

Abstract
HIV-1 reverse transcriptase (RT) is a bi-functional enzyme, having both DNA polymerase (RNA- and DNA-dependent) and ribonuclease H (RNH) activities. HIV-1 RT has been an exceptionally important target for antiretroviral therapeutic development, and nearly half of the current clinically used antiretrovirals target RT DNA polymerase. However, no inhibitors of RT RNH are on the market or in preclinical development. Several drug-like small molecule inhibitors of RT RNH have been described, but little structural information is available about the interactions between RT RNH and inhibitors that exhibit antiviral activity. In this report, we describe NMR studies of the interaction of a new RNH inhibitor, BHMP07, with a catalytically active HIV-1 RT RNH domain fragment. We carried out solution NMR experiments to identify the interaction interface of BHMP07 with the RNH domain fragment. Chemical shift changes of backbone amide signals at different BHMP07 concentrations clearly demonstrate that BHMP07 mainly recognizes the substrate handle region in the RNH fragment. Using RNH inhibition assays and RT mutants, the binding specificity of BHMP07 was compared with another inhibitor, dihydroxy benzoyl naphthyl hydrazone. Our results provide a structural characterization of the ribonuclease H-inhibitor interaction and are likely to be useful for further improvements of the inhibitors.
Keywords: Reverse transcriptase, ribonuclease H, HIV, acylhydrazone, inhibitor, enzyme, protein, NMR
Reverse transcriptase (RT) plays a crucial role in HIV-1 replication and has thus proven to be an attractive target for the development of antiretroviral agents. Indeed, approximately half of the current clinically approved drugs for the treatment of HIV-1 infection target RT, specifically, the DNA polymerase activity of the enzyme (1-6). However, HIV-1 RT is a bifunctional enzyme, possessing ribonuclease H (RNH) activity in addition to its DNA polymerase activity. These two enzymatic activities reside in spatially distinct active sites on RT. Like RT DNA polymerase activity, RT-RNH activity is absolutely required for viral replication (7-10), and, thus, RT-RNH represents a potential target for antiretroviral drug development. Although a number of RT-RNH inhibitors have been described (11-19), there are no clinically approved anti-AIDS drugs targeting this enzyme activity, nor are there any in advanced preclinical development.
One of the reasons for the difficulty in development of HIV RNH inhibitors may be the lack of detailed structural information about the interaction of potent RNH inhibitors with the RT-RNH domain. A previous structural study of RT in the presence of an N-acylhydrazone analogue, DHBNH, demonstrated that this RNH-specific inhibitor bound the RT polymerase domain, and not to the RNH site (20). Recently, crystal structures have been solved for the active site-directed RNH inhibitor β-thujaplicinol bound to the RNH domain of RT and to an isolated RT-RNH domain fragment (21); another RNH active site-directed compound, pyrimidinol carboxylic acid, was found to bind a catalytically active RT-RNH domain fragment (22). While these structures are significant advances, neither of these compounds has antiviral activity, and thus the importance of this structural information to the development of therapeutics targeting RT-RNH is still unclear.
We have been studying acylhydrazones as potential inhibitors of RT-RNH (13, 20, 23, 24). This chemical class is interesting, as a number of the compounds that we have developed possess antiviral activity (13, 20). Thus, understanding how these compounds interact with RT-RNH may provide information for the design of new analogs with potential therapeutic utility. In the present work, we investigated the interaction of several acylhydrazones with RT and with an RT-RNH domain fragment using a variety of methodologies including protein NMR. The RT-RNH fragment used in these studies, p15-EC, is a chimera created by replacing a small loop segment of HIV-1 RT-RNH with a 24 residue α-helical substrate-binding loop derived from Escherichia coli RNHI into the HIV RT RNH domain (Figure 1). This modification confers catalytic activity to the RNH fragment. Since the native isolated RT RNH domain fragment does not exhibit measurable RNH activity, the p15-EC chimeric construct has been widely used to screen RNH inhibitors and to characterize the protein-inhibitor interactions (25-27).
Figure 1.

Primary sequence of p15-EC RNH fragment (1-148 residues). Numbers at the beginning of each line indicate amino acid positions relative to chain A of HIV RT RNH domain sequence. The E.coli sequence introduced into HIV RT RNH domain fragment is underlined. To present the NMR results conducted using the RNH fragment, the fragment residue numbers are described with the RT residues number in parentheses.
RESULTS AND DISCUSSION
Inhibition of HIV-1 RT-RNH by acylhydrazones
Our previous crystal structure of an acylhydrazone (DHBNH) bound to the polymerase domain of RT suggested possible structural alterations of the inhibitor that might provide additional contacts with RT and thus improve inhibitory potency (20). We, therefore, synthesized compounds in which the fused naphthyl ring system of DHBNH was replaced with a flexible and extended biphenyl system possessing a carboxylate moiety in the distal phenyl ring (Table 1), with the hypothesis that this carboxylate might form ionic interactions with the amino group of K223 in the RT polymerase domain. The 3,4-dihydroxy and 3,4,5-trihydroxy benzoyl structures donated by the acylhydrazide (see references in Himmel et al. (20)) were maintained. The new compounds provided interesting inhibition phenotypes. The trihydroxy compound, termed BHMP07, inhibited both RT polymerase and RT-RNH activities, whereas the dihydroxy analog, BHMP03, inhibited RT-RNH activity only (Table 1). Unlike the dihydroxy compounds DHBNH and BHMP03, the trihydroxy BHMP07 showed potent inhibition of the p15-EC RNH. Although both BHPM03 and BHMP07 bound to p15-EC RNH in a saturable manner as determined by the quenching of intrinsic protein fluorescence (Figure 2), the interaction of BHMP07 with the protein was substantially stronger than that of BHMP03. BHMP03 and BHMP07 are more soluble in aqueous solution than the naphthyl-based DHBNH and thus were more readily used for solution NMR studies.
Table 1.
Inhibitory properties of acylhydrazones used in the present study
| PROPERTY |
|

|
|
|---|---|---|---|
| DHBNHa | BHMP03 | BHMP07 | |
|
| |||
| Inhibition of RT DNA polymerase activity (IC50, μM) | > 25 | > 50 | 0.3 ± 0.2 |
|
| |||
| Inhibition of RT RNH (IC50, μM) | 0.5 ± 0.2 | 0.4 ± 0.2 | 0.2 ± 0.1 |
|
| |||
| Inhibition of the p15-EC RNH fragment (IC50, μM) | 18.5 ± 3.4 | > 50 | 0.8 ± 0.06 |
Data were taken from the reference, (20). Enzyme activity was measured as described in Materials and Methods.
Figure 2.

Interaction of BHMP03 (▽) or BHMP07 (▲) with the p15-EC RNH domain fragment monitored by intrinsic protein fluorescence in the presence of 2 mM Mn2+. The observed half maximal interaction values determined by the fluorescence quenching experiment for BHMP03 and BHMP07 were 23.1 and 5.3 μM, respectively.
Characterization of p15-EC RT-RNH for NMR studies
The 1H-15N HSQC spectrum of the p15-EC sample revealed excellent chemical shift dispersion and line widths, indicating stable folding of the recombinant protein. The signal positions in the HSQC spectrum were essentially identical to those observed previously (28) (Figure 3). With the exception of 8 residues that may undergo chemical exchange broadening, amide 15N and 1H signal positions were assigned.
Figure 3.

Overlay of 1H-15N HSQC spectra of the RNH fragment recorded in the absence (black) or presence of 5, 10, and 20 mM of Mg2+ in 40 mM sodium phosphate buffer at 25 °C (black, orange, light blue, and pink, respectively). Residues that showed distinct chemical shift differences in the presence or absence of Mg2+ are labeled in the spectra.
The 1H-15N HSQC spectra recorded in the absence or presence of 20 mM Mg2+ (200:1 molar ratio of Mg2+ to p15-EC RNH protein) showed clear residue specific effects of the divalent metal (Figure 3). Residues exhibiting significant changes in chemical shifts were observed in four clustered regions, all of which were in positions close to the active site (Figure 4, inset). The crystal structure of the HIV-1 RT RNH domain shows two metal ions bound to the active site residues, D17, E52, D72, and D137 (corresponding to catalytically essential RT-RNH residues, D443, E478, D498, and D549, respectively) (29). The 1H-15N HSQC spectra show that the presence of Mg2+ induced significant chemical shift changes in E52 (E478) and D72 (D498) that are located in the α-helix and a loop region. In contrast, significant shift change was not observed for D17 (D443), most likely because the backbone amide of D17 forms a hydrogen bond with a carbonyl of G30 (G456) in a β-sheet structure that is not perturbed upon Mg2+ titration. We were unable to assign the signal of D137 (D549) due to signal overlap or broadening. Overall, Mg2+ affects primarily the active site region (metal binding sites) of RNH and does not lead to global protein structural perturbation, suggesting that the Mg2+ ions are specifically coordinated to the protein as previously observed in the E. coli RNHI and RT RNH crystal structures (29-31). We did not examine effects of Mg2+ at concentrations higher than 20 mM since physiologically relevant intracellular total Mg2+ levels are on the order of 10 mM. These localized effects of Mg2+ on the p15-EC RNH contrast with the solution effects of Mg2+ on the isolated non-chimeric and catalytically inactive RT-RNH domain fragment where the presence of divalent metal cation induces global effects on RT-RNH in solution (32).
Figure 4.

Differences in backbone amide chemical shifts of the RNH fragment in the presence or absence of 20 mM Mg2+. The magnesium-induced shift in the RNH was calculated as the square root of the sum of the square of the 1H and 15N chemical shift difference. The resonances were considered shifted when the difference was greater than 20 Hz, based on resolution and signal broadenings. In the inserted model structure (See Materials and Methods), residues that exhibited significant chemical shift changes (> 20 Hz) are highlighted with pink in the backbone, and previously described metal coordinating side chains of D17(443), E52(478), D72(498), and D137(549) (29-31) are shown in yellow.
Effect of BHMP07 on NMR chemical shift changes of p15-EC RNH
Addition of BHMP07 to p15-EC RNH (titrated up to 4:1 molar ratio of inhibitor to protein) in the absence of Mg2+ resulted in shifts of several protein residue amide peaks in the 1H-15N HSQC spectrum of the RNH domain fragment (Figure 5A and Supplementary Figure S1). The residues affected by BHMP07 included D73 (D499), A76 (A502), I79 (I505), I80 (I506), R90 (in the loop-helix component taken from E. coli RNHI), and I114 (I526) (the corresponding RT RNH residue numbers are listed in parentheses). These shifts demonstrate specific inhibitor-protein interaction. Generally, changes in protein residue chemical shifts upon titration with ligand, rather than generation of a new set of peaks, occur when the weighted sum of the ligand on/off rates is sufficiently faster than the difference in chemical shifts between the ligand-bound and free forms (33). When the on-rate is close to the diffusion limit, the interaction of the RNH fragment with BHMP07 that shows such a fast exchange is expected to be relatively weak.
Figure 5.

Differences in backbone amide chemical shift of the RNH fragment in the presence or absence of four-fold BHMP07 recorded in the absence (A) or presence (B) of 20 mM Mg2+. The BHMP07-induced shift in the RNH was calculated as the square root of the sum of the square of the 1H and 15N chemical shift difference. The resonances were considered shifted when the difference was greater than 20 Hz, based on resolution and signal broadenings. The region inserted from E. Coli RNH is indicated by a solid-line arrow (residues 81 to 103) and the “substrate handle region” is indicated by a dashed arrow (residues 72 to 110).
The residues exhibiting chemical shift perturbation upon BHMP07 addition were mostly localized between residues 72 and 110, which includes the “substrate binding handle” region (residues 81 to 104) (Figures 5A and 6A). In addition to the changes in backbone signals, the Nε proton of W83 in this loop-helix also showed remarkable changes in chemical shift (Supplementary Figure S1B), suggesting hydrophobic interactions of this residue with the inhibitor and is consistent with the effect of the compound on quenching of the intrinsic p15-EC RNH fluorescence (Figure 2). Overall, the observed changes in chemical shifts indicate that the substrate handle region provides an important primary interaction site for BHMP07.
Figure 6.

Ribbon representation of the p15-EC RNH fragment in the absence (A) or presence (B) of 20 mM Mg2+, and of the HIV-1 RT (C). In (A) and (B), the residues that were perturbed by BHMP07 are highlighted red. In (C), the highlighted p15-EC structure (cyan) was superimposed on the RT structure (PDB 2I5J, p51 subunit (gray) and p66 subunit (lime)). The substrate handle region in p15-EC RNH is marked with a dashed circle, and is located at the domain-interface in RT.
BHMP03 also perturbed the NMR signals of the RNH fragment, such as D73 (D499), A76 (A502), I79 (I505), I80 (I506), I114, and V124 (Supplementary Figure S2). This chemical-shift perturbation by BHMP03 is similar to that of BHMP07, consistent with the fact that the chemical structure of BHMP03 is analogous to that of BHMP07 (Table 1). Since BHMP03 exhibits more than 50 times higher IC50 of the p15-EC RNH than BHMP07 (Table 1) and lower binding affinity (Figure 2), we focused our follow-up NMR experiments on the interaction of BHMP07 with p15-EC RNH.
To understand the impact of metal ions on the interaction between BHMP07 and RNH, we repeated our NMR titration studies, in the presence of 20 mM Mg2+. Similar to observations made in the absence of metal ions, significant changes in chemical shifts in the substrate handle region (including residues from 72 to 110) of p15-EC RNH were observed when BHMP07 was titrated in the presence of magnesium (Figures 5B and 6B), demonstrating homologous binding modes of BHMP07 in both the presence and absence of the metal. Close inspection of the changes in chemical shifts in Figure 5 indicates that the magnitude of the chemical shift changes in the presence of Mg2+ are not necessarily the same as those observed in the absence of Mg2+. For example, K88 and I114 (I526) exhibit larger changes in chemical shifts in the presence of Mg2+ whereas W87 and V124 (V536) exhibit an opposite tendency. In addition to these changes in the substrate handle region, residues close to the active site, G18 (444), A19 (445), and K28 (454), also exhibited changes in chemical shifts upon BHMP07 titration in the presence of Mg2+ (Figure 5B). Since the distance from the active site to the center of the substrate binding handle is more than 15 Å, the chemical shift perturbations at these residues may be due to indirect effects, for example, through the active site metals or through repositioning of the inhibitor due to interference with the metals.
BHMP07 - p15-EC RNH dissociation constants
The chemical shift perturbations upon BHMP07 titration were used to determine an approximate inhibitor-dissociation constant (KD), assuming a simple two-state model (Figure 7). Although the observed changes in chemical shifts did not reach complete saturation, KD values were approximately ~200 μM in the absence of 20 mM Mg2+ (Table 2). In general, such weak binding could be due to non-specific interaction of the inhibitor to different hydrophobic surfaces in the protein. However, it is not the case. The NMR titration experiment provides information of the effects of inhibitor binding at individual sites. As described in Table 2, KD values were obtained consistently among different sites in the substrate binding handle for the backbone and for tryptophan side chain protons. The binding appears to be cooperative, demonstrating specific interaction of BHMP07 to the p15-EC RNH fragment.
Figure 7.
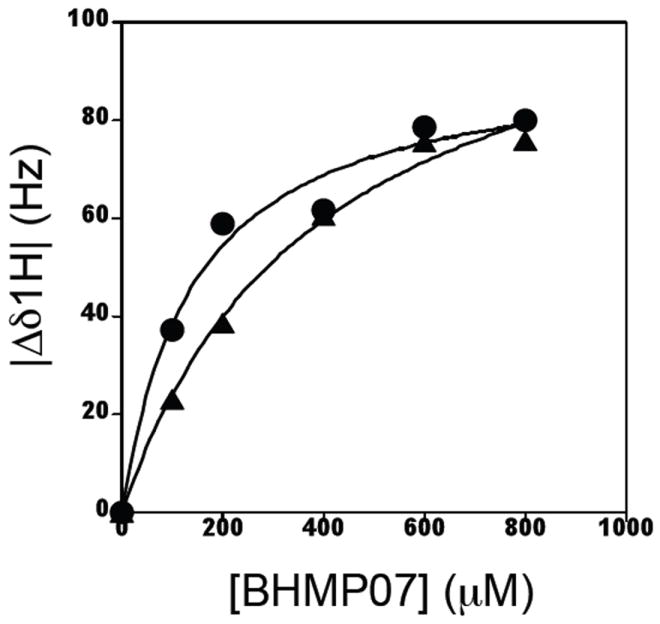
Interaction of BHMP07 with the p15-EC RNH domain fragment monitored by resonance shifts of the NMR signal of W83 in the absence (●, circle) or presence (▲, triangle) of 20 mM Mg2+. Dissociation constants, KD, determined by NMR for BHMP07 were 142.7 ± 35.7 μM and 392.0 ± 69.6 μM in the absence and presence of 20 mM Mg2+, respectively.
Table 2.
Site specific investigation of the BHMP07 dissociation constants against the p15-EC RNH
| RNH fragment residue (RT residue) | Inhibitor dissociation constant, KD (μM)
|
|
|---|---|---|
| No Mg++ | + 20 mM Mg++ | |
|
| ||
| Q74 (500) | - | 246.4 ± 72.8 |
| Y75 (501) | - | 262.4 ± 134.8 |
| A76 (502) | 191.9 ± 28.9 | 311.4 ± 41.5 |
| I79 (505) | 211.6 ± 20.1 | 371.6 ± 60.2 |
| W83a | 142.7 ± 35.7 | 392.0 ± 69.6 |
| K93 | 193.8 ± 18.3 | 454.1 ± 150.6 |
| I114 (526) | 216.8 ± 38.4 | 320.9 ± 26.2 |
| V124 (536) | 242.5 ± 18.2 | - |
Detected for indole amine signal.
The binding became weaker (KD, ~400 μM) in the presence of 20 mM Mg2+ (Figure 7 and Table 2). While the increase in the KD in the presence of Mg2+ suggests that BHMP07 binding may be competitive with Mg2+, we consider this unlikely for two reasons. First, if BHMP07 competes with Mg2+ for binding to p15-EC RNH, then large changes in chemical shifts should be observed in Mg2+ binding sites. However, the residues at or around the four major Mg2+ coordinating residues, including D17 (D443) and E52 (E478), did not show any significant changes upon BHMP07 interaction (Figure 5B). Secondly, the regions that exhibit chemical shift changes upon BHMP07 interaction are very similar, in the presence or absence of Mg2+, showing the major interaction site in the substrate handle region. Thus, the Mg2+ effect on KD is not likely due to direct competition, but could be due to minor interaction or indirect structural changes caused by metal binding.
The KD value determined by NMR (KD, ~400 μM, Figure 7 and Table 2) is 10 fold larger than those estimated by the fluorescence quenching data (Figure 2) and the in vitro inhibitory potency (Table 1). This is not surprising because the NMR experiments were performed at significantly higher protein concentration (~200 μM) at pH 7 than those of fluorescence quenching and in vitro assays (low μM) at pH 8 in the presence of Mn2+. In such NMR titrations that use high inhibitor concentrations, high solubility of the inhibitor, to levels equal or greater than that of the protein, is critical. Even though BHMP07 exhibits higher aqueous solubility than DHBNH, BHMP07 is not likely completely soluble in aqueous solution at the levels used in the NMR studies, and some inhibitor precipitation was noted following overnight incubation. These solubility issues will impact the apparent KD for BHMP07 determined by NMR. Nonetheless, it is important to note that the NMR data demonstrate specific interaction of BHMP07 to p15-EC RNH even at these high inhibitor concentrations.
Effect of mutations of RT residues identified as interacting with acylhydrazones
HIV-1 RT consists of two subunits of 51 kDa (p51) and 66 kDa (p66), with the RNH domain comprising a 15 kDa carboxy-terminus segment of the p66 subunit (Figure 7C). The interaction site for BHMP07 in the p15-EC RNH fragment is primarily conserved in intact p66/p51 heterodimeric HIV-1 RT. We evaluated the impact of mutating acylhydrazone-interacting residues in the latter protein on the inhibitory potency of the compounds. In the p15-EC RNH fragment, residue A76 (which corresponds to A502 in RT) exhibited significant chemical shift upon addition of BHMP07 (Figure 5). The A502G mutation in p66/p51 heterodimeric RT resulted in substantially reduced sensitivity to inhibition of RT-RNH activity by all acylhydrazones tested, as did the A502F mutation (Table 3). Residue I79 in p15-EC (I505 in RT) also showed considerable chemical shifts upon BHMP07 binding. The I505V mutation had no effect on acylhydrazone inhibitory potency. This is perhaps not surprising given the conservative nature of this substitution that would maintain the potential to form hydrophobic interactions with the inhibitor. Mutations of this residue in RT to amino acids other than valine (such as I505F) completely abrogated RT-RNH activity (data not shown).
Table 3.
Effect of mutations of inhibitor-interacting RNH residues in RT on inhibition of RT RNH activity by acylhydrazones.
| INHIBITOR | IC50 for inhibition of RT-RNH activity (μM)
|
||||
|---|---|---|---|---|---|
| WT | A502G (A76)a |
A502F (A76) |
I505Vb (I79) |
A502G+D256G | |
|
| |||||
| DHBNH | 0.9 ± 0.3 | 13.7 ± 5.2 | > 10 | 0.9 ± 0.1 | > 50 |
| BHMP03 | 0.9 ± 0.2 | 4.2 ± 1.1 | > 10 | 1.0 ± 0.2 | 6.5 ± 2.1 |
| BHMP07 | 0.2 ± 0.1 | 1.0 ± 0.7 | > 10 | 0.3 ± 0.3 | 4.0 ± 1.3 |
The residue numbers in the parentheses indicate those in the RNH fragment sequence
Mutations other than I505V eliminated RT RNH activity and thus could not be studied.
NMR studies indicate that both A76 and I79 likely form part of the acylhydrazone interaction site in the RNH domain. However, NMR also shows that residues in the inserted loop-helix region from E. coli RNHI that confers catalytic activity to the fragment also contribute to acylhydrazone binding. The equivalent residues to A76 and I79 (A502 and I505, respectively) in the p66/p51 RT heterodimer are positioned near the p66 – p51 subunit interface. Structural alignment of the p15-EC RNH fragment with the RT structure indicates that the additional component in the fragment may mimic the inter-subunit interaction at the interface of the RT p51 subunit with the RT RNH domain in the p66 subunit (Figure 6C). Residue D256 in the RT p51 subunit is located at the subunit interface with the RNH domain of p66, close to the RNH active site. Introduction of the D256G mutation in the p51 subunit of RT, in addition to the A502G mutation identified from our NMR studies, resulted in 20-fold reduced inhibitory potency for BHMP07 (Table 3), which may also demonstrate importance of the domain interface for the inhibitor interaction.
Comparison of the identified BHMP07 interaction sites with those of other RT RNH inhibitors
Several classes of HIV-1 RNH inhibitors have been reported (6, 34). Biochemical studies have shown that some of these inhibitors directly recognize the active site of RNH, primarily through their metal-binding characteristics (14, 27, 35). Recent crystal structures clearly demonstrate interaction of two such inhibitors, β-thujiplicinol (21) and a pyrimidinol derivative (22) with the active-site metals in RT or in an isolated RNH fragment. These structures are significant advances, but both describe active site-directed inhibitors whereas BHMP07 appears to bind outside of the RNH active site (Figure 6). Furthermore, neither of the compounds used in the crystal studies has antiviral activity, whereas acylhydrazones have been found to possess antiviral activity (13, 20). HIV-1 RT RNH activity is optimal at Mg2+ concentrations above 5 mM, but intracellular free Mg2+ is generally considered to be much lower (36). Therefore, it is perhaps not surprising that the Mg2+-mediated inhibitors might be disadvantaged in vivo. Our data indicate that acylhydrazones are likely to interact with RT RNH in a manner independent of Mg2+ as the primary binding site of the acylhydrazone inhibitor was identified as the substrate handle region rather than the metal binding site. While it is possible that the inhibitor binds to another (allosteric) site, thereby inducing large chemical shifts in the substrate handle region, such significant conformational changes have never been seen in previous studies (21, 22). We thus feel that our present study demonstrates a clear structural distinction in the potential inhibition mechanism of the acylhydrazone inhibitors from that of the active site directed metal-mediated inhibitors.
The binding site found in this study may reasonably explain why an N-acylhydrazone analogue, DHBNH, was not soaked into the RNH specific site in the previous crystal structure, but only into the RT polymerase site (20). In E. Coli RNHI, the substrate handle region is basically rigid but contains dynamics in different time scales (37, 38). The substrate-handle region of p15-EC RNH is expected to have such dynamics. Consistently, the domain interfaces of RT RNH, a part of which the p15-EC RNH mimics, is not rigid in solution either (39). Thus, the flexibility that allows inhibitor binding may be frozen in the RT crystal structure, and thus impact the ability of the acylhydrazone to bind.
CONCLUSIONS
Although small molecule inhibitors of RT RNH have been screened, little structural information is available about the interactions between RT RNH. In this report, we describe NMR studies of the interaction of a new RNH inhibitor, BHMP07, with a catalytically active HIV-1 RT RNH domain fragment. Our data indicate that acylhydrazones are likely to interact with RT RNH in a manner independent of Mg2+ with the primary binding site at the substrate handle region. This demonstrates a clear structural distinction in the potential inhibition mechanism of the acylhydrazone inhibitors from that of the active site directed metal-mediated inhibitors, providing guidance for future experiments for RNH inhibitor design and the structural characterization.
MATERIALS and METHODS
Synthesis of acylhydrazones
Acylhydrazones were synthesized by condensation of the aromatic aldehyde with the corresponding acid hydrazide, essentially as described (40). As an example, BHMP07 was prepared by the drop-wise addition of 3, 4, 5-trihydroxybenzyhydrazide (1.1 mmol in 10 ml ethanol) to a solution of 3’-formyl-[1, 1’-biphenyl]-4-carboxylic acid (1 mmol in 4% acetic acid in ethanol) with stirring, while heating in a boiling water bath. Heating and stirring were continued for 20 minutes following completion of addition of the ethanolic acid hydrazide solution, and then the mixture was allowed to cool to room temperature. The resulting precipitate was collected by filtration, washed with cold ethanol and diethyl ether, and dried. Elemental and mass spectral analyses were consistent with the expected structure. Stock solutions of the acylhydrazones were prepared in dimethyl sulfoxide (DMSO) and used for assays and the NMR experiments.
Protein preparation and assay
Plasmid p6HRT (a gift from Dr. S. Le Grice, NCI-Frederick, Frederick, MD) expresses wild-type p66/p51 heterodimeric HIV-1 RT (Le Grice & Gruniger-Leitch, 1990). Mutations I505V, A502G, and D256G/A502G were introduced into this plasmid using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). The presence of the appropriate mutations was verified by sequencing. Heterodimeric p66/p51 RT (wild type and mutant forms) was expressed and purified essentially as previously described (Fletcher et al., 1996). Plasmid pCSR231 encoding a codon-optimized chimeric HIV-1 RNH domain fragment protein containing an α-helical substrate-binding loop derived from E. coli RNase HI (25-27) was a generous gift from Dr. Daria Hazuda (Merck, West Point, PA). This protein, termed p15-EC, was overexpressed and purified as described below. RT RNA-dependent DNA polymerase activity was measured as described (13). Ribonuclease H activities of the RT p66/p51 heterodimer and the p15-EC RNH domain fragment were determined using a rapid fluorescence assay (24), essentially as described, except that assays with RT p66/p51 used 10 mM Mg2+ as metal cofactor whereas assays with p15-EC used 2 mM Mn2+ as cofactor since this chimeric fragment shows reduced catalytic activity with Mg2+.
Fluorescence quenching measurements
All experiments were done at ambient temperature (23 - 25°C) using a PTI-QM-6/2500 Steady State Spectrofluorometer, with excitation at 295 nm and emission at 350 nm. Briefly, aliquots of solutions of BHMP03 or BHMP07 in DMSO were titrated into 393 μl of a solution of p15-EC RNH (2 μM) in 50 mM Tris buffer (pH 8.0) containing 60mM KCl, and 2mM MnCl2. The final volume after completion of acylhydrazone addition was 400 μl, thus the final DMSO in the sample was less than 2%. The experiment was recorded twice, and data points are the averages of two independent sets of measurements.
Isotopic labeling of p15-EC RNH
Isotopic labeling of proteins for NMR experiments was carried out by growing the pCSR231-transformed bacterial cell culture in a modified M9 minimal media containing 15NH4Cl (1 g/l) as the sole nitrogen source in the absence or in the presence of [u-13C]-glucose as the sole carbon source. Over-expressed protein was purified using HiTrapTM Q SP Sepharose ion exchange and a HiTrap SP columns (GE Healthcare), equilibrated with 25 mM sodium phosphate buffer (pH 7.0), 0.02% sodium azide, and 1 mM dithiothreitol (DTT). Protein was eluted using a linear gradient of 1M NaCl. RNH was further purified by size exclusion chromatography on a Superdex75 26/60 column (GE Healthcare) using buffer containing 25 mM sodium phosphate buffer (pH 7.0), 0.02% sodium azide, 100 mM NaCl and 1 mM DTT. The resulting p15-EC RNH was homogeneous as determined by 20% SDS-PAGE gel electrophoresis. Protein was stored in aliquots at -70°C until use.
NMR experiments
All spectra were recorded on Bruker AVANCE600 or AVANCE700 spectrometers equipped with a 5mm-triple-resonance-z-gradient cryogenic probe (Bruker Biospin, Billerica, MA) at 25°C. The p15-EC RNH sample was prepared in 40 mM sodium phosphate, (pH 6.8) in a 95% H2O/5%D2O ratio in a sample volume of ~350μL (final concentration ~ 200 μM protein) in a 5-mm Shigemi tube (Shigemi, Inc., Allison Park, PA). A series of 1H-15N Heteronuclear Single Quantum Coherence (HSQC) experiments were recorded at different Mg2+ concentrations (0, 5, 10, and 20 mM) to identify the Mg2+ interaction sites. Although the above assay was conducted at 10 mM, we tested various concentrations up to 20 mM Mg2+ in the NMR studies due to the high protein concentrations needed for this approach. Similarly, a series of 1H-15N HSQC experiments were recorded at different concentration of BHMP07 (0, 0.1, 0.2, 0.4, 0.6, 0.8 mM) to identify the inhibitor-interaction site in the presence and absence of 20 mM Mg2+. Each series of the titration experiment was conducted using the same condition and completed within a half day. Changes in chemical shifts were presented as the square root of the sum of the square of the 1H and 15N chemical shift differences, (Hz). Backbone 1H-15N signals were assigned by recording HNCA, CBCAONH and CCONH experiments using 15N/13C-labeled protein at ~500 μM concentration (41). NMR data were processed and analyzed using the nmrDraw and nmrView (42, 43).
Model structure of the p15-EC RNH fragment
A model structure of the p15-EC RNH fragment was generated by inserting the sequence, TQWIHNWKKRGWKTADKKPVKNV (Supplementary Figure S1), to the RT RNH domain coordinates (PDB = 2I5J) using Modeller 9v6 software. This modeled structure was used to indicate residues that exhibited changes in NMR chemical shifts enabling an estimation of the binding location for the acylhydrazone inhibitors. Graphics were generated using VMD 1. 8. 6 (44).
Supplementary Material
Acknowledgments
This study was supported by grants from the National Institutes of Health (AI077424 to R.I. and M.A.P., and AI073975 to M.A.P.), the National Science Foundation (MCB 0814905 to R.I.), and funds from the University of Pittsburgh. We thank Jinwon Jung, Jason A. Concel and Eva Nagy for technical assistance in this work.
Footnotes
SUPPORTING INFORMATION AVAILABLE
Three figures, that include amino acid sequence of p15-EC RNH applied in this study, and HSQC spectra in the absence and presence of BHMP07 and BHMP03, are available as supporting information.
References
- 1.Sarafianos SG, Das K, Hughes SH, Arnold E. Taking aim at a moving target: designing drugs to inhibit drug-resistant HIV-1 reverse transcriptases. Curr Opin Struct Biol. 2004;14:716–30. doi: 10.1016/j.sbi.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 2.De Clercq E. Non-nucleoside reverse transcriptase inhibitors (NNRTIs): past, present, and future. Chem Biodivers. 2004;1:44–64. doi: 10.1002/cbdv.200490012. [DOI] [PubMed] [Google Scholar]
- 3.Basavapathruni A, Anderson KS. Reverse transcription of the HIV-1 pandemic. FASEB J. 2007;21:3795–808. doi: 10.1096/fj.07-8697rev. [DOI] [PubMed] [Google Scholar]
- 4.Sluis-Cremer N, Tachedjian G. Mechanisms of inhibition of HIV replication by non-nucleoside reverse transcriptase inhibitors. Virus Res. 2008;134:147–56. doi: 10.1016/j.virusres.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jochmans D. Novel HIV-1 reverse transcriptase inhibitors. Virus Res. 2008;134:171–85. doi: 10.1016/j.virusres.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 6.Ilina T, Parniak MA. Inhibitors of HIV-1 reverse transcriptase. Adv Pharmacol. 2008;56:121–67. doi: 10.1016/S1054-3589(07)56005-9. [DOI] [PubMed] [Google Scholar]
- 7.Peliska JA, Benkovic SJ. Mechanism of DNA strand transfer reactions catalyzed by HIV-1 reverse transcriptase. Science. 1992;258:1112–8. doi: 10.1126/science.1279806. [DOI] [PubMed] [Google Scholar]
- 8.DeStefano JJ, Bambara RA, Fay PJ. Parameters that influence the binding of human immunodeficiency virus reverse transcriptase to nucleic acid structures. Biochemistry. 1993;32:6908–15. doi: 10.1021/bi00078a014. [DOI] [PubMed] [Google Scholar]
- 9.Cirino NM, Cameron CE, Smith JS, Rausch JW, Roth MJ, Benkovic SJ, et al. Divalent cation modulation of the ribonuclease functions of human immunodeficiency virus reverse transcriptase. Biochemistry. 1995;34:9936–43. doi: 10.1021/bi00031a016. [DOI] [PubMed] [Google Scholar]
- 10.Ghosh M, Williams J, Powell MD, Levin JG, Le Grice SF. Mutating a conserved motif of the HIV-1 reverse transcriptase palm subdomain alters primer utilization. Biochemistry. 1997;36:5758–68. doi: 10.1021/bi963045e. [DOI] [PubMed] [Google Scholar]
- 11.Loya S, Hizi A. The interaction of illimaquinone, a selective inhibitor of the RNase H activity, with the reverse transcriptases of human immunodeficiency and murine leukemia retroviruses. J Biol Chem. 1993;268:9323–8. [PubMed] [Google Scholar]
- 12.Palaniappan C, Fay PJ, Bambara RA. Nevirapine alters the cleavage specificity of ribonuclease H of human immunodeficiency virus 1 reverse transcriptase. J Biol Chem. 1995;270:4861–9. doi: 10.1074/jbc.270.9.4861. [DOI] [PubMed] [Google Scholar]
- 13.Borkow G, Fletcher RS, Barnard J, Arion D, Motakis D, Dmitrienko GI, et al. Inhibition of the ribonuclease H and DNA polymerase activities of HIV-1 reverse transcriptase by N-(4-tert-butylbenzoyl)-2-hydroxy-1-naphthaldehyde hydrazone. Biochemistry. 1997;36:3179–85. doi: 10.1021/bi9624696. [DOI] [PubMed] [Google Scholar]
- 14.Budihas SR, Gorshkova I, Gaidamakov S, Wamiru A, Bona MK, Parniak MA, et al. Selective inhibition of HIV-1 reverse transcriptase-associated ribonuclease H activity by hydroxylated tropolones. Nucleic Acids Res. 2005;33:1249–56. doi: 10.1093/nar/gki268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shaw-Reid CA, Feuston B, Munshi V, Getty K, Krueger J, Hazuda DJ, et al. Dissecting the effects of DNA polymerase and ribonuclease H inhibitor combinations on HIV-1 reverse-transcriptase activities. Biochemistry. 2005;44:1595–606. doi: 10.1021/bi0486740. [DOI] [PubMed] [Google Scholar]
- 16.Tramontano E, Esposito F, Badas R, Di Santo R, Costi R, La Colla P. 6-[1-(4-Fluorophenyl)methyl-1H-pyrrol-2-yl)]-2,4-dioxo-5-hexenoic acid ethyl ester a novel diketo acid derivative which selectively inhibits the HIV-1 viral replication in cell culture and the ribonuclease H activity in vitro. Antiviral Res. 2005;65:117–24. doi: 10.1016/j.antiviral.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 17.Schultz SJ, Champoux JJ. RNase H activity: structure, specificity, and function in reverse transcription. Virus Res. 2008;134:86–103. doi: 10.1016/j.virusres.2007.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wendeler M, Lee HF, Bermingham A, Miller JT, Chertov O, Bona MK, et al. Vinylogous ureas as a novel class of inhibitors of reverse transcriptase-associated ribonuclease H activity. ACS Chem Biol. 2008;3:635–44. doi: 10.1021/cb8001039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sarafianos SG, Marchand B, Das K, Himmel DM, Parniak MA, Hughes SH, et al. Structure and function of HIV-1 reverse transcriptase: molecular mechanisms of polymerization and inhibition. J Mol Biol. 2009;385:693–713. doi: 10.1016/j.jmb.2008.10.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Himmel DM, Sarafianos SG, Dharmasena S, Hossain MM, McCoy-Simandle K, Ilina T, et al. HIV-1 reverse transcriptase structure with RNase H inhibitor dihydroxy benzoyl naphthyl hydrazone bound at a novel site. ACS Chem Biol. 2006;1:702–12. doi: 10.1021/cb600303y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Himmel DM, Maegley KA, Pauly TA, Bauman JD, Das K, Dharia C, et al. Structure of HIV-1 reverse transcriptase with the inhibitor beta-Thujaplicinol bound at the RNase H active site. Structure. 2009;17:1625–35. doi: 10.1016/j.str.2009.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kirschberg TA, Balakrishnan M, Squires NH, Barnes T, Brendza KM, Chen X, et al. RNase H active site inhibitors of human immunodeficiency virus type 1 reverse transcriptase: design, biochemical activity, and structural information. J Med Chem. 2009;52:5781–4. doi: 10.1021/jm900597q. [DOI] [PubMed] [Google Scholar]
- 23.Sluis-Cremer N, Arion D, Parniak MA. Destabilization of the HIV-1 reverse transcriptase dimer upon interaction with N-acyl hydrazone inhibitors. Mol Pharmacol. 2002;62:398–405. doi: 10.1124/mol.62.2.398. [DOI] [PubMed] [Google Scholar]
- 24.Parniak MA, Min KL, Budihas SR, Le Grice S, Beutler JA. A fluorescence-based high-throughput screening assay for inhibitors of human immunodeficiency virus-1 reverse transcriptase-associated ribonuclease H activity. Anal Biochem. 2003;322:33–9. doi: 10.1016/j.ab.2003.06.001. [DOI] [PubMed] [Google Scholar]
- 25.Stahl SJ, Kaufman JD, Vikić-Topić S, Crouch RJ, Wingfield PT. Construction of an enzymatically active ribonuclease H domain of human immunodeficiency virus type 1 reverse transcriptase. Protein Eng. 1994;7:1103–8. doi: 10.1093/protein/7.9.1103. [DOI] [PubMed] [Google Scholar]
- 26.Keck JL, Marqusee S. Substitution of a highly basic helix/loop sequence into the RNase H domain of human immunodeficiency virus reverse transcriptase restores its Mn(2+)-dependent RNase H activity. Proc Natl Acad Sci U S A. 1995;92:2740–4. doi: 10.1073/pnas.92.7.2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shaw-Reid CA, Munshi V, Graham P, Wolfe A, Witmer M, Danzeisen R, et al. Inhibition of HIV-1 ribonuclease H by a novel diketo acid, 4-[5-(benzoylamino)thien-2-yl]-2,4-dioxobutanoic acid. J Biol Chem. 2003;278:2777–80. doi: 10.1074/jbc.C200621200. [DOI] [PubMed] [Google Scholar]
- 28.Kern G, H T, Marqusee S. Characterization of a folding intermediate from HIV-1 ribonuclease H. Protein Sci. 1998;7:2164–74. doi: 10.1002/pro.5560071014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Davies JFn, Hostomska Z, Hostomsky Z, Jordan SR, Matthews DA. Crystal structure of the ribonuclease H domain of HIV-1 reverse transcriptase. Science. 1991;252:88–95. doi: 10.1126/science.1707186. [DOI] [PubMed] [Google Scholar]
- 30.Katayanagi K, Miyagawa M, Matsushima M, Ishikawa M, Kanaya S, Ikehara M, et al. Three-dimensional structure of ribonuclease H from E. coli. Nature. 1990;347:306–9. doi: 10.1038/347306a0. [DOI] [PubMed] [Google Scholar]
- 31.Yang W, Hendrickson WA, Crouch RJ, Satow Y. Structure of ribonuclease H phased at 2 A resolution by MAD analysis of the selenomethionyl protein. Science. 1990;249:1398–405. doi: 10.1126/science.2169648. [DOI] [PubMed] [Google Scholar]
- 32.Pari K, Mueller GA, DeRose EF, Kirby TW, London RE. Solution structure of the RNase H domain of the HIV-1 reverse transcriptase in the presence of magnesium. Biochemistry. 2003;42:639–50. doi: 10.1021/bi0204894. [DOI] [PubMed] [Google Scholar]
- 33.McConnell HM. Reaction Rates by Nuclear Magnetic Resonance. Journal of Chem Phys. 1958;28:430–1. [Google Scholar]
- 34.Klumpp K, Mirzadegan T. Recent progress in the design of small molecule inhibitors of HIV RNase H. Curr Pharm Des. 2006;12:1909–22. doi: 10.2174/138161206776873653. [DOI] [PubMed] [Google Scholar]
- 35.Klumpp K, Hang JQ, Rajendran S, Yang Y, Derosier A, Wong Kai In P, et al. Two-metal ion mechanism of RNA cleavage by HIV RNase H and mechanism-based design of selective HIV RNase H inhibitors. Nucleic Acids Res. 2003;31:6852–9. doi: 10.1093/nar/gkg881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goldschmidt V, Didierjean J, Ehresmann B, Ehresmann C, Isel C, Marquet R. Mg2+ dependency of HIV-1 reverse transcription, inhibition by nucleoside analogues and resistance. Nucleic Acids Res. 2006;34:42–52. doi: 10.1093/nar/gkj411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yamasaki K, Saito M, Oobatake M, Kanaya S. Characterization of the internal motions of Escherichia coli ribonuclease HI by a combination of 15N-NMR relaxation analysis and molecular dynamics simulation: examination of dynamic models. Biochemistry. 1995;34:6587–601. doi: 10.1021/bi00020a003. [DOI] [PubMed] [Google Scholar]
- 38.Mandel AM, Akke M, Palmer AGr. Dynamics of ribonuclease H: temperature dependence of motions on multiple time scales. Biochemistry. 1996;35:16009–23. doi: 10.1021/bi962089k. [DOI] [PubMed] [Google Scholar]
- 39.Seckler JM, Howard KJ, Barkley MD, Wintrode PL. Solution structural dynamics of HIV-1 reverse transcriptase heterodimer. Biochemistry. 2009;48:7646–55. doi: 10.1021/bi900790x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Edward JT, Gauthier M, Chubb FL, Ponka P. Synthesis of New Acylhydrazones as Iron-Chelating Compounds. J Chem Eng Data. 1988;33:538–40. [Google Scholar]
- 41.Cavanagh J, Fairbrother WJ, Palmer AGI, SKelton NJ. Protein NMR Spectroscopy. San Diego: Academic Press; 1996. [Google Scholar]
- 42.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. Nmrpipe - a Multidimensional Spectral Processing System Based on Unix Pipes. Journal of Biomolecular Nmr. 1995;6:277–93. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 43.Johnson BA. Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol Biol. 2004;278:313–52. doi: 10.1385/1-59259-809-9:313. [DOI] [PubMed] [Google Scholar]
- 44.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–8. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.