

Abstract
Direct arylations of pyridine N-oxide (PyO), a convenient method to prepare 2-arylpyridines, catalyzed by Pd(OAc)2 and PtBu3 have been proposed to occur by the generation of a PtBu3-ligated arylpalladium acetate complex (PtBu3)Pd(Ar)(OAc) and the reaction of this complex with PyO. We provide strong evidence that (PtBu3)Pd(Ar)(OAc) does not react directly with pyridine N-oxide. Instead, our data imply that the cyclometallated complex [Pd(OAc)(tBu2PCMe2CH2)]2, which is generated from the decomposition of (PtBu3)Pd(Ar)(OAc), reacts with PyO and serves as a catalyst for the reaction of PyO with (PtBu3)Pd(Ar)(OAc). The reaction of PyO with (PtBu3)Pd(Ar)(OAc) occurs with an induction period, and the reaction of (PtBu3)Pd(Ar)(OAc) with excess PyO in the presence of [Pd(OAc)(tBu2PCMe2CH2)]2 is zero-order in (PtBu3)Pd(Ar)(OAc). Moreover, the rates of reactions of PyO with bromobenzene catalyzed by [Pd(OAc)(tBu2PCMe2CH2)]2 and [Pd(PtBu3)2] depend on the concentration of [Pd(OAc)(tBu2PCMe2CH2)]2, but not on the concentration of [Pd(PtBu3)2]. Finally, the reaction of (PtBu3)Pd(Ar)(OAc) with the model heteroarylpalladium complex containing a cyclometallated phosphine [(PEt3)Pd(2-benzothienyl)(tBu2PCMe2CH2)] rapidly formed the arylated heterocycle. Together, these data imply that the rate-determining C-H bond cleavage occurs between PyO and the cyclometallated [Pd(OAc)(tBu2PCMe2CH2)]2, rather than between PyO and (PtBu3)Pd(Ar)(OAc). In this case, the resulting heteroarylpalladium complex transfers the heteroaryl group to (PtBu3)Pd(Ar)(OAc), and C-C bondformation occurs from (PtBu3)Pd(Ar)(2-pyridyl oxide). This mechanism proposed for the direct arylation of PyO constitutes an example of C-H bond functionalization in which C-H activation occurs at one metal center, and the activated moiety undergoes functionalization after transfer to a second metal center.
Direct arylation, the reaction of aryl halides with arenes or heteroarenes to form biaryl or aryl-heteroaryl products, is an attractive alternative to traditional cross coupling because it occurs without the need to prepare organometallic or main-group reagents.1 For example, the direct arylation of pyridine N-oxide with aryl halides,2 the subject of the mechanistic studies in this paper, leads to the selective formation of 2-aryl pyridine derivatives after reduction to the pyridine without the need for 2-pyridyl organometallic reagents, which are often unstable and difficult to synthesize. The scope of direct arylation has broadened to encompass various heteroarenes,3 arenes with directing groups,4 electron-deficient arenes,5 and even simple arenes such as benzene6 as coupling partners.
However, little experimental information on the mechanism of direct arylations is available, especially on the mechanism by which the aryl or heteroaryl C-H bond is cleaved. Phosphine-ligated arylpalladium carboxylate complexes LPd(Ar)(OC(O)R) are typically proposed to react with heteroarenes or arenes to form biarylpalladium complexes through a concerted metallation-deprotonation (CMD) pathway,7 but we recently reported that a “ligandless” species is involved in the direct arylation of benzene,8 not the proposed phosphine-ligated complex.
In contrast, DFT calculations suggested that phosphine-ligated arylpalladium acetates are competent to undergo C-H bond cleavage with various heteroarenes, including pyridine N-oxides.7c A recent experimental study showed that the reaction of isolated (PtBu3)Pd(Ph)(OAc) with 4-nitropyridine N-oxide formed the arylated product, but the yield of this reaction (48%) was lower than that for the analogous catalytic process.9 Thus, the detailed mechanism of the cleavage of the aromatic C-H bonds in the direct arylation of pyridine N-oxides remains uncertain.
We report the synthesis and full characterization of PtBu3-ligated arylpalladium acetate (PtBu3)Pd(Ar)(OAc),10 and detailed mechanistic studies on the direct arylation of pyridine N-oxide with this isolated complex. Inconsistent with previous proposals, the direct reaction of isolated (PtBu3)Pd(Ar)(OAc) with PyO does not form arylated products. Instead, the cyclometallated complex [Pd(OAc)(tBu2CMe2CH2)]2, generated from the decomposition of the PtBu3-ligated arylpalladium acetate complex, catalyzes the reaction between (PtBu3)Pd(Ar)(OAc) and PyO to form the arylated product. Our data imply that this cyclometallated palladium acetate complex directly reacts with PyO to cleave the aromatic C-H bond. Thus, this reaction occurs by a cooperative C-H bond functionalization process in which one metal fragment cleaves the C-H bond, and a second fragment functionalizes the moiety resulting from this C-H bond cleavage event.
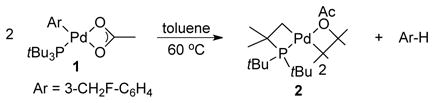
The synthesis of (PtBu3)Pd(Ar)(OAc) is outlined in Scheme 1. The reaction of Pd(PtBu3)2 with 3-fluoromethyl phenyl iodide in the presence of AgOAc formed the PtBu3-ligated arylpalladium acetate 1 in acceptable yield for our studies. Complex 1 was characterized by elemental analysis and NMR spectroscopy. The fluoromethyl moiety allowed us to monitor reactions by 19F NMR spectroscopy. The fluorine atom does not affect the catalytic process; the reaction of 3-fluoromethyl phenyl bromide with pyridine N-oxide catalyzed by Pd(OAc)2 and PtBu3 generated the 2-aryl pyridine oxide in good yield (77%).
Scheme 1.

Synthesis of (PtBu3)Pd(Ar)(OAc) (1) and Reaction of (1) with PyO under the previously reported conditions.a
a Yield of arylated product determined by 19F NMR.
In contrast to the 77% yield of the catalytic reaction, the stoichiometric reaction of isolated complex 1 with pyridine N-oxide at 120 °C in toluene formed the arylated pyridine N-oxide in a lower 52% yield. This observation was consistent with a previous report in which a similar difference in yield was obtained between the stoichiometric reaction of an isolated PtBu3-ligated arylpalladium acetate complex (PtBu3)Pd(Ph)(OAc) with 4-nitropyridine N-oxide and the catalytic reaction of Ph-Br with 4-nitropyridine N-oxide.9
These differences in yield between the catalytic reactions and the stoichiometric reactions led us to study the reaction of isolated complex 1 in more detail. Figure 1 shows the profile of the decay of 1 during the reaction of 1 with pyridine N-oxide at 60 °C in toluene, as measured by 19F NMR spectroscopy. This reaction occurred with an induction period, followed by a relatively linear decay of 1. The yield of the arylated product was 55% at the end of the reaction, as determined by 19F NMR. Further analysis of the NMR data showed that less than 1% of the arylated product was formed during the induction period. Instead, fluoromethylbenzene and the cyclometallated complex [Pd(μ2-OAc)(κ2-tBu2CMe2CH2)]2 (2)11 were formed during this induction period (eq 1). Compound 2 forms from decomposition of 1; heating 1 in toluene for 8 h at 60 °C gave 2 in 92% yield.
Figure 1.

Decay of (PtBu3)Pd(Ar)(OAc) (1) in toluene at 60 °C in the presence of 20 equiv of pyridine N-oxide, 1.0 equiv of PtBu3 and 2.5 equiv of K2CO3 with no additive (black) or with 1 equiv of added complex 2 (blue).
 |
(1) |
The formation of cyclometallated 2 during the induction period suggested that this complex could be involved in the reaction of PtBu3-ligated 1 with pyridine N-oxide. To test this hypothesis, the reaction of 1 with PyO and one equivalent of added 2 was conducted. No induction period was observed during this process, and the 2-aryl pyridine N-oxide was formed in yield (84%) that is as high as that of the catalytic process. Moreover, a zero-order decay of complex 1 was observed for the reaction with added complex 2, suggesting that the rate-determining step12 occurred without the involvement of (PtBu3)Pd(Ar)(OAc). Finally, the concentration of 2 was constant during this reaction, making complex 2 a catalyst for the stoichiometric reaction of PyO with 1.
To assess whether the C-H cleavage step occurs by reaction of pyridine N-oxide with complex 2, the order of the reaction in each reagent was measured. Figure 2 shows plots of the reaction rate versus [PyO] and [2]1/2. A linear fit of the rate vs the concentration of pyridine N-oxide from 0.2 to 0.6 M was obtained, indicating the reaction was first-order in PyO. A linear fit of the rate vs [2]1/2 and a non-linear fit of the rate vs [2] revealed a half-order, or at least partial order, dependence on the concentration of complex 2 from 0.015 to 0.15 M. This partial order in [2] implies that the reaction occurs by a pre-equilibrium between the observed dimeric 2 and a catalytically active monomer before the rate-determining C-H bond cleavage step. These results, along with the zero-order dependence on the concentration of PtBu3-ligated 1, clearly support the proposal that the C-H cleavage occurs by reaction of pyridine N-oxide with complex 2, not the arylpalladium acetate complex 1.
Figure 2.

Plots of rate versus [PyO] and [2]1/2 in toluene at 60 °C for the reaction of complex 1 with PyO.
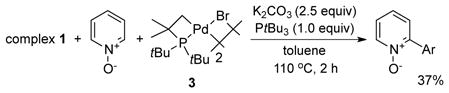
To determine whether the cyclometallated PtBu3 ligand or the acetate ligand is involved in the cleavage of the aromatic C-H bond (Scheme 2, pathway a vs b), an analogous cyclometallated palladium bromide complex, [PdBr(tBu2CMe2CH2)]2 (3), was added to the reaction of complex 1 and PyO (eq 2). The yield (37%) of the arylated product from this reaction was even lower than that from the reaction between complex 1 and PyO without any additives. Moreover, no deuterium incorporation into PtBu3 was observed from the reaction of complex 2 with PyO-d5. These results imply that the acetate ligand in the cyclometallated complex is involved in the C-H bond cleavage process.
Scheme 2.

Two Possible Modes for C-H Bond Cleavage by Cyclometallated Palladium Acetate Complex.
 |
(2) |
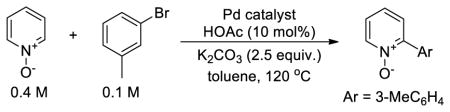
If the rate-determining, C-H bond cleavage step occurs between pyridine N-oxide and complex 2, then the catalytic reactions of bromoarenes with pyridine N-oxide should depend on the concentration of 2, but not on the concentration of 1. To test this prediction, reactions of 3-bromotoluene with pyridine N-oxide catalyzed by complex 2 and Pd(PtBu3)2 were conducted with different amounts of 2 and Pd(PtBu3)2 (Table 1). The Pd(PtBu3)2 component, along with the combination of HOAc and base, provides access to intermediate 1. A series of reactions was conducted with identical amounts of Pd(PtBu3)2 and varying amounts of 2 (entries 1–3). The reactions with higher concentrations of 2 were faster than those with lower concentrations of 2, and the dependence on [2] was half-order. A series of reactions was also conducted with the same amount of 2, but varying amounts of Pd(PtBu3)2 (entries 3–5). The rate of these reactions did not depend measurably on the concentration of Pd(PtBu3)2. These results further support our assertion that the C-H bond cleavage occurs by the reaction of PyO with the monomeric form of the cyclometallated palladium acetate complex 2.
Table 1.
Reactions of PyO with 3-bormotoluene with Varied Amounts of the two Pd cocatalysts.
 | ||
|---|---|---|
| entry | Pd catalyst | initial rate (M/s)a |
| 1 | 2 (1 mol%), Pd(PtBu3)2 (5 mol%) | 2.8 × 10−6 |
| 2 | 2 (2 mol%), Pd(PtBu3)2 (5 mol%) | 5.3 × 10−6 |
| 3 | 2 (5 mol%), Pd(PtBu3)2 (5 mol%) | 8.5 × 10−6 |
| 4 | 2 (5 mol%), Pd(PtBu3)2 (2 mol%) | 7.7 × 10−6 |
| 5 | 2 (5 mol%), Pd(PtBu3)2 (1 mol%) | 8.2 × 10−6 |
Calculated by monitoring the reaction by GC during the first hour (estimated errors are ±0.3 X 10−6 M/s)
To assess the differences in barriers for cleavage of the C-H bond in pyridine N-oxide by the PtBu3–ligated arylpalladium acetate complex 1 and the cyclometallated palladium acetate complex 2, we used density functional theory (DFT) to compute the barriers for the reaction of PyO with (PtBu3)Pd(Ph)(OAc) and PdOAc(tBu2PCMe2CH2). The calculated ΔG‡ for the reaction of Pd(OAc)(tBu2PCMe2CH2) with PyO was found to be 27 kcal/mol at 25 °C, whereas the calculated ΔG‡ for the reaction of (PtBu3)Pd(Ph)(OAc) with PyO was found to be 33 kcal/mol at 25 °C (see the Supporting Information for details). The calculated barrier for the reaction of PyO with 2 matches well with the experimental barrier (27 kcal/mol) obtained from the kinetic data, and is much lower than the barrier computed for the reaction of (PtBu3)Pd(Ph)(OAc) with PyO. These computed barriers are, again, consistent with our proposal that PyO reacts with the cyclometallated palladium acetate species 2, not with complex 1.
Based on these studies, we propose that the reaction of PyO with 1 occurs by the catalytic cycle shown in Scheme 3. In this cycle, PyO reacts with cyclometallated palladium acetate complex A, which is the monomer of 2, to generate a heteroarylpalladium intermediate B. This intermediate then reacts with 1 to transfer the aryl group to form aryl heteroaryl complex C. In this mechanism complex C is the species that undergoes reductive elimination to form the Pd(0) product and the 2-aryl pyridine.
Scheme 3.

Reaction of PyO with 1 Catalyzed by 2.
To assess further the proposed pathway, we prepared an example of the proposed heteroarylpalladium intermediate containing the cyclometallated phosphine. With such a complex, we could evaluate whether the reaction of such a complex with the arylpalladium carboxylate complex 1 was sufficiently fast to be part of the catalytic cycle. Because of the known difficulty in generating 2-metallated pyridine oxides,13 which would be used to generate 2-pyridyloxide palladium complexes, in high yield, we conducted studies on 2-benzothienylpalladium complexes. Benzothiophene undergoes catalytic direct arylation under conditions similar to those of PyO.3f To assess more specifically whether studies on the benzothienyl complexes would be relevant to the mechanism of the reaction of PyO, we first assessed whether the mechanistic data implying that the reactions of PyO involves two metal complexes also would be obtained from studies of the reactions of benzothiophene (BT). Indeed, the reaction of arylpalladium acetate 1 with BT occurred in yields (64%) that were lower than those for the catalytic direct arylation of benzothiophene (85%), but the rate and yield of the reaction of 1 with BT, like those of 1 with PyO, were much faster and higher (88%) in the presence of added complex 2. The yield of the reaction of 1 with BT in the presence of 2 matched that of the direct arylation catalyzed by Pd(OAc)2 and PtBu3. Thus, studies on the transmetallation between 1 and a 2-benzothienylpalladium complex should assess whether transmetallation between two palladium complexes could be part of the reaction of PyO and would assess the scope of our mechanistic conclusions.
PEt3-stabilized, benzothienylpalladium complex 4 containing the cyclometallated phosphine was prepared in acceptable isolated yield, as shown in Scheme 4, by the reaction of cyclometallated bromide complex 3 with 2-benzothienyllithium in the presence of PEt3 and Ag2CO3. This complex was characterized by 1H and 31P NMR spectroscopy. A species we assign to be the analogous 2-benzothieneyl complex lacking PEt3 was generated in ca 70% yield by the reaction of complex 3 with 2-benzothienyllithium in the absence of PEt3 and Ag2CO3. This complex was characterized in situ by 1H and 31P NMR spectroscopy.
Scheme 4.

Reaction of Benzothienylpalladium Complex 4 and 5 with arylpalladium acetate complex 1.
The reaction of 2-benzothienyl complex 4 with arylpalladium acetate 1 formed the 2-arylbenzothiophene product within 5 min at 50 °C in 84% yield (Scheme 4). The analogous reaction with in situ generated complex 5 occurred even faster and formed the arylated product in 74% yield within 5 min at room temperature. The fast rate and high yield of the reaction of acetate 1 with 2-benzothienyl complexes 4 and 5 containing the cyclometallated phosphine indicate that transmetallation between a heteroarylpalladium intermediate and complex 1 is kinetically and chemically competent to be part of the mechanism for the stoichiometric reaction of 1 with PyO and BT and the catalytic arylations of these heteroarenes in the presence of Pd(OAc)2 and PtBu3.
The pathway in Scheme 5 for the direct arylation of pyridine N-oxide catalyzed by Pd(OAc)2 and PtBu3 is consistent with our mechanistic data on the stoichiometric reaction of PyO with 1. This catalytic network comprises the cooperation of two distinct metal fragments in two merging catalytic cycles, The left cycle begins with oxidative addition of the aryl bromide to (PtBu3)Pd(0) to form an arylpalladium(II) intermediate D. Exchange of acetate for bromide would generate the arylpalladium acetate intermediate (complex 1) proposed previously to cleave the heteroaryl C-H bond. Instead, our data imply that an arylpalladium(II) species undergoes transmetallation with cyclometallated intermediate B to form the aryl heteroaryl palladium species C. C-C bond-forming reductive elimination would then form the 2-arylpyridine oxide product and regenerate the active Pd(0) catalyst to complete the cycle shown at the left of Scheme 5. In a second cycle, shown at the right of Scheme 5, the monomeric Pd(OAc)(tBu2CMe2CH2) reacts with pyridine N-oxide to cleave the C-H bond and form the 2-pyridylpalladium complex B containing the cyclometallated phosphine.
Scheme 5.

Revised Mechanism for the Direct Arylation of PyO.
Our kinetic data imply that the turnover-limiting step in the catalytic direct arylation of PyO occurs between PyO and the monomeric Pd(OAc)(tBu2CMe2CH2). The equilibrium between the observed dimer 2 and the active monomeric complex A is consistent with the partial-order behavior in palladium, and the overall scheme is consistent with the first-order behavior in PyO and zero-order behavior in arylpalladium acetate complex 1. Calculations of the barrier of the reactions of pyridine N-oxide with (PtBu3)Pd(Ph)(OAc) and Pd(OAc)(tBu2CMe2CH2) with DFT also support the conclusion that the cyclometallated palladium acetate species is more reactive towards C-H bond cleavage than is the arylpalladium acetate intermediate. These data are also consistent with the kinetic data previously obtained on the catalytic process9 and resolve the inconsistency in the conclusion from these studies of the partial order in palladium and the monomeric nature of the proposed catalyst resting state and the species proposed to react with the pyridine oxide in the turnover-limiting step. Analogous data on the reaction of benzothiophene imply that our conclusions pertain to direct arylation reactions beyond pyridine N-oxide.
In summary, detailed kinetic studies on the reactions of the isolated arylpalladium acetate complex 1 with pyridine N-oxide in the presence of added cyclometallated palladium acetate complex 2, along with studies on isolated heteroarylpalladium complex 4 containing a cyclometallated phosphine, have revealed an unexpected mechanism in which the C-H bond of pyridine N-oxide is cleaved by the reaction with cyclometallated complex 2. We propose that the constrained ring in 2 makes the metal center less hindered and, thereby, allows the reaction of PyO with 2 to occur with a lower barrier than the reaction of PyO with 1. By the proposed mechanism, one palladium fragment containing P(tBu)3 as ligand adds the aryl halide and forms the C-C bond, but a second fragment containing a cyclometallated phosphine undergoes the C-H bond cleavage step. One can envision many processes for C-H bond functionalization in which one metal cleaves a C-H bond and transfers the resulting hydrocarbyl ligand to a second metal that leads to functionalization, but the demonstration of such a process, other than reactions in which a second metal deprotonates an acidic substrate, is rarely documented.14 New processes that are based on such a design will be the subject of future studies.
Supplementary Material
Acknowledgments
We thank the NIH (GM-58108) for financial support and Dale Pahls for helpful guidance and suggestions with the DFT calculations.
Footnotes
Supporting Information Available: Experimental procedures, spectra for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.For recent reviews on direct arylations, see: Alberico D, Scott ME, Lautens M. Chem Rev. 2007;107:174. doi: 10.1021/cr0509760.Campeau LC, Stuart DR, Fagnou K. Aldrichimica Acta. 2007;40:35.Ackermann L, Vicente R, Kapdi AR. Angew Chem, Int Ed. 2009;48:9792. doi: 10.1002/anie.200902996.
- 2.Campeau LC, Rousseaux S, Fagnou K. J Am Chem Soc. 2005;127:18020. doi: 10.1021/ja056800x. [DOI] [PubMed] [Google Scholar]
- 3.(a) Akita Y, Itagaki Y, Takizawa S, Ohta A. Chem Pharm Bull. 1989;37:1477. [Google Scholar]; (b) Pivsa-Art S, Satoh T, Kawamura Y, Miura M, Nomura M. Bull Chem Soc Jpn. 1998;71:467. [Google Scholar]; (c) McClure MS, Glover B, McSorley E, Millar A, Osterhout MH, Roschangar F. Org Lett. 2001;3:1677. doi: 10.1021/ol0158866. [DOI] [PubMed] [Google Scholar]; (d) Kobayashi K, Sugie A, Takahashi M, Masui K, Mori A. Org Lett. 2005;7:5083. doi: 10.1021/ol052063y. [DOI] [PubMed] [Google Scholar]; (e) Lane BS, Brown MA, Sames D. J Am Chem Soc. 2005;127:8050. doi: 10.1021/ja043273t. [DOI] [PubMed] [Google Scholar]; (f) Rene O, Lapointe D, Fagnou K. Org Lett. 2009;11:4560. doi: 10.1021/ol901799p. [DOI] [PubMed] [Google Scholar]
- 4.(a) Satoh T, Kawamura Y, Miura M, Nomura M. Angew Chem, Int Ed Engl. 1997;36:1740. [Google Scholar]; (b) Satoh T, Kametani Y, Terao Y, Miura M, Nomura M. Tetrahedron Lett. 1999;40:5345. [Google Scholar]; (c) Daugulis O, Zaitsev VG. Angew Chem, Int Ed. 2005;44:4046. doi: 10.1002/anie.200500589. [DOI] [PubMed] [Google Scholar]; (d) Lazareva A, Daugulis O. Org Lett. 2006;8:5211. doi: 10.1021/ol061919b. [DOI] [PubMed] [Google Scholar]
- 5.(a) Lafrance M, Rowley CN, Woo TK, Fagnou K. J Am Chem Soc. 2006;128:8754. doi: 10.1021/ja062509l. [DOI] [PubMed] [Google Scholar]; (b) Lafrance M, Shore D, Fagnou K. Org Lett. 2006;8:5097. doi: 10.1021/ol0619967. [DOI] [PubMed] [Google Scholar]
- 6.Lafrance M, Fagnou K. J Am Chem Soc. 2006;128:16496. doi: 10.1021/ja067144j. [DOI] [PubMed] [Google Scholar]
- 7.(a) Biswas B, Sugimoto M, Sakaki S. Organometallics. 2000;19:3895. [Google Scholar]; (b) Davies DL, Donald SMA, Macgregor SA. J Am Chem Soc. 2005;127:13754. doi: 10.1021/ja052047w. [DOI] [PubMed] [Google Scholar]; (c) Gorelsky SI, Lapointe D, Fagnou K. J Am Chem Soc. 2008;130:10848. doi: 10.1021/ja802533u. [DOI] [PubMed] [Google Scholar]
- 8.Tan Y, Hartwig JF. J Am Chem Soc. 2011;133:3308. doi: 10.1021/ja1113936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun HY, Gorelsky SI, Stuart DR, Campeau LC, Fagnou K. J Org Chem. 2010;75:8180. doi: 10.1021/jo101821r. [DOI] [PubMed] [Google Scholar]
- 10.The first synthesis of (PtBu3)Pd(Ph)(OAc) was reported in Barrios-Landeros, F. Oxidative Addition of Haloarenes by Pd(0) Complexes of Bulky Alkyl Phosphines: Synthesis of Intermediates and Mechanistic Studies. Dissertation. Yale University; 2007.
- 11.For independent synthesis of complex 2, see: Wu L, Hartwig JF. J Am Chem Soc. 2005;127:15824. doi: 10.1021/ja053027x.
- 12.For results on primary KIEs, see ref 2 and 9. In addition, we monitored the catalytic reaction by 31P NMR spectroscopy, and complex 2 was observed as the catalyst’s resting state.
- 13.Andersson H, Gustafsson M, Olsson R, Almqvist F. Tetrahedron Lett. 2008;49:6901. [Google Scholar]
- 14.For a recent example of cooperative catalysis between palladium and copper in direct arylation of acidic arenes, see: Huang J, Chan J, Chen Y, Borths CJ, Baucom KD, Larsen RD, Faul MM. J Am Chem Soc. 2010;132:3674. doi: 10.1021/ja100354j.For a recent example of cooperative catalysis between palladium and copper in direct allylation, see: Fan S, Chen F, Zhang X. Angew Chem, Int Ed Engl. 2011;50:5918. doi: 10.1002/anie.201008174.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.