

Summary
ICEBs1 is a mobile genetic element found in the chromosome of Bacillus subtilis. Excision and transfer of ICEBs1 is regulated by the global DNA damage response and intercellular peptide signalling. We identified and characterized a repressor, ImmR (formerly YdcN), encoded by ICEBs1. ImmR represses transcription of genes required for excision and transfer, and both activates and represses its own transcription. ImmR regulates transcription within ICEBs1 by binding to several sites in the region of DNA that contains promoters for both immR and xis (encoding excisionase). In addition, we found that ImmR confers immunity from acquisition of additional copies of ICEBs1. ImmR-mediated regulation serves to keep a single copy of ICEBs1 stably maintained in the absence of induction, allows a rapid response to inducing signals, and helps limit acquisition of additional copies of ICEBs1.
Introduction
Mobile genetic elements play an important role in horizontal gene transfer and the evolution of bacterial species (reviewed in Ochman et al., 2000; Brussow et al., 2004; Burrus and Waldor, 2004; Frost et al., 2005). ICEBs1 is an integrative and conjugative element (also known as a conjugative transposon) found in the Bacillus subtilis chromosome (Burrus et al., 2002; Auchtung et al., 2005). ICEBs1 is normally propagated passively by the host cell through chromosomal replication and cell division. When the host cell undergoes DNA damage or ICEBs1-containing cells are surrounded by cells lacking ICEBs1, ICEBs1 can excise from the chromosome and transfer to recipient cells (Auchtung et al., 2005). Excision requires the site-specific recombinase, Int, and an accessory protein, Xis (C.A. Lee, J.M. Auchtung, R.E. Monson and A.D. Grossman, in preparation). Transfer requires several ICEBs1-encoded conjugation proteins (C.A. Lee, M.B. Berkmen, and A.D. Grossman, unpublished results).
Most of the genes in ICEBs1 are located downstream from xis (Fig. 1) and transcription of most of these genes (xis through yddJ) appears to be co-regulated (Auchtung et al., 2005). The global DNA damage response stimulates expression of these genes, and leads to excision and mating of ICEBs1 (Auchtung et al., 2005). Thus, ICEBs1 has a mechanism to sense host cell genomic stress and initiate its escape from distressed cells.
Fig. 1. Organization of open reading frames in ICEBs1.
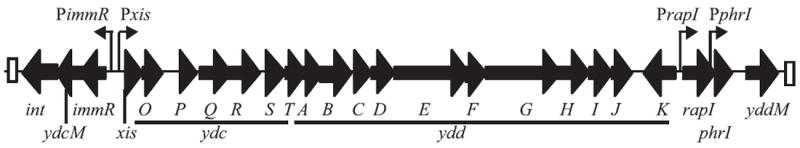
The 24 open reading frames of ICEBs1 are indicated by thick black arrows, oriented in the direction of transcription, with the name of each gene indicated below the arrow. ydcO-ydcT and yddA-yddK are indicated by the terminal unique letter directly under each arrow and the appropriate initial three-letter designation indicated underneath each underlined section of arrows. ydc and ydd indicate genes with unknown function. The positions of the promoters for xis, immR, rapI (Jarmer et al., 2001; Auchtung et al., 2005) and phrI (McQuade et al., 2001) are indicated by vertical lines with small arrows pointed in the direction of transcription. The 60 bp direct repeats marking the ends of the element are indicated by white boxes.
Intercellular peptide signalling also regulates transcription of xis through yddJ, and excision and transfer of ICEBs1 (Auchtung et al., 2005). This regulation occurs independently of the global DNA damage response and is mediated by the rapI-phrI signalling cassette encoded in ICEBs1. RapI stimulates expression of xis through yddJ and this stimulatory activity is antagonized by the secreted signalling peptide, PhrI. PhrI is a pentapeptide that is re-imported through the oligopeptide permease (Auchtung et al., 2005). As rapI and phrI are contained in ICEBs1, they function in self-recognition, inhibiting excision and transfer of ICEBs1 when other cells containing ICEBs1 are present. It is currently unknown how the global DNA damage response and RapI regulate transcription of xis through yddJ.
ICEBs1 encodes a putative repressor, ImmR, which was recognized based on homology to some bacteriophage repressors (Burrus et al., 2002; Auchtung et al., 2005). We investigated the role of immR (formerly ydcN) in regulating the activity of ICEBs1. We found that ImmR represses transcription of the promoter that drives expression of xis. This repression is critical for preventing excision of ICEBs1 and expression of most of its genes (xis through yddJ). In addition, we found that ImmR activates and represses its own transcription and transcription of the two genes downstream, ydcM and int. Autoregulation likely allows the levels of ImmR to be maintained at concentrations high enough to repress transcription of xis-yddJ and low enough to respond efficiently to inducing signals. Furthermore, we found that ImmR functions as an immunity repressor. That is, when expressed in a potential recipient, it greatly reduces the frequency of acquisition of ICEBs1 via conjugation. This serves to limit acquisition of additional copies of ICEBs1 by cells already containing the element. Immunity conferred by ImmR can be suppressed by increased expression of int, indicating that ImmR mediates immunity by limiting expression or activity of integrase. Thus, ImmR functions to ensure stable maintenance of a single copy of ICEBs1 and to limit acquisition of additional copies.
Results
Characterization of the xis promoter
xis encodes a protein required for excision of ICEBs1 (C.A. Lee, J.M. Auchtung and A.D. Grossman, unpubl. results). It is the first gene in a putative operon (xis through yddJ) involved in excision and transfer of ICEBs1 (Fig. 1). Expression of these genes (xis-yddJ) increases dramatically in response to inducing conditions, such as overproduction of RapI or treatment of the cells with the DNA-damaging agent mitomycin C (MMC) (Auchtung et al., 2005).
We mapped the 5′ end of the xis transcript using primer extension analysis (Fig. 2A and B). In cells overexpressing rapI or treated with MMC, we detected an abundant RNA species with a 5′ end 39 nucleotides upstream of the xis start codon (Fig. 2B). Similar results were obtained using a different primer in the extension reactions (data not shown). We did not detect any primer extension product specific to xis using RNA from uninduced cells as template. These results indicate that there is a strong transcription start site upstream of xis that is induced by overproduction of RapI or treatment with MMC.
Fig. 2. Characterization of the xis promoter and its activation by RapI overexpression and the global DNA damage response.
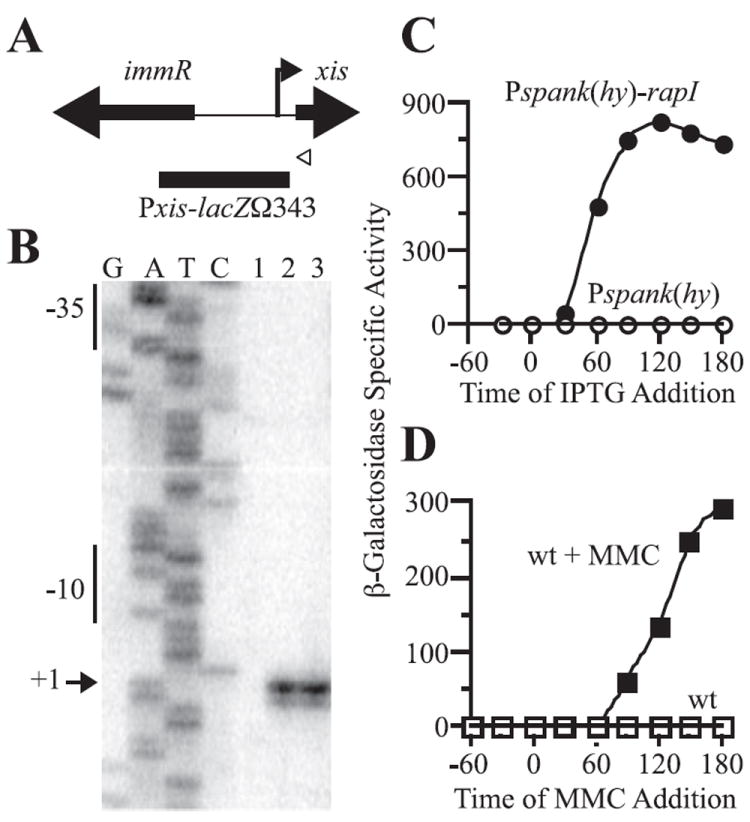
A. Schematic of xis, immR, and the shared intergenic region. The thick arrows indicate the locations of the xis and immR genes, and show the orientation of transcription of each. The white arrowhead indicates the position of the primer used for the primer extension assay in (B). The 5′ end of the xis transcript identified in (B) is indicated by a vertical line and arrow pointing to the right. The black box indicates the region upstream of xis fused to lacZ and used to monitor xis expression in (C) and (D).
B. The 5′ end of the xis transcripts was determined by primer extension assays. G, A, T and C indicate the lanes containing dideoxynucleotide sequencing reactions with the indicated nucleotide. RNA was isolated from untreated wild-type cells (JH642, lane 1); from wild-type cells 1 h after treatment with mitomycin C (lane 2); and from Pspank(hy)-rapI cells (JMA28) 30 min after treatment with IPTG (lane 3). Results of reverse transcription reactions with the primer indicated in (A) are shown; similar results were seen when reverse transcription reactions were carried out with a primer more proximal to +1 (data not shown). The sequences complementary to the consensus −10 and −35 regions are indicated on the left of the gel. The arrow indicates the nucleotide complementary to the end of the major transcript.
C and D. Cells containing a Pxis–lacZΩ343 fusion were grown in minimal medium and samples for β-galactosidase assays were collected at the times indicated. Cells were treated with 1 mM IPTG (C) or 1 μg ml−1 MMC (D) in mid-exponential phase (OD600 = 0.4–0.6). β-Galactosidase-specific activities were calculated relative to the cell densities (OD600) of the cultures. β-Galactosidase-specific activities are plotted relative to the time (in minutes) before and after addition of IPTG or MMC. In these graphs, β-galactosidase specific activity in wild-type cells appears to be at or near background (zero) levels. However, there is a low level of activity above background (Fig. 3A and B).
C. xis expression in cells (KLG126) containing Pxis–lacZΩ343 and Pspank(hy)-rapI (●) and cells (KLG125) containing Pxis–lacZΩ343 and Pspank(hy) empty vector (○).
D. xis expression in Pxis–lacZΩ343 cells (JMA201) (□) and Pxis–lacZΩ343 cells treated with MMC (■).
Just upstream of the position of the 5′ end of the xis mRNA there are potential −10 and −35 recognition elements for B. subtilis RNA polymerase containing the major sigma factor, sigma-A. The −10 sequence, TATAAT, is a perfect match to the consensus (Helmann, 1995). The −35 sequence, TTGACT, differs from the consensus in only one position (underlined). Together, the primer extension and DNA sequence indicate that transcription of xis likely initiates from a sigma-A-dependent promoter located upstream of xis and that transcription from this promoter increases dramatically under conditions known to induce ICEBs1 gene expression and excision.
To further study the xis promoter (Pxis), we constructed a transcriptional fusion of the region upstream of xis (from −343 to −6 bp upstream of xis start codon; Fig. 2A) to lacZ and integrated this fusion at an ectopic site in the chromosome. We found that Pxis–lacZΩ343 was expressed at very low levels in wild-type cells (Figs 2 and 3) and that expression increased substantially in response to overexpression of rapI (Fig. 2C) or treatment of cells with MMC (Fig. 2D). These results are in accordance with the results of transcriptional profiling (Auchtung et al., 2005) and primer extension analysis (Fig. 2B), and indicate that the region of DNA contained in the fusion contains the sequences necessary for appropriate regulation of transcription of xis.
Fig. 3. ImmR regulates excision through transcription of xis.
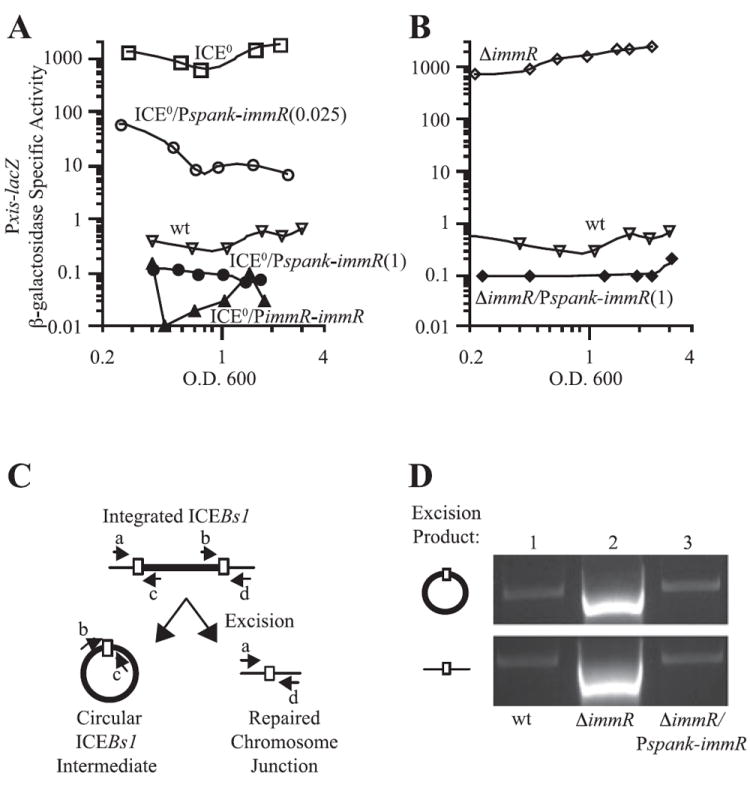
A and B. Pxis–lacZΩ343 expression was monitored throughout exponential growth in minimal medium as described in Fig. 2. IPTG (0.025 or 1 mM, as indicated) was present throughout growth.
A. Pxis–lacZΩ343 expression in wild-type (ICEBs1+) cells (JMA201, ▽, wt); ICEBs10 (JMA264, □, ICE0); ICEBs10 Pspank-immRΩ26 (JMA362) cells grown in the presence of 0.025 mM [○, ICE0/Pspank-immR(0.025)] or 1 mM IPTG [●, ICE0/Pspank-immR(1)]; and ICEBs10 PimmR-immRΩ267 (JMA421, ▲, ICE0/PimmR-immR) cells.
B. Pxis–lacZΩ343 expression in wild-type cells (JMA201, ▽, wt; same data as in A); ΔimmR cells (JMA214, ◊, ΔimmR); and ΔimmR Pspank-immRΩ26 cells in the presence of 1 mM IPTG (JMA541, ◆, ΔimmR/Pspank-immR).
C. Schematic representation of the excision assay performed in (D). Upon excision of ICEBs1 from the chromosome, two products are formed, an ICEBs1 circular intermediate and a repaired chromosomal junction. These products can be detected through PCR using primers b + c and a + d (respectively), which are represented by small arrows in the diagram. The sequences of these primers [oJMA93 (a), oJMA95 (b), oJMA97 (c) and oJMA100 (d)] were described previously (Auchtung et al., 2005).
D. Excision was monitored in wild-type (JMA201, lane 1), ΔimmR (JMAM214, lane 2) and ΔimmR Pspank-immRΩ26 (JMA541, grown in the presence of 1 mM IPTG, lane 3) cells. DNA was extracted from cells in exponential phase and 100 ng was used as template to amplify the indicated products described in (C). Quantitative PCR performed on DNA extracted from a population of ΔimmR cells revealed that excision occurred in ~97% of cells whereas excision occurred in ~0.003% of wild-type cells.
Characterization of ImmR
ImmR (YdcN) is similar to some bacteriophage repressors (Burrus et al., 2002; Auchtung et al., 2005). We found that ImmR represses transcription from the xis promoter. Expression of Pxis–lacZΩ343 was low in cells containing ICEBs1 (Fig. 3A and B). In contrast, expression increased >1000-fold in ICEBs10 cells, that is, in cells cured of ICEBs1 (Fig. 3A). Expression of Pxis–lacZΩ343 in the ICEBs10 strain was repressed upon ectopic expression of immR (Fig. 3A). This repression was observed when immR was expressed ectopically from either the isopropyl-β-d-thiogalactopyranoside (IPTG)-inducible promoter, Pspank, or its native promoter, PimmR (Fig. 3A; see below). With Pspank-immR, the amount of repression of Pxis–lacZΩ343 was dependent on the concentration of IPTG (Fig. 3A). Strong repression was observed at a high concentration of inducer (1 mM) and partial repression was observed at a lower concentration of inducer (0.025 mM). These results demonstrate that ImmR is the only ICEBs1-encoded protein required for repression of the xis promoter and that ImmR represses xis transcription in a dose-dependent manner.
We also analysed the phenotype caused by an immR null mutation (Auchtung et al., 2005). This mutation caused an ~1000-fold increase in β-galactosidase-specific activity from Pxis–lacZΩ343 (Fig. 3B). This expression was reduced to near wild-type levels in the presence of Pspank-immR (Fig. 3B). These complementation results indicate that the derepression of Pxis–lacZΩ343 observed in the immR null mutant was due to loss of immR and not a secondary or polar effect. In combination, these results indicate that ImmR is both necessary and sufficient to repress transcription from the xis promoter.
In addition to affecting expression of Pxis–lacZΩ343, deletion of immR caused excision of ICEBs1 in most cells (Fig. 3C and D). Increased excision in the immR mutant is likely due to increased expression of xis, because increased expression of xis alone is sufficient to stimulate ICEBs1 excision (C.A. Lee, J.M. Auchtung, R. Monson and A.D. Grossman, in preparation). The increase in excision in the immR mutant was suppressed by expression of Pspank-immR, indicating that the increased excision was due to loss of immR (Fig. 3D). Furthermore, because of increased excision, ICEBs1 is unstable in the immR null mutant and is lost at a relatively high frequency; approximately 10% of cells in a population of the immR null mutant have lost ICEBs1 (Auchtung et al., 2005). We suspect that ICEBs1 is not lost from all cells due to replication of the excised element. The increase in Pxis–lacZΩ343 expression observed for the ΔimmR mutant reflects the expression in the ~90% of cells in the population that are ΔimmR and contain the element as well as the ~10% of cells that have lost ICEBs1. Together, these data indicate that ImmR represses expression from the xis promoter, thereby repressing excision of ICEBs1.
In the course of constructing and growing the immR null mutant, we noticed that there was increased lysis of mutant colonies during prolonged growth on agar plates (data not shown). Cells that were missing immR due to complete loss of ICEBs1 did not have increased lysis, indicating that the increased lysis was due to loss of immR combined with the presence of ICEBs1 and not due to some additional function of immR outside of the context of ICEBs1.
The phenotypes caused by loss of immR appear to be complicated. The loss of immR causes constitutive expression of almost all of the genes in ICEBs1 and we suspect that this leads to accumulation of toxic levels of one or more gene products, perhaps components of the ICEBs1 mating pore. In addition, loss of immR causes increased excision of ICEBs1 and the excised element can replicate (J.D. Wang and A.D. Grossman, unpublished data). We suspect that this might also contribute to the cellular phenotypes caused by loss of immR.
Identification of the immR promoter
immR is transcribed divergently from xis (Fig. 1) in a putative operon that also contains ydcM (encoding a protein of unknown function) and int (encoding the integrase). A fragment containing the immR promoter was identified through cloning and functional analysis. We introduced immR along with 267 bp of upstream sequence (PimmRimmRΩ267, Fig. 4A) into an ectopic chromosomal locus in ICEBs10 cells containing a Pxis–lacZΩ343 fusion and assayed xis expression. xis transcription was repressed (Figs 3A and 4B) indicating that this sequence is sufficient for expression of ImmR. Two other constructs that contain immR and some upstream sequences were evaluated for the ability to repress transcription of Pxis–lacZΩ343. A clone that contained 141 bp upstream of the immR open reading frame was able to repress Pxis (PimmR-immRΩ141, Fig. 4A and B). In contrast, a clone that contained only 26 bp upstream of the immR open reading frame was not able to repress Pxis (Pspank-immRΩ26, Fig. 4A and B). This clone contained a functional copy of immR because induction of transcription from Pspank (contained in the clone) established repression (Fig. 3A). These results indicate that there is a functional promoter between 26 and 141 bp upstream of immR that is driving expression of immR.
Fig. 4. ImmR represses and activates its own transcription.
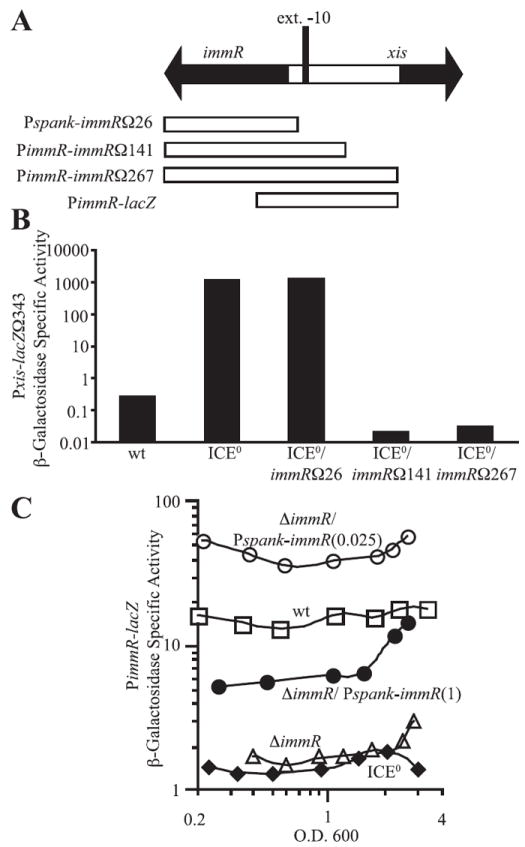
A. Schematic of xis, immR, and the intergenic region. xis, immR, and their direction of transcription are indicated by the large black arrows. The location of the putative extended −10 recognition sequence for EσA is indicated by a vertical black rectangle (ext. −10). The sequences present in the immR constructs used in (B) and (C) are indicated by white boxes underneath the diagram.
B. Functional analysis of the immR promoter. Constructs containing the entire immR open reading frame and segments of its upstream sequence (diagrammed in A) were tested for their ability to reconstitute ImmR function by repressing transcription of Pxis–lacZΩ343 in ICEBs10 cells. β-Galactosidase-specific activity in cells grown to mid-late exponential phase (OD600 ~ 1) in minimal medium was determined. Pxis–lacZΩ343 containing cells assayed were wild type (JMA201, wt); ICEBs10 (JMA264, ICE0); ICEBs10 Pspank-immRΩ26 (JMA362, ICE0/immRΩ26); ICEBs10 PimmR-immRΩ141 (JMA266, ICE0/immRΩ141); and ICEBs10 PimmR-immRΩ267 (JMA421, ICE0/immRΩ267).
C. Expression of a PimmR–lacZ fusion was monitored throughout exponential growth in minimal medium in otherwise wild-type cells (JMA309, □), ΔimmR (JMA310, Δ), ICEBs10 (JMA576, ◆) and ΔimmR Pspank-immRΩ26 cells (JMA638) grown in the in the presence of 0.025 (○) or 1 mM IPTG (●). IPTG, when used, was present throughout growth. β-Galactosidase-specific activities are plotted relative to the OD600 of the cultures.
Within the sequence between 26 and 141 bp upstream of immR, there is an almost perfect match [TG(N)TATTAT, differs from consensus at the underlined position] to the consensus for the extended −10 recognition region for RNA polymerase containing the major sigma subunit, sigma-A (Helmann, 1995). There is not a sequence that is readily recognizable as matching the consensus for the −35 recognition element. Based on the sequence and functional analyses, we conclude that transcription of immR most likely initiates from this putative sigma-A-dependent promoter.
ImmR represses and activates its own transcription
We constructed a transcriptional fusion of lacZ to the region upstream of and including the 5′ end of the immR open reading frame (Fig. 4A). This fusion, PimmR–lacZ, was expressed at a moderate level in wild-type cells throughout growth (Fig. 4C). Expression was not significantly affected by overexpression of rapI or treatment with MMC (data not shown), consistent with previous transcriptional profiling experiments (Auchtung et al., 2005).
Expression of the PimmR–lacZ fusion was decreased >10-fold in the immR null mutant or in cells missing ICEBs1 entirely (Fig. 4C). These results indicated that ImmR likely activates its own expression.
Complementation of the immR null mutation with Pspank-immR revealed that ImmR both activates and represses transcription from PimmR, depending on the amount of ImmR (Fig. 4C). When cells were grown in the presence of a high concentration of inducer (1 mM IPTG), PimmR–lacZ expression was repressed and was about half that in wild-type (ICEBs1+ immR+) cells. At a lower concentration of inducer (0.025 mM IPTG), PimmR–lacZ expression increased approximately threefold relative to that in wild-type cells (ICEBs1 + immR+). These results indicate that ImmR activates its own transcription at lower concentrations and represses its own transcription at higher concentrations.
ImmR binds to the immR-xis intergenic region
The results described above demonstrated that the promoters for xis and immR are divergent and located in the immR-xis intergenic region, and that these promoters are regulated by ImmR. It seemed likely that ImmR regulates these promoters by binding to sites in this region. To test this, we overexpressed and purified recombinant ImmR-his6 from Escherichia coli and tested the ability of this protein to bind to DNA from the intergenic region. Addition of the C-terminal his6 did not disrupt function of ImmR. That is, ImmR-his6 was active in vivo as judged by its ability to repress expression of Pxis–lacZΩ343 (data not shown). Using mobility shift assays, we found that purified ImmR bound specifically to the DNA from the xis and immR promoter regions but not to DNA from the rapI promoter (data not shown).
We used DNase I protection assays (footprinting) to further characterize the binding of ImmR to the xis-immR intergenic region (Figs 5 and 6). We found that at relatively low concentrations (~5–30 nM), ImmR protected six regions in the xis-immR intergenic region. We refer to the three regions and the binding sites within these regions that are proximal to immR as R1, R2 and R3, and the three regions and sites proximal to xis as X1, X2 and X3 (Figs 5 and 6). Three of these six regions were protected at lower concentrations of ImmR (~5–15 nM), whereas the other three regions were protected at somewhat higher concentrations of ImmR (~30 nM) (Fig. 5B and C). Two additional regions were protected at even higher concentrations of ImmR (~100 nM). These protected regions are adjacent to R3 and X1 and labelled ‘a’ and ‘b’ respectively.
Fig. 5. ImmR binds to the xis and immR intergenic region.
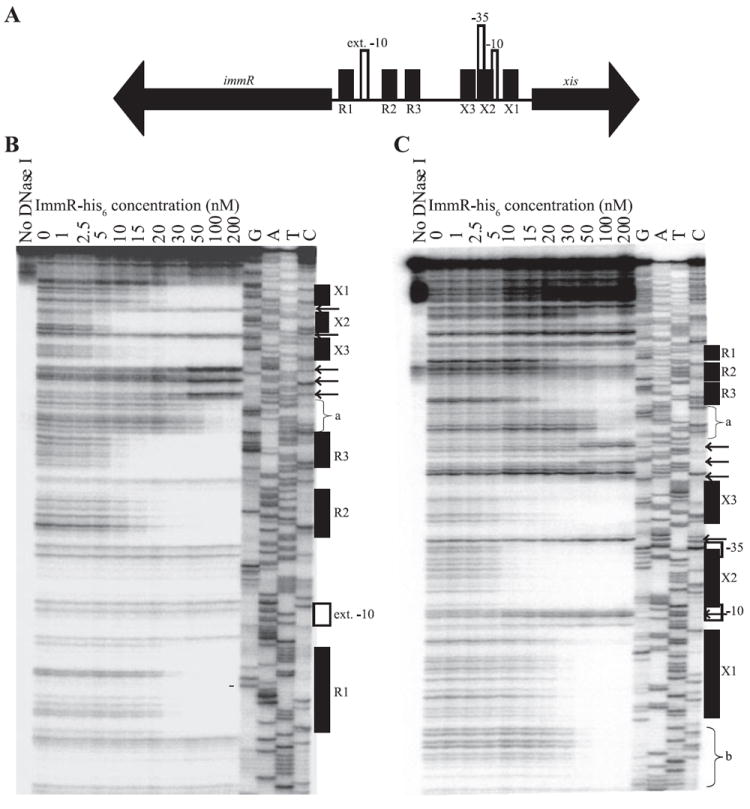
A. Detailed schematic of xis, immR, and the intergenic region. xis, immR, and the directions of transcription are indicated by the big black arrows. The location of the putative extended −10 recognition sequence for sigma-A-containing RNA polymerase in the immR promoter, and the −10 and −35 recognition sequences for sigma-A-containing RNA polymerase in the xis promoter are indicated by vertical white rectangles. The positions of the six regions protected by ImmR (sites X1, X2, X3, R1, R2 and R3) are indicated by shorter vertical black boxes.
B and C. Binding of ImmR to the xis-immR intergenic region was monitored through DNase I protection assays. Increasing concentrations (1–200 nM) of purified ImmR-his6 protein were incubated with radiolabelled DNA from the xis-immR intergenic region. DNase I was added to each reaction to digest DNA not protected by ImmR. Reactions were analysed by electrophoresis along with dideoxynucleotide sequencing reactions of the xis-immR intergenic region. The concentrations of ImmR used in each reaction are indicated above each lane of the gel. G, A, T and C indicate the dideoxynucleotide used in each sequencing reaction. Positions of the extended −10 recognition sequence for EσA in the immR promoter, and the −10 and −35 recognition sequences for EσA in the xis promoter, the six protected regions described in (A), and the two additional sites of ImmR protection (a and b) are indicated to the right of each gel image. The arrows indicate the positions of DNase I hypersensitive sites. In (B), the 5′ end of DNA at the immR end (the top strand) was labelled; in (C), the 5′ end of DNA near xis (the bottom strand) was labelled.
Fig. 6. Identification of a conserved ImmR binding motif.
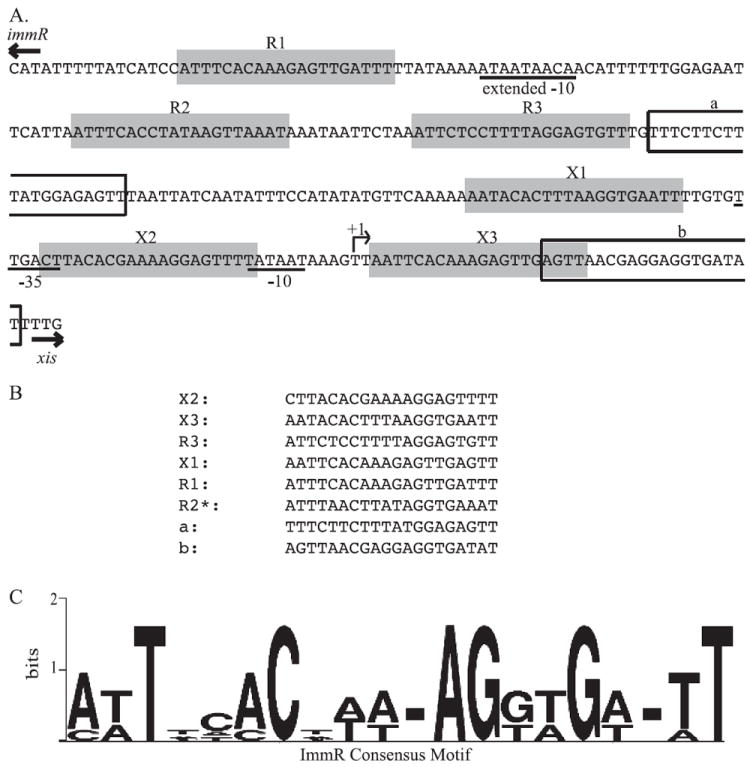
A. The sequence of a single strand of the xis-immR intergenic region is shown. The positions of the immR and xis start codons are indicated by arrows above the appropriate sequences. The positions of the PimmR extended −10 and the Pxis −35 and −10 recognition sequences for RNA polymerase containing sigma-A are indicated by the underlined nucleotides. The positions of R1, R2 and R3, and X1, X2 and X3 are indicated by grey boxes. R1, R3, and X1, X2 and X3 are all on the same strand of DNA whereas R2 is on the complementary strand. The positions of the a and b sites are indicated by white boxes.
B. An alignment of the nucleotide sequences of all eight sites protected by ImmR listed in order of ImmR affinities observed in the DNase I protection experiments (Fig. 5).
C. A representation of the consensus motif for the ImmR-binding sequence was generated using Weblogo (Crooks et al., 2004). The size of each nucleotide corresponds to the frequency with which that nucleotide was observed in that position; dashes at a position indicate lack of consensus.
The regions protected at lower ImmR concentrations are located between the −10 and −35 of Pxis (X2), upstream of the −35 of Pxis (X3), and upstream of the extended −10 of PimmR (R3). The regions protected at higher ImmR concentrations were downstream of the −10 for Pxis (X1), downstream of the −10 of PimmR (R1), and between R1 and R3, adjacent to R3 (R2) (Figs 5 and 6).
We used two motif identification programs, meme (Bailey and Elkan, 1994) and bioprospector (Liu et al., 2001), to search for conserved motifs in these protected regions. We analysed all eight binding regions and also just the six regions with the strongest binding. Based on analysis of the six regions with strongest binding, we identified a 20 bp conserved motif that was present in each protected region (Fig. 6), which is likely the binding sequence for ImmR (Fig. 6). Five of the six binding sites were identified on the same strand of DNA, whereas R2 was on the opposite strand. There is an imperfect inverted repeat contained in this sequence. Inclusion of the two weaker binding regions (a and b) in the sequence analysis resulted in a similar, but more degenerate consensus motif as these putative sites contain several differences from the others. Mutational analyses of all of the proposed sites will be necessary to discern the relative contributions of each to repression from Pxis and activation and repression from PimmR and the roles of cooperativity in binding at each site.
Based on their proximities to the sigma-A recognition sequence of Pxis, we think it is likely that binding of ImmR at X1, X2 and X3 are primarily responsible for repressing transcription of Pxis. Consistent with this hypothesis, we observed that a shorter Pxis–lacZ fusion (Pxis–lacZΩ136, containing DNA from −136 to −6 bp upstream of the xis start codon) that contains X1, X2 and X3, and lacks R1, R2 and R3, was repressed significantly by ImmR, although not to the same level as the Pxis–lacZΩ343 fusion containing all six binding sites. Basal expression was three- to fourfold higher in Pxis–lacZΩ136 (the shorter fusion) than in Pxis–lacZΩ343; both promoter fusions were derepressed to similar levels in response to MMC induction (data not shown). These findings indicate that the region of DNA containing X1, X2 and X3 is primarily responsible for repression of Pxis. However, additional upstream sequence elements appear to be required for full repression of Pxis. This requirement is presumably due to the presence of R1, R2 or R3 and probably involves cooperative interactions with ImmR bound at one or more of these sites.
Activation of the PimmR promoter does not require X1, X2 or X3, as immR is expressed at levels sufficient for Pxis repression when transcribed from a promoter region that contains only R1, R2 and R3 (Fig. 4A and B). Given the positions of R1, R2 and R3 relative to the sigma-A recognition sequence for PimmR and the relative affinities of these sites for ImmR in vitro, we think it is likely that binding of ImmR to R3 is required for activation of PimmR and that binding at R1 and R2 may mediate repression. However, it is also possible that repression of PimmR could involve cooperative interactions between ImmR bound at R1 and/or R2 and X1, X2 or X3. Further work will be needed to distinguish the specific roles for the ImmR-binding sequences and the role of cooperativity.
Identification of other potential sites of ImmR binding
We used the consensus binding motif (Fig. 6) to search the B. subtilis genome for additional ImmR binding sites using the Motif Alignment and Search Tool (Bailey and Gribskov, 1998). The P-values calculated by MAST for the six ImmR binding sites in the xis-immR intergenic region range from 4.1 × 10−9 to 7.1 × 10−8. Using MAST, we searched for additional sites in the B. subtilis chromosome with a P-value ≤ 1.0 × 10−7. We identified four sites: 1420 bp upstream of yeeC, 231 bp upstream of yhjM on the strand opposite of its transcription, 808 bp upstream of fliJ on the strand opposite of its transcription, and 24 bp upstream of yonB. Induction of ICEBs1 does not induce expression of any of these genes, nor does the absence or presence of ICEBs1 in the genome (data not shown). Therefore, we think it is unlikely that ImmR binding to DNA in these regions significantly influences transcription.
ImmR in recipient cells mediates immunity
We previously observed that acquisition of ICEBs1 by conjugal transfer is greatly reduced in cells that already contain the element (Auchtung et al., 2005). A similar level of inhibition is observed when excision and transfer is induced by overproduction of RapI or by induction of the DNA damage response (Auchtung et al., 2005, and J.M. Auchtung, C.A. Lee, and A.D. Grossman, unpublished results). This seemed similar to the repressor-mediated immunity to superinfection exhibited by various bacteriophage lysogens (Gottesman and Weisberg, 2004; Oppenheim et al., 2005).
We found that expression of ImmR in recipient cells inhibits acquisition (i.e. reduces the mating frequency) of ICEBs1. We compared transfer of ICEBs1 from donor cells into recipient cells that either did or did not express immR. Acquisition of ICEBs1 by cells already containing a copy of ICEBs1 (expressing immR from this element) was ~0.2% of that of cells without a copy of ICEBs1 (Table 1, line 1 versus 2). Furthermore, ectopic expression of immR, from its own promoter, in cells lacking ICEBs1 inhibited acquisition to a similar extent (Table 1, lines 1, 2, 3). These results indicate that production of ImmR in recipient cells is sufficient to confer immunity to acquisition of ICEBs1.
Table 1.
ImmR inhibits acquisition of ICEBs1 by recipient cells and is bypassed by expression of int.
| Recipienta (strain No.) | Mating frequencyb |
|---|---|
| 1. ICEBs10 (CAL419) | (5.1 ± 2.4) × 10−3 |
| 2. ICEBs1+ (JMA875) | (9.2 ± 4.0) × 10−6 |
| 3. ICEBs10; PimmR-immR (JMA872) | (2.4 ± 0.62) × 10−6 |
| 4. ICEBs10; PimmR-immR; Pspank-int (JMA873) | (2.0 ± 0.84) × 10−3 |
| 5. ICEBs10; Pspank-int (JMA878) | (8.0 ± 2.3) × 10−3 |
| 6. ICEBs1(Δint); Pspank-int (JMA882) | (2.2 ± 0.78) × 10−4 |
All recipients contained a comK null mutation and were streptomycin resistant (str). Recipient cells containing Pspank-int were grown in the presence of IPTG to allow production of Int.
Mating assays were performed as described (Auchtung et al., 2005). The mating frequency is the mean number of transconjugants per donor cell (±standard error of the mean), calculated from at least two experiments. The donor strain was JMA168 [ICEBs1(ΔrapIphrI∷kan); Pspank(hy)-rapI]. Expression of Pspank(hy)-rapI was induced with IPTG 1 h before mating. Production of RapI causes excision of ICEBs1 and expression of its conjugation genes.
ImmR-mediated immunity can be bypassed by increased levels of Int
In wild-type ICEBs1, int appears to be transcribed from the immR promoter: immR, ydcM and int are co-oriented and there are no predicted transcriptional terminators present. Thus, the presence of ImmR in mating recipients could potentially limit expression of int on the incoming element (by binding to PimmR and repressing transcription) and prevent acquisition of that element. In addition, ImmR in the recipient could have an effect on some other aspect of ICEBs1 function.
We monitored acquisition of ICEBs1 in recipients expressing int. Expression of int (from Pspank) in recipients lacking ICEBs1 had no significant effect on acquisition (Table 1, line 1 versus 5). However, in cells with reduced acquisition due to expression of immR (Table 1, line 1 versus 3), expression of int restored acquisition to levels similar to those in cells without immR (Table 1, line 1, 3, 4). These results indicate that the presence of ImmR in the recipient inhibits expression and/or function of Int and can be bypassed by overproduction of Int.
We also tested for relief of immunity in cells containing ICEBs1Δint and expressing int from Pspank. Expression of int in the recipient substantially restored acquisition, but not up levels in cells cured of ICEBs1. Acquisition of the incoming ICEBs1 was ~20-fold more efficient than in the absence of ectopic expression of int (Table 1, line 6 versus 2), but less efficient (~20-fold) than that of cells cured of ICEBs1 (Table 1, line 1). This ~20-fold inhibition of acquisition indicates that there is likely to be another ICEBs1-dependent mechanism that confers immunity independently of the effects of ImmR on Int.
Discussion
Our results show that expression of immR in potential recipients is sufficient to confer immunity to acquisition of ICEBs1. More importantly, ImmR promotes stable maintenance of a resident copy of ICEBs1 by functioning as a primary regulator of transcription. Inactivation or loss of ImmR, in otherwise ICEBs1 + cells, causes increased excision and loss of ICEBs1 from host cells (Auchtung et al., 2005). ImmR regulates transcription within ICEBs1 by binding to sites in the intergenic region shared by the immR and xis promoters. ImmR represses transcription of genes that mediate excision (xis) and transfer and both activates and represses transcription from its own promoter. These properties of ImmR from ICEBs1 are similar to those of other repressor proteins from many mobile elements, including the well-studied coliphage lambda (Ptashne, 2004; Dodd et al., 2005; Oppenheim et al., 2005).
Mechanism of ImmR-mediated immunity
Many mobile genetic elements possess mechanisms to prevent acquisition of additional copies of the same or a related element. One example is superinfection immunity mediated by the lambda repressor CI (reviewed in Echols and Guarneros, 1983; Ptashne, 2004; Oppenheim et al., 2005). CI binds to and prevents expression of lytic promoters, thereby preventing lytic development of incoming homoimmune phages. Low levels of int expression in an established lysogen also limits the ability of an incoming phage to integrate and form a double lysogen.
ImmR appears to mediate immunity from incoming ICEBs1 by limiting production or activity of ICEBs1 Int. Expression of immR in potential recipients is sufficient to limit acquisition of ICEBs1 and this limitation is almost completely suppressed by ectopic expression of int. We do not know how ImmR limits production or activity of Int. ImmR activates and represses its own expression, and presumably that of int as well. However, there appears to be enough Int produced to allow excision of a resident ICEBs1 without further significant increase in int mRNA levels (Auchtung et al., 2005). Perhaps more Int is needed for integration than for excision. Int could also function preferentially in cis, such that the Int that is produced by a resident ICEBs1 is much less effective at mediating integration of an incoming element. It is also possible that ImmR somehow inhibits binding of Int to the attachment sites and that increased production of Int overcomes this. If this is the case, then ImmR is likely binding sequences different from the operator sites to which it binds to mediate transcriptional regulation as there are no obvious matches to the consensus operator sequences near the attachment sites.
In addition to limiting stable acquisition of an incoming element, we suspect that ImmR in the recipient will repress transcription from Pxis on the incoming element, analogous to lambda CI-mediated repression of pL and pR that results in immunity to superinfection. Such repression by ImmR would block expression of xis and the downstream conjugation functions, allowing the resident element to remain stably integrated and preventing the incoming element from being transferred to a second recipient.
Our results indicate that there is an additional mechanism or mechanisms contributing to immunity that is independent of the effects of ImmR on Int. ImmR-independent expression of int in ICEBs1+ cells was not sufficient to restore mating frequencies to the levels observed in cells cured of ICEBs1, as mating occurred ~20-fold less efficiently (Table 1, line 6 versus 1). However, mating into ICEBs10 recipient cells that express immR as well as int is approximately fourfold less efficient than mating into ICEBs10 recipient cells that express only int (Table 1, line 4 versus 5). Therefore, there may be a small Int-independent role for ImmR in mediating immunity as well as other potential immunity mechanisms functioning in ICEBs1.
Based on comparisons to other mobile elements, at least three additional types of regulation seem possible: (i) inhibition of contact between donor and recipient cells (surface exclusion; Achtman et al., 1977; Anthony et al., 1999), (ii) inhibition of uptake of DNA into the cells (entry exclusion; Achtman et al., 1977; Anthony et al., 1999), and (iii) inhibition of integration into DNA near an established element (target immunity; Skelding et al., 2003, and references therein). Further work will be needed to determine if any of these mechanisms also contribute to ICEBs1 immunity and what role, if any, ImmR plays in mediating this regulation.
Organization of the promoter regions regulated by ImmR
The organization of the promoters regulated by ImmR (Fig. 1) is similar to that observed in several bacteriophage, with the promoter for the repressor transcribed divergently from genes involved in lytic development (Lucchini et al., 1999; Ptashne, 2004; Waldor and Friedman, 2005). The most well-known of these is bacteriophage lambda. The gene for lambda repressor, cI, is transcribed divergently from an operon encoding several genes involved in lytic development (cro, O, P, Q) (summarized in Ptashne, 2004). This organization likely contributes to the regulatory circuits that control stability and induction of the integrated element.
Role of autoregulation of immR
Autoregulation of immR transcription likely serves at least two functions: (i) it maintains the concentration of ImmR at a level high enough to prevent expression of the ICEBs1 excision and conjugation genes, and (ii) it maintains ImmR at a level sufficiently low to respond to the appropriate inducing signals.
Similarly, lambda repressor is also autoregulated. CI activates its own transcription at lower concentrations and represses its own transcription at higher concentrations (reviewed in Dodd et al., 2005). In the case of lambda, negative autoregulation is needed to maintain low enough concentrations for proper lysogenic induction, whereas positive autoregulation is required to maintain levels of CI sufficient for maintenance of lysogeny (Dodd et al., 2005; Michalowski and Little, 2005).
Transcription from Pxis also appears to be affected by one or more genes in ICEBs1 other than immR. The existence of the additional regulator(s) is evidenced by the higher levels of ImmR-mediated repression of Pxis that occur in ICEBs10 cells containing PimmR-immR expressed ectopically than in wild-type ICEBs1+ cells (Figs 3A and 4B). This regulation could be affecting transcription from Pxis directly, although we favour the notion that it affects the activity of ImmR. Further work will be needed to determine how additional genes in ICEBs1 affect transcription.
Mechanisms of induction of ICEBs1
There are two mechanisms known to activate ICEBs1 gene expression and excision, and both are likely to involve relief from ImmR-mediated repression (Auchtung et al., 2005). One mechanism involves activation of the RecA-dependent SOS response and the other is independent of RecA and requires RapI. Both mechanisms require at least one other ICEBs1 gene product in addition to ImmR (J.M. Auchtung, C.A. Lee and A.D. Grossman, unpubl. results).
Activation of ICEBs1 by RapI
RapI is normally produced as cells become crowded and enter stationary phase. Its activity is inhibited by the pentapeptide PhrI that is secreted and then imported via the oligopeptide permease (Auchtung et al., 2005). Most of the experiments presented here bypass this regulation due to ectopic overproduction of RapI to induce excision and mating.
Normal production of RapI, when the concentration of PhrI is low, causes increased ICEBs1 gene expression and subsequent excision (Auchtung et al., 2005). Because both rapI and phrI are contained in ICEBs1, this regulation normally provides a mechanism for cells to recognize whether neighbours contain ICEBs1. If the neighbours do not contain a copy of ICEBs1, then excision and transfer can occur. Homologues of rapI and phrI are found in other mobile elements, indicating that this type of recognition of self might be conserved (Auchtung et al., 2005).
RapI likely stimulates increased expression of the ICEBs1 excision and conjugation genes by inhibiting the activity of ImmR (Auchtung et al., 2005). Consistent with this, loss of immR is epistatic to loss or overexpression of rapI (data not shown). Despite the derepression of Pxis upon overproduction of RapI, there is little if any obvious effect on transcription from PimmR, as measured in DNA microarray experiments (Auchtung et al., 2005) and in the assays monitoring β-galactosidase activity from a PimmR–lacZ fusion. In both cases, we were monitoring expression in the bulk population of cells. If immR expression had increased in some cells due to loss of ImmR-mediated repression and decreased in other cells due to loss of ImmR-mediated activation, these changes would not have been reflected in our assays. Such effects could be observed by monitoring gene expression in single cells.
In addition, significant derepression of Pxis occurs at levels of ImmR that still cause transcriptional activation at PimmR (compare the derepression of Pxis–lacZΩ343 expression at 0.025 mM ImmR in Fig. 3A with the increased expression of PimmR–lacZ at 0.025 mM in Fig. 4C). Therefore, the levels of active ImmR may drop sufficiently low upon overproduction of RapI to allow derepression of Pxis without losing activation of PimmR.
Activation of ICEBs1 by the SOS response
The RecA-dependent SOS response causes induction of many mobile elements (Walker, 1996; Gottesman and Weisberg, 2004; Dodd et al., 2005; McCabe et al., 2005; Quinones et al., 2005). In many cases, this activation involves the RecA-stimulated autocleavage of a repressor protein. For example, in lambda, activated RecA stimulates autocleavage of CI (Little, 1984) and subsequent derepression of pL and pR, excision, and expression of the lytic pathway (Ptashne, 2004; Oppenheim et al., 2005).
In B. subtilis, the RecA-dependent SOS response causes induction of expression of ICEBs1 genes (Auchtung et al., 2005; Goranov et al., 2006) and subsequent excision, most likely by causing the inactivation of ImmR (Auchtung et al., 2005). This RecA-dependent (RapI-independent) inactivation of ImmR requires at least one other gene in ICEBs1 (J.M. Auchtung, C.A. Lee, and A.D. Grossman, unpublished results). This requirement is in marked contrast to the well-characterized mobile elements in which activation of RecA during the SOS response stimulates the autocleavage of the repressor. It will be interesting to characterize the ICEBs1 gene responsible for this regulation and to determine its mechanism of action.
Experimental procedures
Media
For standard genetic manipulation, E. coli and B. subtilis were grown at 37°C in LB (Sambrook and Russell, 2001). For experiments, B. subtilis was grown in S7 minimal salts medium (Vasantha and Freese, 1980) (containing 50 mM MOPS instead of 100 mM) with 1% glucose, 0.1% glutamate, with required amino acids as needed [trp and phe (40 μg ml−1), and thr (120 μg ml−1)], or LB medium, as indicated. Antibiotics were used at the following concentrations: ampicillin (100 μg ml−1, unless otherwise indicated); chloramphenicol (5 μg ml−1); kanamycin (5 μg ml−1); spectinomycin (100 μg ml−1); streptomycin (100 μg ml−1); and erythromycin (0.5 μg ml−1) and lincomycin (12.5 μg ml−1) together to select for macrolide-lincosamide-streptogramin B (MLS) resistance. IPTG (Sigma) was used at the concentrations indicated, MMC (Sigma) was used at a final concentration of 1 μg ml−1, and l-arabinose (Sigma) was used at a final concentration of 0.2%.
Strains and alleles
Strains used in this study are listed in Table 2. Standard techniques were used for cloning and strain construction (Harwood and Cutting, 1990; Sambrook and Russell, 2001). The BL21-AI (Invitrogen) strain was used for overexpression of immR-his6 in E. coli. The ICEBs10 strain, the spontaneous streptomycin (str) resistant allele (likely in rpsL), and the Δint205∷cat, ΔimmR208∷cat and amyE∷Pspank(hy)-rapI alleles were previously described (Auchtung et al., 2005). All bp positions are indicated relative to the positions of the start codons annotated in the B. subtilis 168 genome sequence.
Table 2.
Strains used in this study.
| Strain | Relevant genotypea |
|---|---|
| JH642 | trpC2 pheA1 |
| CAL419 | ICEBs10 comK∷cat str |
| JMA28 | amyE∷[(Pspank(hy)-rapI) spc] |
| JMA168 | Δ(rapI-phrI)342∷kan amyE∷[(Pspank(hy)-rapI) spc] |
| JMA201 | thrC∷[(Pxis–lacZΩ343) erm] |
| JMA214 | ΔimmR208∷cat thrC∷[(Pxis–lacZΩ343) erm] |
| JMA264 | ICEBs10 thrC∷[(Pxis–lacZΩ343) erm] |
| JMA266 | ICEBs10 amyE∷[(PimmR-immRΩ141) spc] thrC∷[(Pxis–lacZΩ343) erm] |
| JMA309 | thrC∷[(PimmR–lacZ) erm] |
| JMA310 | ΔimmR208∷cat thrC∷[(PimmR–lacZ) erm] |
| JMA362 | ICEBs10 amyE∷[(Pspank-immRΩ26) spc] thrC∷[(Pxis–lacZΩ343) erm] |
| JMA421 | ICEBs10 cgeD∷[(PimmR-immRΩ267) kan] thrC∷[(Pxis–lacZΩ343) erm] |
| JMA541 | ΔimmR208∷cat amyE∷[(Pspank-immRΩ26) spc] thrC∷[(Pxis–lacZΩ343) erm] |
| JMA576 | ICEBs10 thrC∷[(PimmR–lacZ) erm] |
| JMA631 | ICEBs10 cgeD∷[(PimmR-immR-his6 cat) kan] thrC∷[(Pxis–lacZΩ343) erm] |
| JMA638 | ΔimmR208∷cat amyE∷[(Pspank-immRΩ27) spc] thrC∷[(immR–lacZ) erm] |
| JMA683 | thrC∷[(Pxis–lacZΩ136) erm] |
| JMA870 | ICEBs10 cgeD∷[(Pspank(hy)-rapI) kan] thrC∷[(Pxis–lacZΩ343) erm] |
| JMA872 | ICEBs10 amyE∷[(PimmR-immRΩ141) spc] comK∷cat str |
| JMA873 | ICEBs10 amyE∷[(PimmR-immRΩ141) spc] thrC∷[(Pspank-int) erm] comK∷cat str |
| JMA875 | comK∷cat str |
| JMA878 | ICEBs10 thrC∷[(Pspank-int) erm] comK∷cat str |
| JMA882 | Δint205∷cat∷tet thrC∷[(Pspank-int) erm] comK∷cat str |
| KLG125 | amyE∷[(Pspank(hy)) spc] thrC∷[(Pxis–lacZΩ343) erm] |
| KLG126 | amyE∷[(Pspank(hy)-rapI) spc] thrC∷[(Pxis–lacZΩ343) erm] |
All B. subtilis strains are derived from JH642 and contain trpC2 and pheA1 (Perego et al., 1988).
Δint205∷cat∷tet was created by integrating the drug resistance conversion plasmid pCm∷Tet (Steinmetz and Richter, 1994) into the Δint205∷cat allele. Double-cross-over insertion was confirmed by selecting for resistance to tetracycline and screening for sensitivity to chloramphenicol.
Pxis–lacZΩ343 (Fig. 2A) was generated by cloning DNA from 343 to 6 bp upstream of the xis start codon upstream of a promoterless lacZ in the vector pDG793 (Guerout-Fleury et al., 1996).
PimmR–lacZ (Fig. 4A) was generated by cloning the same sequence in the opposite orientation in pDG793. Both vectors were integrated into the chromosome at thrC by double-cross-over homologous recombination, which was verified by conversion to threonine auxotrophy.
Pxis–lacZΩ136 was generated by cloning DNA from 136 to 6 bp upstream of the xis start codon upstream of lacZ in pDG793 followed by integration into the chromosome at thrC by double-cross-over homologous recombination.
Several fusions to the LacI-repressible IPTG-inducible promoter Pspank were generated by cloning into the vector pDR110 (Rokop et al., 2004). Fusions were then recombined into the chromosome in single copy at amyE. Pspank-immRΩ26 was generated by cloning from −26 to +383 of immR into pDR110.
Pspank-int was generated by cloning −49 to +1128 of int into the pCAL215, which contains lacI, Pspank, and the multiple cloning site from pDR110, ligated into the thrC integration vector pDG795 (Guerout-Fleury et al., 1996). This vector was integrated into the chromosome by double-cross-over homologous recombination, which was verified by conversion to threonine auxotrophy.
PimmR-immRΩ141 was generated by cloning from −141 to +383 of immR into pDR110. immR was under control of its own promoter and Pspank. Expression of immR from this construct occurred independently of IPTG.
PimmR-immRΩ267 was generated by cloning the sequence from −267 of immR to +383 of immR into the integration vector, pMMB124, which was a generous gift from M.B. Berkmen. pMMB124 contains segments of cgeD flanking the kanamycin resistance gene in pGK67 (Lemon et al., 2001). This allows for integration of DNA by double-cross-over homologous recombination into cgeD. Double-cross-over integrants were identified by screening for sensitivity to chloramphenicol, which is encoded on the plasmid backbone outside of the cgeD regions.
immR-his6 was created by cloning the immR coding sequence (+1 to +380) along with an optimized ribosome-binding sequence (rbs) and spacer region (AGGAGGAAAAACAT, rbs is underlined) downstream of the T7 promoter in the pET21-cat vector to create plasmid pJMA605. pET21-cat contains the chloramphenicol resistance gene from pJH101 (Ferrari et al., 1983) in the SphI site of pET21 (Novagen). pJMA605 was introduced into the B. subtilis chromosome by single-cross-over homologous recombination to generate immR:immR-his6 cat.
β-Galactosidase assays
β-Galactosidase-specific activity was determined as described (Jaacks et al., 1989). Specific activity was calculated relative to the optical density at 600 nm (OD600) of thesamples. Results shown are from single experiments that are representative of results observed in at least two independent experiments.
Excision assays
Qualitative assays
Total DNA was prepared using Qiagen DNeasy Tissue Kit as described (Auchtung et al., 2005). PCR reactions were performed using the previously described primers and conditions (Auchtung et al., 2005), using 100 ng of DNA as a template in each reaction. PCR products were visualized by electrophoresis on 2% agarose gels stained with Ethidium bromide. Gel images were captured with the ChemiImager gel documentation system (Alpha Innotech).
Quantitative assay
Quantitative PCR reactions were performed as described, using twofold dilutions of template to obtain PCR products amplified in the linear range (Auchtung et al., 2005). Gel images were captured and spot densitometry was performed with the ChemiImager gel documentation system (Alpha Innotech). In order to determine the percentage of cells in which excision had occurred, the ratio of the PCR signal from repaired chromosomal junction as a fraction of the PCR signal from a control site for the experimental sample (wild type or ΔimmR) was normalized to the ratio of repaired chromosomal junction to the control site in cells lacking ICEBs1 (JMA222), which simulates 100% excision. The repaired chromosomal junction was used rather than the circular ICEBs1 excision product to avoid overestimates due to replication of the excised product.
Mating assays
Donor and recipient cells were grown in LB medium. Matings were performed on filter paper essentially as described (Auchtung et al., 2005). Briefly, IPTG (1 mM) was added to donor cells in mid-exponential growth (OD600 ~ 0.2) to induce expression of Pspank(hy)-rapI; RapI then causes induction of ICEBs1 gene expression and excision. One hour after expression of RapI, donors were mixed with recipients, filtered to allow mating, and transconjugants were identified and mating frequencies were calculated. Recipient cells containing Pspank-int were grown in the presence of IPTG (1 mM).
Primer extension assays
The 5′ end of the xis transcript was determined by primer extension analysis. RNA was isolated using the RNeasy kit from Qiagen according to the manufacturer’s protocol. Ten micrograms of RNA from untreated wild-type cells, wild-type cells treated with MMC, and cells overexpressing rapI was reverse transcribed as described (Auchtung et al., 2005), except that ~2 pmol of specific 32P-labelled oligonucleotide was used as a primer. Oligonucleotides oJMA102, complementary to −6 to −17 relative to the xis start codon, and oJMA240, complementary to +22 to +49 of xis, were end-labelled with [γ-32P]-ATP (Perkin-Elmer) using T4 polynucleotide kinase (New England Biolabs). Labelled olignucleotides were separated from unincorporated ATP using Qiagen’s Nucleotide Removal Kit. Products of the primer extension reactions were compared with the products of dideoxynucleotide sequencing reactions performed with the fmol DNA Cycle Sequencing System (Promega) using labelled oJMA102 or oJMA240 as primers and PCR products corresponding to −6 to −131 or +22 to −131 of xis as template. Primer extension and dideoxynucleotide sequencing reactions were electrophoresed on 6% polyacrylamide 7 M urea gels. Radioactivity was detected through phosphorimaging using the Typhoon imager 9400 (Amersham Biosciences).
Purification of ImmR-his6
Escherichia coli BL21 cells containing an arabinose-inducible copy of the T7 RNA polymerase (BL21-AI, Invitrogen) and a plasmid encoding immR-his6 under the control of a LacI-repressible/IPTG-inducible T7 polymerase-dependent promoter were grown in LB (containing 200 mg ml−1 ampicillin) at 37°C with shaking. At OD600 ~ 0.4, l-arabinose and 1 mM IPTG were added to induce expression of the T7 polymerase and derepress expression of immR-his6. One hundred millilitres of cells were collected 4 h after induction, pelleted by centrifugation, decanted and stored at −80°C.
The cell pellet was thawed on ice, re-suspended in 0.2 volumes lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, pH 8.0) and lysed by sonication on ice for 4 × 30 s. Supernatant was separated by centrifugation at 10 000 g at 4°C for 20 min. ImmR-his6 was purified by Ni-NTA column chromatography (Qiagen) according to the manufacturer’s protocol for batch purification under native conditions, except that proteins were eluted by increased imidazole concentrations (50, 100, 200 and 400 mM imidazole).
Elution fractions were analysed by SDS-PAGE. ImmR was present in the 400 mM elution fraction and was ~95% pure. ImmR was dialysed into buffer (50 mM NaH2PO4, 300 mM NaCl, pH 8.0) at 4°C overnight, glycerol was added to 50% and concentration was determined by Bradford assay (Bio-Rad). Protein was stored at −80°C.
DNase I protection assays
DNA fragments from the xis-immR intergenic region were labelled at one end using 32P-labelled primers oJMA109 or oJMA240 in PCR reactions along with an unlabelled primer (oJMA240 or oJMA109). Labelled PCR products were purified on polyacrylamide gels and eluted into buffer containing 0.5 M ammonium acetate, 10 mM magnesium acetate and 2 mM EDTA at 37°C overnight. Labelled DNA was ethanol-precipitated and re-suspended in buffer (10 mM Tris-HCl, 20 mM NaCl, 0.1 mM EDTA pH 8).
ImmR-his6 (1–200 nM) was allowed to bind the labelled DNA (≤ 0.4 μM) in binding buffer (20 mM Tris-HCl, 50 mM KCl, 3 mM MgCl2, 5% glycerol, 2.5 mM CaCl2, 50 ng μl−1 poly (dI/dC), 100 ng μl−1 BSA, pH 7.5) in 25 μl of reactions at 37°C for 10 min, followed by incubation at room temperature incubation for 20 min. DNase I (0.5 units) (Ambion) was added to each reaction (total reaction volume = 30 μl), and incubated for 1 min at room temperature prior to the addition of 7.5 μl of stop solution (3.3% SDS, 60 mM EDTA, 0.5 mM Tris-HCl pH 9.5).
DNA was isolated from each reaction by phenol-chloroform extraction followed by ethanol precipitation in the presence of yeast tRNA as carrier and 3 mM sodium acetate. DNA was re-suspended in 3 μl of buffer (10 mM Tris, 10 mM EDTA, pH 8). An equal volume of formamide running buffer (90% formamide, 10 mM EDTA, 0.04% xylene cyanol, 0.04% bromphenol blue) was added to each sample.
In order to determine which regions were protected from DNase I digestion by ImmR binding, dideoxynucleotide sequencing reactions of the xis-immR intergenic region were also performed (as described above) using the corresponding radiolabelled primer (oJMA109 or oJMA240). DNase I protection and dideoxynucleotide sequencing reactions were analysed by electrophoresis on 6% polyacrylamide 7 M urea gels. Radioactivity was detected through phosphorimaging using the Typhoon imager 9400 (Amersham Biosciences).
Acknowledgments
We thank M.B. Berkmen for providing plasmid pMMB124, K. MacIsaac for advice about motif searching, and A.L. Sonen-shein, F. Solomon, G. Walker, R. Sauer and J.D. Wang for comments on the manuscript. This work was supported in part by Public Health Service Grant GM50895 from the NIH to A.D.G.
References
- Achtman M, Kennedy N, Skurray R. Cell–cell interactions in conjugating Escherichia coli: role of traT protein in surface exclusion. Proc Natl Acad Sci USA. 1977;74:5104–5108. doi: 10.1073/pnas.74.11.5104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony KG, Klimke WA, Manchak J, Frost LS. Comparison of proteins involved in pilus synthesis and mating pair stabilization from the related plasmids F and R100-1: insights into the mechanism of conjugation. J Bacteriol. 1999;181:5149–5159. doi: 10.1128/jb.181.17.5149-5159.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auchtung JM, Lee CA, Monson RE, Lehman AP, Grossman AD. Regulation of a Bacillus subtilis mobile genetic element by intercellular signaling and the global DNA damage response. Proc Natl Acad Sci USA. 2005;102:12554–12559. doi: 10.1073/pnas.0505835102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol. 1994;2:28–36. [PubMed] [Google Scholar]
- Bailey TL, Gribskov M. Combining evidence using p-values: application to sequence homology searches. Bioinformatics. 1998;14:48–54. doi: 10.1093/bioinformatics/14.1.48. [DOI] [PubMed] [Google Scholar]
- Brussow H, Canchaya C, Hardt WD. Phages and the evolution of bacterial pathogens: from genomic rearrangements to lysogenic conversion. Microbiol Mol Biol Rev. 2004;68:560–602. doi: 10.1128/MMBR.68.3.560-602.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrus V, Waldor MK. Shaping bacterial genomes with integrative and conjugative elements. Res Microbiol. 2004;155:376–386. doi: 10.1016/j.resmic.2004.01.012. [DOI] [PubMed] [Google Scholar]
- Burrus V, Pavlovic G, Decaris B, Guedon G. The ICESt1 element of Streptococcus thermophilus belongs to a large family of integrative and conjugative elements that exchange modules and change their specificity of integration. Plasmid. 2002;48:77–97. doi: 10.1016/s0147-619x(02)00102-6. [DOI] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodd IB, Shearwin KE, Egan JB. Revisited gene regulation in bacteriophage lambda. Curr Opin Genet Dev. 2005;15:145–152. doi: 10.1016/j.gde.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Echols H, Guarneros G. Control of integration and excision. In: Hendrix RW, Roberts JW, Stahl FW, Weisberg RA, editors. Lambda II. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1983. pp. 75–92. [Google Scholar]
- Ferrari FA, Nguyen A, Lang D, Hoch JA. Construction and properties of an integrable plasmid for Bacillus subtilis. J Bacteriol. 1983;154:1513–1515. doi: 10.1128/jb.154.3.1513-1515.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost LS, Leplae R, Summers AO, Toussaint A. Mobile genetic elements: the agents of open source evolution. Nat Rev Microbiol. 2005;3:722–732. doi: 10.1038/nrmicro1235. [DOI] [PubMed] [Google Scholar]
- Goranov AI, Kuester-Schoeck E, Wang JD, Grossman AD. Characterization of the global transcriptional responses to different types of DNA damage and disruption of replication in Bacillus subtilis. J Bacteriol. 2006;188:5595–5605. doi: 10.1128/JB.00342-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesman ME, Weisberg RA. Little lambda, who made thee? Microbiol Mol Biol Rev. 2004;68:796–813. doi: 10.1128/MMBR.68.4.796-813.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerout-Fleury AM, Frandsen N, Stragier P. Plasmids for ectopic integration in Bacillus subtilis. Gene. 1996;180:57–61. doi: 10.1016/s0378-1119(96)00404-0. [DOI] [PubMed] [Google Scholar]
- Harwood CR, Cutting SM. Molecular Biological Methods for Bacillus. Chichester: John Wiley & Sons; 1990. [Google Scholar]
- Helmann JD. Compilation and analysis of Bacillus subtilis sigma A-dependent promoter sequences: evidence for extended contact between RNA polymerase and upstream promoter DNA. Nucleic Acids Res. 1995;23:2351–2360. doi: 10.1093/nar/23.13.2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaacks KJ, Healy J, Losick R, Grossman AD. Identification and characterization of genes controlled by the sporulation-regulatory gene spo0H in Bacillus subtilis. J Bacteriol. 1989;171:4121–4129. doi: 10.1128/jb.171.8.4121-4129.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarmer H, Larsen TS, Krogh A, Saxild HH, Brunak S, Knudsen S. Sigma A recognition sites in the Bacillus subtilis genome. Microbiology. 2001;147:2417–2424. doi: 10.1099/00221287-147-9-2417. [DOI] [PubMed] [Google Scholar]
- Lemon KP, Kurtser I, Grossman AD. Effects of replication termination mutants on chromosome partitioning in Bacillus subtilis. Proc Natl Acad Sci USA. 2001;98:212–217. doi: 10.1073/pnas.011506098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little JW. Autodigestion of LexA and phage lambda repressors. Proc Natl Acad Sci USA. 1984;81:1375–1379. doi: 10.1073/pnas.81.5.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Brutlag DL, Liu JS. BioProspector: discovering conserved DNA motifs in upstream regulatory regions of co-expressed genes. In: Altman RB, Dunker RK, Hunter L, Lauderdale K, Klein TE, editors. Pacific Symposium on Biocomputing. Hackensack, N.J: World Scientific Press; 2001. pp. 127–138. [PubMed] [Google Scholar]
- Lucchini S, Desiere F, Brussow H. Similarly organized lysogeny modules in temperate Siphoviridae from low GC content gram-positive bacteria. Virology. 1999;263:427–435. doi: 10.1006/viro.1999.9959. [DOI] [PubMed] [Google Scholar]
- McCabe BC, Pawlowski DR, Koudelka GB. The bacteriophage 434 repressor dimer preferentially undergoes autoproteolysis by an intramolecular mechanism. J Bacteriol. 2005;187:5624–5630. doi: 10.1128/JB.187.16.5624-5630.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuade RS, Comella N, Grossman AD. Control of a family of phosphatase regulatory genes (phr) by the alternate sigma factor sigma-H of Bacillus subtilis. J Bacteriol. 2001;183:4905–4909. doi: 10.1128/JB.183.16.4905-4909.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalowski CB, Little JW. Positive autoregulation of cI is a dispensable feature of the phage lambda gene regulatory circuitry. J Bacteriol. 2005;187:6430–6442. doi: 10.1128/JB.187.18.6430-6442.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochman H, Lawrence JG, Groisman EA. Lateral gene transfer and the nature of bacterial innovation. Nature. 2000;405:299–304. doi: 10.1038/35012500. [DOI] [PubMed] [Google Scholar]
- Oppenheim AB, Kobiler O, Stavans J, Court DL, Adhya S. Switches in bacteriophage lambda development. Annu Rev Genet. 2005;39:409–429. doi: 10.1146/annurev.genet.39.073003.113656. [DOI] [PubMed] [Google Scholar]
- Perego M, Spiegelman GB, Hoch JA. Structure of the gene for the transition state regulator, abrB: regulator synthesis is controlled by the spo0A sporulation gene in Bacillus subtilis. Mol Microbiol. 1988;2:689–699. doi: 10.1111/j.1365-2958.1988.tb00079.x. [DOI] [PubMed] [Google Scholar]
- Ptashne M. A Genetic Switch: Phage Lambda Revisited. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2004. [Google Scholar]
- Quinones M, Kimsey HH, Waldor MK. LexA cleavage is required for CTX prophage induction. Mol Cell. 2005;17:291–300. doi: 10.1016/j.molcel.2004.11.046. [DOI] [PubMed] [Google Scholar]
- Rokop ME, Auchtung JM, Grossman AD. Control of DNA replication initiation by recruitment of an essential initiation protein to the membrane of Bacillus subtilis. Mol Microbiol. 2004;52:1757–1767. doi: 10.1111/j.1365-2958.2004.04091.x. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- Skelding Z, Queen-Baker J, Craig NL. Alternative interactions between the Tn7 transposase and the Tn7 target DNA binding protein regulate target immunity and transposition. EMBO J. 2003;22:5904–5917. doi: 10.1093/emboj/cdg551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmetz M, Richter R. Plasmids designed to alter the antibiotic resistance expressed by insertion mutations in Bacillus subtilis, through in vivo recombination. Gene. 1994;142:79–83. doi: 10.1016/0378-1119(94)90358-1. [DOI] [PubMed] [Google Scholar]
- Vasantha N, Freese E. Enzyme changes during Bacillus subtilis sporulation caused by deprivation of guanine nucleotides. J Bacteriol. 1980;144:1119–1125. doi: 10.1128/jb.144.3.1119-1125.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldor MK, Friedman DI. Phage regulatory circuits and virulence gene expression. Curr Opin Microbiol. 2005;8:459–465. doi: 10.1016/j.mib.2005.06.001. [DOI] [PubMed] [Google Scholar]
- Walker GC. The SOS response of Escherichia coli. In: Neidhardt FC, Curtiss R III, Ingraham JL, Lin EC, Low KB, Magasanik B, et al., editors. Escherichia coli and Salmonella: Cellular and Molecular Biology. Washington, DC: American Society for Microbiology; 1996. pp. 1400–1416. [Google Scholar]