 (2 H, s), 1.29 ppm (9 H, s); [M+H]+=343; HRMS C23H22N2O calcd: 343.1805, obsd: 343.1806.
3,3-Dimethyl-1-(3-(pyridin-4-yl)-1H-indazol-1-yl)butan-2-one (65): 1H NMR ([D6]DMSO, 500 MHz): δ=8.69 (2 H, dd, J=1.6, 6.1 Hz), 8.22 (1 H, d, J=8.3 Hz), 7.97 (2 H, dd, J=1.6, 6.2 Hz), 7.59 (1 H, d, J=8.5 Hz), 7.48 (1 H, t, J=7.5 Hz), 7.32 (1 H, t, J=7.5 Hz), 5.82 (2 H, s), 1.28 ppm (9 H, s); [M+H]+=294; HRMS C18H19N3O calcd: 294.1601, obsd: 294.1614.
1-(3-(Furan-2-yl)-1H-indazol-1-yl)-3,3-dimethylbutan-2-one (66): 1H NMR ([D6]DMSO, 500 MHz): δ=8.10 (1 H, d, J=8.2 Hz), 7.87 (1 H, d, J=1.7 Hz), 7.52 (1 H, d, J=8.5 Hz), 7.56 (2 H, m), 7.44 (1 H, m), 7.26 (1 H, m), 5.72 (2 H, s), 1.27 ppm (9 H, s); [M+H]+=283; HRMS C17H18N2O2 calcd: 283.1441, obsd: 283.1452.
1-(3-(3-Pyridyl)-1H-indazol-1-yl)-3,3-dimethylbutan-2-one (67): 1H NMR ([D6]DMSO, 500 MHz): δ=9.17 (1 H, m), 8.63 (1 H, dd, J=1.6, 4.8 Hz), 8.35 (1 H, dt, J=1.9 7.9 Hz), 8.12 (1 H, d, J=8.3 Hz), 7.56 (2 H, m), 7.47 (1 H, m), 7.29 (1 H, m), 5.77 (2 H, s), 1.28 ppm (9 H, s); [M+H]+=294; HRMS C18H19N3O calcd: 294.1601, obsd: 294.1616.
3,3-Dimethyl-1-(3-(thiophen-3-yl)-1H-indazol-1-yl)butan-2-one (68): 1H NMR ([D6]DMSO, 500 MHz): δ=8.13 (2 H, m), 7.69 (2 H, m), 7.49 (1 H, d, J=8.5 Hz), 7.42 (1 H, m), 7.25 (1 H, m), 5.70 (2 H, s), 1.27 ppm (9 H, s); [M+H]+=299; HRMS C17H18N2OS calcd: 299.1213, obsd: 299.1219.
1-(3-(1-Isobutyl-1H-pyrazol-4-yl)-1H-indazol-1-yl)-3,3-dimethylbutan-2-one (69): 1H NMR ([D6]DMSO, 500 MHz): δ=8.39 (1 H, s), 8.04 (1 H, d, J=8.1 Hz), 7.99 (1 H, s), 7.45 (1 H, d, J=8.4 Hz), 7.39 (1 H, m), 7.19 (1 H, t, J=7.1 Hz), 5.66 (2 H, s), 4.01 (2 H, d, J=7.2 Hz), 2.20 (1 H, m), 1.26 (9 H, s), 0.88 ppm (6 H, d, J=6.6 Hz); [M+H]+=339; HRMS C20H26N4O calcd: 339.2179, obsd: 339.2183.
1-(3-(3-Chlorophenyl)-1H-indazol-1-yl)-3,3-dimethylbutan-2-one (70): 1H NMR ([D6]DMSO, 500 MHz): δ=8.11 (1 H, d, J=8.2 Hz), 7.97 (1 H, m), 7.95 (1 H, m), 7.56 (2 H, m), 7.47 (2 H, m), 7.28 (1 H, t, J=7.5 Hz), 5.77 (2 H, s), 1.30 ppm (9 H, s); [M+H]+=327/329; HRMS C19H19ClN2O calcd: 327.1259, obsd: 327.1262.
Compound 59 was obtained from ChemDiv.
3-(tert-Butylsulfonylmethyl)-1-(3-fluorophenyl)-1H-indazole (73): To a stirred suspension of indazole-3-carboxylic acid (5 g, 30.8 mmol) in MeOH (125 mL) was added concd H2SO4 (0.5 mL) and the reaction heated at reflux for 16 h. The reaction mixture was cooled to room temperature and the solvent removed in vacuo. The resultant crude residue was taken up in CH2Cl2, washed (saturated NaHCO3 solution), dried (MgSO4) and concentrated in vacuo to give methyl 1H-indazole-3-carboxylate (4.95 g, 91 %) as a white solid; [M+H]+=177.
To a stirred solution of methyl 1H-indazole-3-carboxylate (881 mg, 5 mmol) in CH2Cl2 (125 mL), was added 3-fluorophenylboronic acid (1.40 g, 10 mmol), pyridine (0.81 mL, 10 mmol), copper(II) acetate (1.36 g, 7.5 mmol) and molecular sieves (4 Å, 3.82 g) and the reaction stirred (open to air) for 46 h. The reaction mixture was then filtered through a pad of Celite and concentrated in vacuo to give a crude residue which was purified by column chromatography, eluting with 0–50 % Et2O/hexane, to give methyl 1-(3-fluorophenyl)-1H-indazole-3-carboxylate (332 mg, 25 %) as a white solid; [M+H]+=271.
To a stirred solution of methyl 1-(3-fluorophenyl)-1H-indazole-3-carboxylate (329 mg, 1.2 mmol) in THF (10 mL) at 0 °C, under an inert atmosphere, was added LiAlH4 (185 mg, 4.9 mmol), the reaction warmed to room temperature and stirred for 16 h. The reaction mixture was quenched (Na2SO4⋅10 H2O), filtered through a pad of Celite and concentrated in vacuo. The resultant crude residue was taken up in 3:1 THF/CH2Cl2 (12 mL), PPh3 (479 mg, 1.826 mmol) and N-chlorosuccinimide (NCS; 244 mg, 1.8 mmol) added and the reaction stirred for 24 h. Extra PPh3 (479 mg, 1.8 mmol) and NCS (244 mg, 1.8 mmol) were then added and stirring continued for 24 h. The reaction mixture was then concentrated in vacuo to give a crude residue which was taken up in EtOAc, washed (saturated NaHCO3 solution, H2O, and brine), dried (MgSO4) and concentrated in vacuo. The resultant crude residue was purified by column chromatography, eluting with 0–50 % Et2O/hexane, to give 3-(chloromethyl)-1-(3-fluorophenyl)-1H-indazole (143 mg, 45 %) as a straw-coloured oil; [M+H]+=261/263.
To a stirred solution of 3-(chloromethyl)-1-(3-fluorophenyl)-1H-indazole (46 mg, 0.18 mmol) in CH3CN (3 mL) was added Cs2CO3 (63 mg, 0.19 mmol) and tert-butylthiol (0.022 mL, 0.19 mmol) and the reaction stirred for 16 h. Extra Cs2CO3 (32 mg, 0.097 mmol) and tert-butylthiol (0.011 mL, 0.097 mmol) were then added and stirring continued for 6 h. The reaction mixture was taken up in CH2Cl2, washed (H2O, brine), dried (MgSO4) and concentrated in vacuo. The resultant crude residue was purified by column chromatography, eluting with 0–50 % Et2O/hexane, to give 3-(tert-butylthiomethyl)-1-(3-fluorophenyl)-1H-indazole (45 mg, 81 %) as a colourless oil; [M+H]+=315.
To a stirred solution of 3-(tert-butylthiomethyl)-1-(3-fluorophenyl)-1H-indazole (45 mg, 0.14 mmol) in CH2Cl2 (5 mL) was added 70 % mCPBA (141 mg, 0.57 mmol) and the reaction stirred for 2 h. NaHSO3 solution (2 m aq) was added, the layers separated, the organic layer washed (saturated NaHCO3 solution) and filtered through a pad of silica, eluting with CH2Cl2 and EtOAc. The filtrate was concentrated in vacuo to give a crude residue which was purified by reversed-phase HPLC (method A), to give title compound 73 (35 mg, 71 %) as a white solid: 1H NMR ([D6]DMSO, 500 MHz): δ=7.97 (1 H, dt, J=0.9, 8.1 Hz), 7.95 (1 H, dt, J=1.0, 8.5 Hz), 7.67 (3 H, m), 7.57 (1 H, m), 7.36 (1 H, m), 7.30 (1 H, m), 5.00 (2 H, s), 1.44 ppm (9 H, s); [M+H]+=347; HRMS C18H19FN2O2S calcd: 347.1224, obsd: 347.1236.
3-(tert-Butylsulfonylmethyl)-1-phenyl-1H-indazole (72): By proceeding in a similar manner to 3-(tert-butylsulfonylmethyl)-1-(3-fluorophenyl)-1H-indazole (see compound 73), except using phenylboronic acid, title compound 72 (35 mg, 81 %) was obtained as a white solid. 1H NMR ([D6]DMSO, 500 MHz): δ=7.96 (1 H, dt, J=0.9, 8.1 Hz), 7.87 (1 H, dt, J=0.9, 8.6 Hz), 7.79 (2 H, m), 7.63 (2 H, m), 7.54 (1 H, m), 7.45 (1 H, m), 7.33 (1 H, m), 5.00 (2 H, s), 1.44 ppm (9 H, s); 13C NMR (125 MHz, CDCl3): δ=23.9, 48.0, 61.1, 110.5, 121.9, 122.3, 123.0, 124.8, 127.1, 127.7, 129.5, 135.8, 139.7, 140.1 ppm; [M+H]+=329; HRMS C18H20N2O2S calcd: 329.1318, obsd: 329.1302.
1-(tert-Butylsulfonylmethyl)-3-(3-fluorophenyl)-1H-indazole (74): 3-(3-Fluorophenyl)-1H-indazole (see compound 71) (0.106 g, 0.5 mmol) was alkylated with tert-butyl(chloromethyl)sulfane (0.104 g, 0.75 mmol), as in accordance with reported procedures28 to give the intermediate 1-(tert-butylthiomethyl)-3-(3-fluorophenyl)-1H-indazole (0.055 g, 35 %). 1H NMR (CDCl3, 300 K) δ=8.03 (1 H, dt, J=7.5 and 0.8 Hz), 7.79 (1 H, dt, J=7.7 and 1.2 Hz), 7.71 (1 H, ddd, J=10.0, 2.5 and 1.0 Hz), 7.65 (1 H, dt, J=8.5 and 0.8 Hz), 7.51-7.47 (2 H, m), 7.29 (1 H, m), 7.12 (1 H, tdd, J=8.5, 2.5 and 0.8 Hz), 5.68 (2 H, s), 1.34 ppm (9 H, s); [M+H]+=315.
A solution of this sulfide in CH2Cl2 (2 mL) was treated with mCPBA (Aldrich, ≤77 %; 0.11 g, ≤0.5 mmol) and stirred at room temperature for 90 min. The reaction was quenched with 2 m aqueous NaHSO3 (10 mL) and saturated aqueous NaHCO3 (10 mL). The organic phase was applied to a plug of silica and eluted with CH2Cl2/EtOAc (0→100 %) to give the title compound 74 as a white solid (0.054 g, 89 %): 1H NMR (CDCl3, 300 K): δ=8.01 (1 H, dt, J=8.2 and 0.9 Hz), 7.78 (1 H, dt, J=8.5 and 0.8 Hz), 7.76 (1 H, ddd, J=11.7, 1.4 and 1.0 Hz), 7.67 (1 H, ddd, J=9.9, 2.5 and 1.5 Hz), 7.55 (1 H, ddd, J=8.4, 7.0 and 1.0 Hz) 7.50 (1 H, td, J=8.1 and 5.9 Hz), 7.34 (1 H, ddd, J=8.2, 6.8 and 0.8 Hz), 7.15 (1 H, tdd, J=8.4, 2.6 and 0.9 Hz), 5.76 (2 H, s), 1.43 ppm (9 H, s); 13C NMR (125 MHz, CDCl3): δ=23.4, 61.2, 64.6, 110.0, 114.4, 115.5, 121.1, 122.3, 123.2, 127.9, 130.5, 134.8, 142.2, 145.1, 161.2, 164.1 ppm; [M+H]+=347; HRMS C18H19FN2O2S calcd: 347.1224, obsd: 347.1214.
2-(3-(3-Fluorophenyl)-1H-indazol-1-yl)-N-(3-(4-methylpiperazin-1-yl)propyl)acetamide (80): To a stirred suspension of 60 % NaH (754 mg, 18.9 mmol) in DMF (20 mL) under an inert atmosphere, was added 3-(3-fluorophenyl)-1H-indazole (1.0 g, 4.7 mmol) in DMF (10 mL) and the reaction was stirred for 15 min. To the reaction mixture was added ethyl bromoacetate (2.09 mL, 18.9 mmol), the reaction was warmed to 75 °C and stirring continued for 66 h. The reaction mixture was cooled to room temperature, taken up in EtOAc, washed (H2O, brine), dried (MgSO4) and concentrated in vacuo. The crude residue was taken up in 1:1 THF/H2O (30 mL) and 2 m (aq) NaOH (9.43 mL, 18.9 mmol), the reaction was heated at 50 °C and stirred for 3 h. The reaction was cooled to room temperature, diluted with H2O, extracted (EtOAc), the aqueous layer acidified (1 m (aq) HCl, pH 2), extracted (EtOAc). The organic layer was dried (MgSO4) and concentrated in vacuo to give 2-(3-(3-fluorophenyl)-1H-indazol-1-yl)acetic acid (980 mg, 77 %) as a white foam; [M+H]+=471.
By proceeding in a similar manner to 26, except using 2-(3-(3-fluorophenyl)-1H-indazol-1-yl)acetic acid and 1-(2-aminopropyl)-4-methylpiperazine, the title compound 80 (65 mg, 43 %) was obtained as a white solid: 1H NMR ([D6]DMSO, 500 MHz): δ=8.25 (1 H, t, J=5.4 Hz), 8.11 (1 H, d, J=8.4 Hz), 7.85 (1 H, d, J=7.9 Hz), 7.72 (1 H, m), 7.68 (1 H, d, J=8.9 Hz), 7.58 (1 H, m), 7.47 (1 H, t, J=7.7 Hz), 7.27 (2 H, m), 5.16 (2 H, s), 3.12 (2 H, m), 2.51 (8 H, m), 2.27 (2 H, m), 2.12 (3 H, s), 1.58 ppm (2 H, m); [M+H]+=410; HRMS C23H28FN5O calcd: 410.2351, obsd: 410.2363.
By proceeding in a similar manner to 26, except using 2-(3-(3-fluorophenyl)-1H-indazol-1-yl)acetic acid and the appropriate amine, the following compounds were prepared:
N-(3-(1H-Imidazol-1-yl)propyl)-2-(3-(3-fluorophenyl)-1H-indazol-1-yl)acetamide (81): 1H NMR ([D6]DMSO, 500 MHz): δ=8.41 (1 H, t, J=5.4 Hz), 8.25 (1 H, s), 8.12 (1 H, d, J=8.3 Hz), 7.84 (1 H, d, J=7.9 Hz), 7.72 (2 H, m), 7.58 (1 H, m), 7.48 (1 H, t, J=7.7 Hz), 7.44 (1 H, m), 7.27 (2 H, m), 7.23 (1 H, m), 5.20 (2 H, s), 4.10 (2 H, m), 3.09 (2 H, m), 1.93 ppm (2 H, m); [M+H]+=378; HRMS C21H20FN5O calcd: 378.1725, obsd: 378.1739.
tert-Butyl-4-(2-(2-(3-(3-fluorophenyl)-1H-indazol-1-yl)acetamido)ethyl)piperazine-1-carboxylate (82): 1H NMR ([D6]DMSO, 500 MHz) δ mixture of rotamers δ=8.12 (2 H, m), 7.86 (1 H, d, J=7.6 Hz), 7.72 (2 H, m), 7.59 (1 H, m), 7.48 (1 H, m), 7.28 (2 H, m), 5.22 (2 H, s), 3.20 (2 H, m), 2.51 (8 H, m), 2.37 (2 H, m), 1.40(5 H, s), 1.39 ppm (4 H, s); [M+H]+=482; HRMS C26H32FN5O3 calcd: 482.2562, obsd: 482.2570.
N-(2-(Dimethylamino)ethyl)-2-(3-(3-fluorophenyl)-1H-indazol-1-yl)acetamide (83): NMR 1H NMR ([D6]DMSO, 500 MHz): δ=8.22 (1 H, t, J=5.5 Hz), 8.12 (1 H, d, J=8.2 Hz), 7.85 (1 H, d, J=7.9 Hz), 7.72 (1 H, m), 7.69 (1 H, d, J=8.5 Hz), 7.59 (1 H, m), 7.47 (1 H, m), 7.26 (2 H, m), 5.19 (2 H, s), 3.20 (2 H, m), 2.31 (2 H, m), 2.15 ppm (6 H, s); [M+H]+=341; HRMS C19H21FN4O calcd: 341.1772, obsd: 341.1767.
N-(3-(Dimethylamino)propyl)-2-(3-(3-fluorophenyl)-1H-indazol-1-yl)acetamide (84): 1H NMR ([D6]DMSO, 500 MHz): δ=8.26 (1 H, t, J=5.3 Hz), 8.13 (1 H, d, J=8.1 Hz), 7.85 (1 H, d, J=7.8 Hz), 7.71 (1 H, m), 7.68 (1 H, d, J=8.6 Hz), 7.59 (1 H, m), 7.48 (1 H, m), 7.26 (2 H, m), 5.16 (2 H, s), 3.13 (2 H, m), 2.20 (2 H, m), 2.01 (6 H, s), 1.54 ppm (2 H, m); 13C NMR (125 MHz, CDCl3): δ=24.4, 40.7, 44.5, 52.6, 59.4, 109.5, 114.2, 115.1, 121.2, 122.1, 123.0, 127.3, 130.5, 135.3, 141.7, 144.0, 162.2, 164.2, 167.3 ppm; [M+H]+=355; HRMS C20H23FN4O calcd: 355.1856, obsd: 355.1862.
N-Methyl-(3-(dimethylamino)propyl)-2-(3-(3-fluorophenyl)-1H-indazol-1-yl)acetamide (85): 1H NMR ([D6]DMSO, 500 MHz) mixture of rotamers; δ=8.12 (1 H, d, J=8.4 Hz), 7.85 (1 H, d, J=7.9 Hz), 7.71 (1 H, m), 7.60(2 H, m), 7.44 (1 H, m), 7.25 (2 H, m), 5.60 (1 H, s), 5.50 (1 H, s), 3.48 (1 H, m), 3.30 (1 H, m), 3.14 (1.5 H, s), 2.84 (1.5 H, s), 2.27 (1 H, m), 2.20 (3 H, s), 2.16 (1 H, m), 2.09 (3 H, s), 1.77 (1 H, m), 1.60 ppm (1 H, m); [M+H]+=369; HRMS C21H25FN4O calcd: 369.2085, obsd: 369.2097.
N-(4-Chlorobenzyl)-2-(3-(3-fluorophenyl)-1H-indazol-1-yl)acetamide (86): 1H NMR ([D6]DMSO, 500 MHz): δ=8.78 (1 H, t, J=5.7 Hz), 8.12 (1 H, d, J=8.1 Hz), 7.85 (1 H, d, J=7.8 Hz), 7.71 (2 H, m), 7.59 (1 H, m), 7.48 (1 H, t, J=7.4 Hz), 7.38 (2 H, d, J=8.2 Hz), 7.32 (2 H, d, J=8.3 Hz), 7.28 (2 H, m), 5.26 (2 H, s), 4.32 ppm (2 H, d, J=6.0 Hz); [M+H]+=394; HRMS C22H17ClFN3O calcd: 394.1117, obsd: 394.1111.
2-(3-(3-Fluorophenyl)-1H-indazol-1-yl)-N-(2-(4-methylpiperazin-1-yl)ethyl)acetamide (90): 1H NMR ([D6]DMSO, 500 MHz): δ=8.12 (1 H, d, J=8.1 Hz), 8.02 (1 H, t, J=5.4 Hz), 7.86 (1 H, d, J=8.1 Hz), 7.72 (1 H, m), 7.71 (1 H, d, J=8.6 Hz), 7.59 (1 H, m), 7.47 (1 H, t, J=7.8 Hz), 7.27 (2 H, m), 5.19 (2 H, s), 3.20 (2 H, m), 2.51 (8 H, m), 2.34 (2 H, m), 2.10 ppm (3 H, s); [M+H]+=396; HRMS C22H26FN5O calcd: 396.2194, obsd: 396.2212.
N,N-Diethyl-2-(3-(3-fluorophenyl)-1H-indazol-1-yl)acetamide (87): 3-(3-Fluorophenyl)-1H-indazole (0.106 g, 0.5 mmol), prepared as in 71, was alkylated with 2-chloro-N,N-diethylacetamide (0.102 mL, 0.74 mmol) to give the title compound 87 (0.073 g, 45 %) as a white solid: 1H NMR (CDCl3, 300 K): δ=8.02 (1 H, d, J=8.2 Hz), 7.78 (1 H, d, J=7.8 Hz), 7.70 (1 H, ddd, J=10.1 2.2 and 1.5 Hz), 7.53 (1 H, d, J=8.5 Hz), 7.50-7.45 (2 H, m), 7.27 (1 H, m), 7.11 (1 H, td, J=7.5 and 2.0 Hz), 5.30 (2 H, s), 3.54 (2 H, q, J=7.1 Hz), 3.43 (2 H, q, J=7.1 Hz), 1.17 ppm (6 H, m); [M+H]+=326; HRMS C19H20FN3O calcd: 326.1663, obsd: 326.1663.
2-(3-(3-Fluorophenyl)-1H-indazol-1-yl)-N,N-dimethylacetamide (88): 3-(3-Fluorophenyl)-1H-indazole (0.106 g, 0.5 mmol), prepared as in 71, was alkylated with 2-chloro-N,N-dimethylacetamide (0.191 g, 1.6 mmol) to give the title compound 88 (0.114 g, 77 %) as a white solid. 1H NMR (CDCl3, 300 K): δ=1 H, d, J=8.2 Hz), 7.78 (1 H, d, J=7.7 Hz), 7.70 (1 H, ddd, J=10.1, 2.4 and 1.6 Hz), 7.52-7.45 (3 H, m), 7.27 (1 H, m), 7.10 (1 H, ddt, J=8.5, 2.6 and 0.8 Hz), 5.32 (2 H, s), 3.18 (3 H, s), 3.01 ppm (3 H, s); [M+H]+=298; HRMS C17H16FN3O calcd: 298.1350, obsd: 298.1348.
2-(3-(3-Fluorophenyl)-1H-indazol-1-yl)-1-(piperidin-1-yl)ethanone (89): Synthesis previously described.20 3-(3-Fluorophenyl)-1H-indazole (0.106 g, 0.50 mmol), prepared as in 71, was alkylated with 2-chloro-1-piperidin-1-ylethanone (0.350 g, 2.2 mmol) to give the title compound (0.05 g, 30 %) as a white solid: 1H NMR (CDCl3, 300 K): δ=8.03 (1 H, d, J=8.2 Hz), 7.77 (1 H, d, J=7.8 Hz), 7.69 (1 H, dt, J=10.0 and 1.7 Hz), 7.55 (1 H, d, J=8.5 Hz), 7.50-7.45 (2 H, m), 7.27 (1 H, m), 7.11 (1 H, td, J=8.4 and 1.4 Hz), 5.32 (2 H, s), 3.60 (4 H, m), 1.64 (2 H, m), 1.54 (2 H, m), 1.48 ppm (2 H, m); [M+H]+=338; HRMS C20H20FN3O calcd: 338.1663, obsd: 338.1660.
Biological assays
Trypanothione synthetase assay: The TryS enzyme assays were conducted as reported.20
Cell-based assays: Proliferation assays using bloodstream form T. brucei and human MRC5 fibroblasts were conducted as reported.17
Abbreviations
Glutathione (GSH); human African trypanosomiasis (HAT); spermidine (Spd); single knockout (SKO); Trypanothione synthetase (TryS).
Acknowledgments
We thank Irene Hallyburton and Bhavya Rao for technical support in performing the cell potency assays, Daniel James for data management support, and Suzanne Norval for technical support in performing the metabolic stability experiments. This work was funded by the Wellcome Trust (WT 077705, WT 079838, and WT 083481).
References
- 1.Stuart KD, Brun R, Croft SL, Fairlamb AH, Gurtler RE, McKerrow JH, Reed S, Tarleton RL. J. Clin. Invest. 2008;118:1301–1310. doi: 10.1172/JCI33945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fairlamb AH. Trends Parasitol. 2003;19:488–494. doi: 10.1016/j.pt.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 3.Barrett MP, Fairlamb AH. Parasitol. Today. 1999;15:136–140. doi: 10.1016/s0169-4758(99)01414-3. [DOI] [PubMed] [Google Scholar]
- 4.El-Sayed NM, Myler PJ, Blandin G, Berriman M, Crabtree J, Aggarwal G, Caler E, Renauld H, Worthey EA, Hertz-Fowler C, Ghedin E, Peacock C, Bartholomeu DC, Haas BJ, Tran AN, Wortman JR, Alsmark UC, Angiuoli S, Anupama A, Badger J, Bringaud F, Cadag E, Carlton JM, Cerqueira GC, Creasy T, Delcher AL, Djikeng A, Embley TM, Hauser C, Ivens AC, Kummerfeld SK, Pereira-Leal JB, Nilsson D, Peterson J, Salzberg SL, Shallom J, Silva JC, Sundaram J, Westenberger S, White O, Melville SE, Donelson JE, Andersson B, Stuart KD, Hall N. Science. 2005;309:404–409. [Google Scholar]
- 5.Fairlamb AH, Cerami A. Annu. Rev. Microbiol. 1992;46:695–729. doi: 10.1146/annurev.mi.46.100192.003403. [DOI] [PubMed] [Google Scholar]
- 6.Krauth-Siegel RL, Meiering SK, Schmidt H. Biol. Chem. 2003;384:539–549. doi: 10.1515/BC.2003.062. [DOI] [PubMed] [Google Scholar]
- 7.Müller S, Liebau E, Walter RD, Krauth-Siegel RL. Trends Parasitol. 2003;19:320–328. doi: 10.1016/s1471-4922(03)00141-7. [DOI] [PubMed] [Google Scholar]
- 8.Augustyns K, Amssoms K, Yamani A, Rajan PK, Haemers A. Curr. Pharm. Des. 2001;7:1117–1141. doi: 10.2174/1381612013397564. [DOI] [PubMed] [Google Scholar]
- 9.Fairlamb AH, Blackburn P, Ulrich P, Chait BT, Cerami A. Science. 1985;227:1485–1487. doi: 10.1126/science.3883489. [DOI] [PubMed] [Google Scholar]
- 10.Oza SL, Ariyanayagam MR, Aitcheson N, Fairlamb AH. Mol. Biochem. Parasitol. 2003;131:25–33. doi: 10.1016/s0166-6851(03)00176-2. [DOI] [PubMed] [Google Scholar]
- 11.Comini M, Menge U, Flohé L. Biol. Chem. 2003;384:653–656. doi: 10.1515/BC.2003.072. [DOI] [PubMed] [Google Scholar]
- 12.Krieger S, Schwarz W, Ariyanayagam MR, Fairlamb AH, Krauth-Siegel RL, Clayton C. Mol. Microbiol. 2000;35:542–552. doi: 10.1046/j.1365-2958.2000.01721.x. [DOI] [PubMed] [Google Scholar]
- 13.Wilkinson SR, Horn D, Prathalingam SR, Kelly JM. J. Biol. Chem. 2003;278:31640–31646. doi: 10.1074/jbc.M303035200. [DOI] [PubMed] [Google Scholar]
- 14.Comini MA, Guerrero SA, Haile S, Menge U, Lunsdorf H, Flohé L. Free Radical Biol. Med. 2004;36:1289–1302. doi: 10.1016/j.freeradbiomed.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 15.Wyllie S, Oza SL, Patterson S, Spinks D, Thompson S, Fairlamb AH. Mol. Microbiol. 2009;74:529–540. doi: 10.1111/j.1365-2958.2009.06761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frearson JA, Wyatt PA, Gilbert IH, Fairlamb AH. Trends Parasitol. 2007;23:589–595. doi: 10.1016/j.pt.2007.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Spinks D, Shanks EJ, Cleghorn LAT, McElroy S, Jones D, James D, Fairlamb AH, Frearson JA, Wyatt PG, Gilbert IH. ChemMedChem. 2009;4:2060–2069. doi: 10.1002/cmdc.200900262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fyfe PK, Oza SL, Fairlamb AH, Hunter WN. J. Biol. Chem. 2008;283:17672–17680. doi: 10.1074/jbc.M801850200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oza SL, Chen S, Wyllie S, Coward JK, Fairlamb AH. FEBS J. 2008;275:5408–5421. doi: 10.1111/j.1742-4658.2008.06670.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Torrie LS, Wyllie S, Spinks D, Oza SL, Thompson S, Harrison JR, Gilbert IH, Wyatt PG, Fairlamb AH, Frearson JA. J. Biol. Chem. 2009;284:36137–36145. doi: 10.1074/jbc.M109.045336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hopkins AL, Groom CR, Alex A. Drug Discovery Today. 2004;9:430–431. doi: 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- 22.Dunn D, Husten J, Ator MA, Chatterjee S. Bioorg. Med. Chem. Lett. 2007;17:542–545. doi: 10.1016/j.bmcl.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 23.Mochizuki M, Miura S. 2009. (Takeda Pharmaceutical Co., Ltd.), WO 2009119088.
- 24.Goldfarb DS. 2009. (University of Rochester, NY, USA), US 20090163545.
- 25.Edwards ML, Cox PJ, Amendola S, Deprets SD, Gillespy TA, Edlin CD, Morley AD, Gardner CJ, Pedgrift B, Bouchard H, Babin DGL, Le Brun A, Majid TN, Reader JC, Payne LJ, Khan NM, Cherry M. 2003. (Aventis Pharmaceuticals Inc.), WO 2003035065.
- 26.Jones DC, Hallyburton I, Stojanovski L, Read KD, Frearson JA, Fairlamb AH. Biochem Pharmacol. 2010;80:1478–1486. doi: 10.1016/j.bcp.2010.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ariyanayagam MR, Oza SL, Guther ML, Fairlamb AH. Biochem. J. 2005;391:425–432. doi: 10.1042/BJ20050911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beight DW, Mehdi S, Koehl JR, Flynn GA. Bioorg. Med. Chem. Lett. 1996;6:2053–2058. [Google Scholar]
- 29.Fujimura Y, Nagano H, Matcunga I, Shindo M. Chem. Pharm. Bull. 1984;32:3252–3254. [Google Scholar]