

Rationale for prophylactic replacement therapy
The rationale for prophylactic replacement therapy with factor VIII (FVIII) or factor IX (FIX) concentrates is based on the observation, made in 1965 by Ahlberg et al.1, that arthropathy was associated with baseline clotting factor activity levels (Figure 1). From his observation in 242 patients, including 102 with severe haemophilia (<1% FVIII/FIX activity) and 60 with moderate haemophilia (1–5% FVIII/IX activity), it was clear that patients with moderate and mild haemophilia had much less arthropathy, expressed as joint scores. In fact, this observation has recently been corroborated for the association of joint bleed frequency with baseline FVIII levels, reproducing almost the same Figure2. The original idea of Nilsson, to convert the bleeding pattern of a severe haemophiliac into that of a moderate haemophiliac by regular prophylactic replacement therapy, does, therefore, make perfect sense3. In fact, it has proven very successful, as shown by long-term data from Sweden and The Netherlands4,5 and short-term data from many cohorts and two randomised trials in children6,7. The long-term efficacy of prophylaxis in preventing haemophilic arthropathy appears to be dependent on starting the prophylaxis early8,9 as well as on the type of regimen used. However, the optimum dose and desired trough level have not yet been established. There is now broad consensus that prophylaxis should be provided to all boys with severe haemophilia, if the necessary resources are available. In theory, it is very logical to continue providing prophylaxis to our adult patients, as bleeding and subsequent arthropathy are still likely to occur. In fact, the evidence to support the effectiveness of prophylaxis in adults is increasing10–12. The logical conclusion is, therefore, to prescribe prophylaxis to all patients with repeated bleeding or life-threatening bleeds, irrespective of their baseline FVIII/IX levels13.
Figure 1.

Haemophilic arthropathy (joint score) according to FVIII/FIX activity level1.
Data from 210 patients in Sweden in the 1960s clearly show a trend towards lower joint scores (i.e. less arthropathy) with increasing baseline FVIII/FIX levels. However, a wide variation in joint scores according to baseline levels was observed.
Prophylaxis in adults: apparently not all patients need it
However, is it really necessary to treat all our adult patients with severe haemophilia with prophylaxis? There are two main drivers to this question: one is financial, the other concerns the burden of treatment for the patient. First, can we afford to prescribe life-long prophylaxis to all our patients? As dosing is usually dependent on body weight, the annual costs for prophylaxis are extremely high (€130,000–162,00014) and are likely to remain constant for the average 50 years until death. Secondly, do patients want to continue giving themselves frequent intravenous infusions throughout life? Maintaining this prophylactic treatment is a heavy burden for the patient, and adherence to treatment is likely to include periods of reduced compliance. Several studies have reported variable adherence rates, and a trend towards lower adherence in adult patients15–17. In a study by de Moerloose et al., 180 patients from different centres in Europe were interviewed about their adherence to treatment and its determinants17. Patient-reported barriers to adherence with prophylaxis are shown in Figure 2. It is striking that the most frequently reported reason for non-adherence was "reduction, fluctuation or disappearance of symptoms", followed by "forgetfulness" and "lack of time". Interestingly, haemophilia treaters have reported that bleeding phenotype is one of the most important reasons for considering changing prophylaxis in an adult patient18.
Figure 2.

Reasons for non-adherence with prophylaxis according to age17.
In fact, a considerable proportion of adults with severe haemophilia apparently discontinue prophylaxis on their own accord. Richards et al. performed a survey of 19 treatment centres throughout Europe including 218 patients aged 16–22 years with severe haemophilia, who were followed for 3–70 months18. It was reported that 70% of these patients modified their prophylaxis in early adulthood: 5% of patients tapered their prophylaxis, but had to revert to their former dose due to increased bleeding, but 22% were able to taper prophylaxis without experiencing very frequent bleeding. In addition, 12% attempted to discontinue but had to resume prophylaxis due to frequent bleeding, and 30% were able to switch to on demand treatment for longer periods. Apparently, these patients were not bleeding frequently, even if they had severe haemophilia. Similar observations were made in two studies on discontinuing prophylaxis in the Netherlands and Denmark. In the first study in 49 patients born between 1970 and 1980, who began prophylaxis at a median age of 14 years, 67% of patients attempted to discontinue prophylaxis on their own account19. At a median age of 21 years, 24% of patients had discontinued prophylaxis while maintaining a low bleeding frequency. Tapering of prophylaxis was not considered in this study. In an analysis of patients' characteristics associated with successful switching to on demand treatment, it was apparent that this subgroup of patients had a milder bleeding phenotype. A prediction rule based on age at starting prophylaxis and the annual number of joint bleeds on prophylaxis and weekly dose of prophylaxis (IU/kg) was able to identify these patients (AUC 0.95). In a subsequent study, data from Denmark (Århus and Copenhagen) and the Netherlands (Utrecht) were combined, including 80 patients with severe haemophilia born between 1970 and 1981, who were evaluated at a median age of 26 years20. Overall, 33% of young adults discontinued prophylactic treatment at a median age of 21 years. The incidence and consequences of tapering prophylaxis were not studied. With a limited median follow-up of 3.6 years, median clinical scores were similar in patients who discontinued prophylaxis (4 points, IQR 0–6) and those who continued (3 points, IQR 1–6), as were median Pettersson scores at 13 (IQR 1–24) and 13 points (IQR 5–23), respectively. The annual number of joint bleeds was only slightly but significantly increased (median 3.2, IQR 0.9–6.0) compared to patients still on prophylaxis (median 1.8, IQR 0–3.0; p=0.04). In this study Dutch patients switched to on-demand treatment had required lower prophylactic doses to prevent bleeding as compared to those who did not discontinue prophylaxis.
It is very likely that the ability to discontinue prophylaxis is associated with both external and patient-related factors. The importance of external factors is illustrated by the striking fact that patients consistently discontinued prophylaxis during early adulthood: at 21 years in Denmark and the Netherlands20, and between 16 and 22 years in the European survey18. This may be associated with life-style factors such as taking on a job and more regulation of physical activities. This theory is corroborated by the finding that adults on prophylaxis show increased bleeding during the summer months, whereas young children do not21. Patient-related factors include joint status and bleeding phenotype. Target joints with frequent bleeding are clinical representations of synovial hypertrophy and early haemophilic arthropathy. It is obvious that a patient can only consider tapering or discontinuing prophylaxis in the absence of synovial hypertrophy or joint instability. In theory, the ideal 'candidate' for successful discontinuation of prophylaxis in adulthood has received early prophylaxis and regular exercise, resulting in an optimal joint status, combined with a mild bleeding phenotype.
Heterogeneity in bleeding phenotype: identification and explanation
This phenomenon of a varying bleeding phenotype is well-known but not yet explained22. Even in the study by Ahlberg (Figure 1) there were some patients with severe haemophilia who had very low joint scores1, and many have reported that 10–15% of patients with severe haemophilia seem to experience joint bleeds only very rarely10,11. On the other hand, there is also a proportion of patients who bleed more frequently than their peers. These patients will need more intensive treatment, as illustrated by the 12–29% of patients with moderate haemophilia who need prophylaxis23,24.
So far, laboratory studies have not been able to identify the determinants of variation in the clinical phenotype of haemophilia. Several studies have established that the presence of the prothrombotic mutations FV 91691A and FII G20210A may affect the phenotype of severe haemophilia. A large paediatric study showed that patients with prothrombotic mutations had their first symptomatic bleeding much later (median age 1.6 years; range 0.5–7.1 years) than patients without risk factors (median age 0.9 years; range 0.1–4.0; p=0.01)25. In a subsequent study from Germany, the presence of these mutations was associated with significantly less bleeding and less radiologically determined arthropathy (Pettersson scores)26. However, these mutations are present in only 5–10% of patients and can, therefore, only partly explain differences in phenotype. An extensive search for coagulation factors explaining phenotype in severe haemophilia only found a trend towards increased fibrinolysis in patients with a more severe phenotype, but no association with other parameters of coagulation27,28. In a group of 21 patients with a mild bleeding phenotype and 50 controls, Santagostino et al. were able to identify patients with a mild phenotype by thrombin generation tests in platelet-rich plasma. Moreover, they found an association between mild phenotypes and the presence of non-null mutations29. So far, thrombin generation tests have mainly been used to monitor treatment, especially in patients with inhibitors. The role of platelets in relation to thrombin generation may be very important, especially at FVIII levels of 1–5%, as was recently reported by a group from Finland30.
Bleeding frequency is the most logical identifier for the clinical identification of phenotype. Joint bleeds are preferred to the total number of bleeds, as they are less susceptible to misclassification, especially in patients on home treatment. In the case of prophylaxis, however, it is impossible to consider the annual number of joint bleeds without considering treatment intensity. Especially if the prophylactic dose is adjusted according to bleeding frequency, variations in treatment requirement (i.e. annual clotting factor use) may reflect variations in the underlying bleeding tendency19,31. However, variations in clotting factor consumption are also dependent on changes in prophylactic treatment. Over the last three decades continuously fewer joint bleeds were tolerated while on prophylaxis, resulting in gradually more intensified treatment, which is shown by an increased weekly dose, increased frequency and earlier start of prophylaxis. Still, in those who do not start prophylaxis before the onset of joint bleeding, the variation in onset of joint bleeding remains considerable, with a median age around 1.8 years and a range from 0.2–5.8 years31. The hypothesis that the age at onset of joint bleeding may be used as a marker for phenotype is corroborated by the fact that late onset of joint bleeding was associated with a consistent and significant reduction in prophylactic dose (Figure 3)31 in severe haemophilia. In moderate haemophilia, onset of joint bleeding, together with baseline FVIII/FIX levels, predicted the need for prophylactic treatment24.
Figure 3.
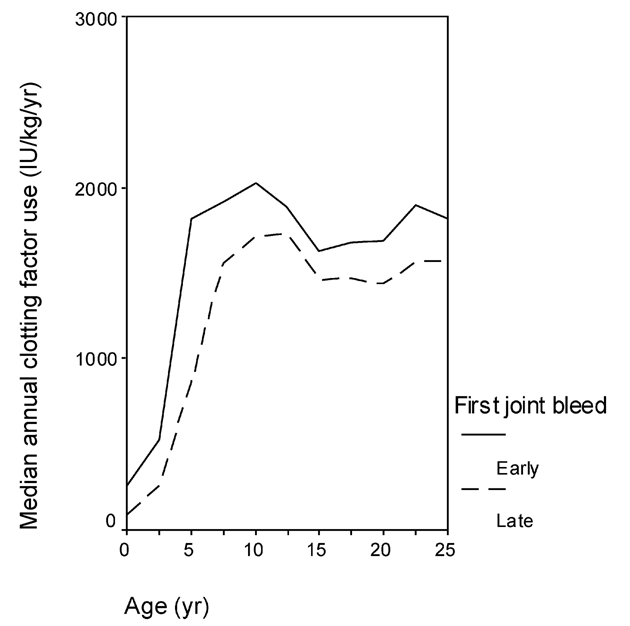
Annual clotting factor use according to onset of joint bleeding33.
Patients who had their first joint bleed early (i.e. before 1.8 years, median age at onset of bleeding) consistently used more clotting factor for prophylaxis than those who had their first joint bleed later.
Individualising prophylactic treatment in adults
While waiting for the results of future research, the physician who wants to individualise treatment still has to work with the clinical bleeding pattern. If an adult patient with good joint health wants to taper or discontinue prophylaxis, his physician should consider bleeding pattern, treatment intensity, and joint status. The only available prediction score for discontinuing prophylaxis requires full documentation of previous treatment and may, therefore, be difficult to apply19. Another scoring system for assessment of phenotype was designed by Schulman et al.32. This score is based on bleeding frequency, clinical joint score, and clotting factor consumption adjusted for age at start of prophylaxis and weight. However, this scoring system compares annual clotting factor consumption with the 'benchmark' of 6,000 IU/Kg/year, which Is likely to reduce its validity for patients on lower dosed regimes. Until better instruments are available, and if prophylaxis was started after the first joint bleed, the clinician may use the age at onset of joint bleeding as a first indicator (i.e. >1.8 years points towards a milder phenotype). In the case of good joint health and little or no bleeding on prophylaxis, one may consider tapering prophylaxis (dose and/or frequency, based on activities and pharmacokinetic information). Tapering may be progressive in several steps towards discontinuation if the patient does not experience frequent bleeding. Evidently, close monitoring of bleeding and prompt reinstitution of prophylaxis if necessary, are warranted to maintain joint health. So far, there is no guidance on the acceptable number of bleeds for patients receiving on-demand treatment, but modelling studies are underway33. The clinical challenge is to tailor prophylactic treatment to the individual patients' needs, the "one size fits all" approach is too simple and will result in overtreatment, high costs and/or reduced adherence.
Footnotes
Conflicts of interest disclosure
Kathelijn Fischer has received speaker's fees from Baxter, Wyeth/Pfizer, NovoNordisk and Biotest, has provided consultancy for Baxter, Biogen and NovoNordisk and has received research support from Bayer, Wyeth/Pfizer, Baxter and Novo Nordisk.
References
- 1.Ahlberg A. Haemophilia in Sweden VII. Incidence, treatment and prophylaxis of arthropathy and other musculo-skeletal manifestations of haemophilia A and B. Acta Orthop Scand. 1965;77(Suppl):5–99. doi: 10.3109/ort.1965.36.suppl-77.01. [DOI] [PubMed] [Google Scholar]
- 2.den Uijl I, Fischer K, Van der Bom JG, et al. Analysis of low frequency bleeding data: the association of joint bleeds according to baseline FVIII activity levels. Haemophilia. 2011;17:41–4. doi: 10.1111/j.1365-2516.2010.02383.x. [DOI] [PubMed] [Google Scholar]
- 3.Nilsson IM, Blomback M, Ahlberg A. Our experience in Sweden with prophylaxis on haemophilia. Bibl Haematol. 1970;34:111–24. [PubMed] [Google Scholar]
- 4.Löfqvist T, Nilsson IM, Berntorp E, Pettersson H. Haemophilia prophylaxis in young patients- a long-term follow-up. J Intern Med. 1997;241:395–400. doi: 10.1046/j.1365-2796.1997.130135000.x. [DOI] [PubMed] [Google Scholar]
- 5.Van den Berg HM, Fischer K, Mauser-Bunschoten EP, et al. Long term outcome of individualised prophylactic treatment of children with severe haemophilia. Br J Haematol. 2001;107:561–5. doi: 10.1046/j.1365-2141.2001.02580.x. [DOI] [PubMed] [Google Scholar]
- 6.Manco-Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007;357:535–44. doi: 10.1056/NEJMoa067659. [DOI] [PubMed] [Google Scholar]
- 7.Gringeri A, Lundin B, von Mackensen S, et al. A randomized clinical trial of prophylaxis in children with hemophilia A (the ESPRIT Study) J Thromb Haemost. 2011;9:700–10. doi: 10.1111/j.1538-7836.2011.04214.x. [DOI] [PubMed] [Google Scholar]
- 8.Astermark J, Petrini P, Tengborn L, et al. Primary prophylaxis in severe haemophilia should be started at an early age but can be individualized. Br J Haematol. 1999;105:1109–13. doi: 10.1046/j.1365-2141.1999.01463.x. [DOI] [PubMed] [Google Scholar]
- 9.Fischer K, Van der Bom JG, Mauser-Bunschoten EP, et al. Effects of postponing prophylactic treatment on long-term outcome in patients with severe haemophilia. Blood. 2002;99:2337–41. doi: 10.1182/blood.v99.7.2337. [DOI] [PubMed] [Google Scholar]
- 10.Aledort LM, Haschmeyer RH, Pettersson H. A longitudinal study of orthopaedic outcomes for severe factor-VIII-deficient haemophiliacs. The Orthopaedic Outcome Study Group. J Intern Med. 1994;236:391–9. doi: 10.1111/j.1365-2796.1994.tb00815.x. [DOI] [PubMed] [Google Scholar]
- 11.Schramm W, Royal S, Kroner B, et al. Clinical outcomes and resource utilization associated with haemophilia care in Europe. Haemophilia. 2002;8:33–43. doi: 10.1046/j.1365-2516.2002.00580.x. [DOI] [PubMed] [Google Scholar]
- 12.Collins P, Faradji A, Morfini M, et al. Efficacy and safety of secondary prophylactic vs. on-demand sucrose-formulated recombinant factor VIII treatment in adults with severe hemophilia A: results from a 13-month crossover study. J Thromb Haemost. 2010;8:83–9. doi: 10.1111/j.1538-7836.2009.03650.x. [DOI] [PubMed] [Google Scholar]
- 13.Valentino LA. Secondary prophylaxis therapy: what are the benefits, limitations and unknowns? Haemophilia. 2004;10:147–57. doi: 10.1111/j.1365-2516.2003.00870.x. [DOI] [PubMed] [Google Scholar]
- 14.Steen-Carlsson K, Hojgard S, Lindgren A, et al. Costs of on-demand and prophylactic treatment for severe haemophilia in Norway and Sweden. Haemophilia. 2004;10:515–26. doi: 10.1111/j.1365-2516.2004.00952.x. [DOI] [PubMed] [Google Scholar]
- 15.Collins PW, Blanchette VS, Fischer K, et al. Breakthrough bleeding in relation to predicted factor VIII levels in patients receiving prophylactic treatment for severe hemophilia A. J Thromb Haemost. 2009;7:413–20. doi: 10.1111/j.1538-7836.2008.03270.x. [DOI] [PubMed] [Google Scholar]
- 16.Walsh CE, Valentino LA. Factor VIII prophylaxis for adult patients with severe haemophilia A: results of a US survey of attitudes and practices. Haemophilia. 2009;15:1014–21. doi: 10.1111/j.1365-2516.2009.02036.x. [DOI] [PubMed] [Google Scholar]
- 17.de Moerloose P, Urbancik W, Van den Berg HM, Richards M. A survey of adherence to haemophilia therapy in six European countries: results and recommendations. Haemophilia. 2008;14:931–8. doi: 10.1111/j.1365-2516.2008.01843.x. [DOI] [PubMed] [Google Scholar]
- 18.Richards M, Altisent C, Batorova A, et al. Should prophylaxis be used in adolescent and adult patients with severe haemophilia? An European survey of practice and outcome data. Haemophilia. 2007;13:473–9. doi: 10.1111/j.1365-2516.2007.01478.x. [DOI] [PubMed] [Google Scholar]
- 19.Fischer K, Van der Bom JG, Prejs R, et al. Discontinuation of prophylactic therapy in severe haemophilia: incidence and effects on outcome. Haemophilia. 2001;7:544–50. doi: 10.1046/j.1365-2516.2001.00560.x. [DOI] [PubMed] [Google Scholar]
- 20.van Dijk K, Fischer K, Van der Bom JG, et al. Can long-term prophylaxis for severe haemophilia be stopped in adulthood? Results from Denmark and the Netherlands. Br J Haematol. 2005;130:107–12. doi: 10.1111/j.1365-2141.2005.05546.x. [DOI] [PubMed] [Google Scholar]
- 21.Fischer K, Collins P, Bjorkman S, et al. Trends in bleeding patterns during prophylaxis for severe haemophilia: observations from a series of prospective clinical trials. Haemophilia. 2011;17:433–8. doi: 10.1111/j.1365-2516.2010.02450.x. [DOI] [PubMed] [Google Scholar]
- 22.Van den Berg HM, De Groot PH, Fischer K. Phenotypic heterogeneity in severe hemophilia. J Thromb Haemost. 2007;5(Suppl 1):151–6. doi: 10.1111/j.1538-7836.2007.02503.x. [DOI] [PubMed] [Google Scholar]
- 23.Plug I, Van der Bom JG, Peters M, et al. Thirty years of hemophilia treatment in the Netherlands, 1972–2001. Blood. 2004;104:3494–500. doi: 10.1182/blood-2004-05-2008. [DOI] [PubMed] [Google Scholar]
- 24.den Uijl Ingrid EM. Variation in FVIII/FIX activity in haemophilia:classification and clinical implications. University of Utrecht; Utrecht, the Netherlands: 22-3-2011. Dissertation. [Google Scholar]
- 25.Escuriola-Ettingshausen C, Halimeh S, Kurnik K, et al. Symptomatic onset of severe haemophilia A in childhood is dependent on the presence of prothrombotic risk factors. Thromb Haemost. 2001;85:218–20. [PubMed] [Google Scholar]
- 26.Kurnik K, Kreuz W, Horneff S, et al. Effects of the factor V G1691A mutation and the factor II G20210A variant on the clinical expression of severe hemophilia A in children-results of a multicenter studys. Haematologica. 2007;92:982–5. doi: 10.3324/haematol.11161. [DOI] [PubMed] [Google Scholar]
- 27.van Dijk K, Van der Bom JG, Fischer K, et al. Phenotype of severe hemophilia A and plasma levels of risk factors for thrombosis. J Thromb Haemost. 2007;5:1062–4. doi: 10.1111/j.1538-7836.2007.02447.x. [DOI] [PubMed] [Google Scholar]
- 28.Grunewald M, Siegemund A, Grunewald A, et al. Paradoxical hyperfibrinolysis is associated with a more intensely haemorrhagic phenotype in severe congenital haemophilia. Haemophilia. 2002;8:768–75. doi: 10.1046/j.1365-2516.2002.00686.x. [DOI] [PubMed] [Google Scholar]
- 29.Santagostino E, Mancuso ME, Tripodi A, et al. Severe hemophilia with mild bleeding phenotype: molecular characterization and global coagulation profile. J Thromb Haemost. 2010;8:737–43. doi: 10.1111/j.1538-7836.2010.03767.x. [DOI] [PubMed] [Google Scholar]
- 30.Wartiovaara-Kautto U, Joutsi-Korhonen L, Ilveskero S, et al. Platelets significantly modify procoagulant activities in haemophilia A. Haemophilia. 2011;17:743–51. doi: 10.1111/j.1365-2516.2011.02601.x. [DOI] [PubMed] [Google Scholar]
- 31.Van Dijk K, Fischer K, Van der Bom JG, et al. Variability in clinical phenotype of severe haemophilia: the role of the first joint bleed. Haemophilia. 2005;11:438–43. doi: 10.1111/j.1365-2516.2005.01124.x. [DOI] [PubMed] [Google Scholar]
- 32.Schulman S, Eelde A, Holmstrom M, et al. Validation of a composite score for clinical severity of hemophilia. J Thromb Haemost. 2008;6:1113–21. doi: 10.1111/j.1538-7836.2008.03001.x. [DOI] [PubMed] [Google Scholar]
- 33.Fischer K, Pouw ME, Lewandowski D, et al. A modeling approach to evaluate long-term outcome of prophylactic and on demand treatment strategies for severe hemophilia A. Haematologica. 2011;96:738–43. doi: 10.3324/haematol.2010.029868. [DOI] [PMC free article] [PubMed] [Google Scholar]